

SUMMARY
We present a novel approach for fluorescent in situ detection of short, single-copy sequences within genomic DNA in human cells. The single copy sensitivity and single base specificity of our method is achieved due to the combination of three components. First, a peptide nucleic acid (PNA) probe locally opens a chosen target site, which allows a padlock DNA probe to access the site and become ligated. Second, rolling circle amplification (RCA) generates thousands of single-stranded copies of the target sequence. Finally, fluorescent in situ hybridization (FISH) is used to visualize the amplified DNA. We validate this new technique by successfully detecting six unique target sites on human mitochondrial and autosomal DNA. We also demonstrate the high specificity of this method by detecting X- and Y- specific sequences on human sex chromosomes and by simultaneously detecting three unique target sites. Finally, we discriminate two target sites that differ by two nucleotides. The PNA-RCA-FISH approach is a unique in situ hybridization method capable of multi-target visualization within human chromosomes and nuclei that does not require DNA denaturation and is extremely sequence specific.
INTRODUCTION
Variations in the human genome are indicators of a number of diseases, predisposition to certain conditions, and abnormal responses to environmental factors. Therefore, sensitive techniques for detecting genomic mutations are critical for improvement of clinical diagnostics, and tremendous efforts have been invested into the development of molecular assays that analyse all ranges of genomic variations (Albertson and Pinkel, 2003).
The sizes of genomic variations range from millions of base pairs to single nucleotide polymorphisms (SNPs). Methods to study genome variations are as diverse as the mutations. They vary from PCR and high-throughput sequencing to microarray analysis and fluorescence in situ hybridization (FISH). Each of these methods has its advantages and limitations. Among other methods, FISH analysis has a unique and important place as an essential cytogenetic tool used in many areas of biological and biomedical research as well as in routine medical diagnostics. In conventional FISH techniques, specific DNA sequences are labelled with fluorescent dyes through denaturation of chromosome or interphase cells and hybridization with the complementary probes. Over the past years, there has been significant improvement in sensitivity and specificity of FISH (Volpi and Bridger, 2008). The resolution has also been enhanced due to advances in fluorescence microscopy and digital imaging (Hell, 2007). However, even with these improvements, FISH is limited to the detection of large genomic changes such as duplications, amplifications, deletions, and translocations that are at least 1–2 kilobases long (Halling and Kipp, 2007). This implies that FISH cannot be used to resolve small insertions and deletions that span several tens of base pairs, not to mention single nucleotide polymorphisms (SNPs), the most common source of genetic variation.
Padlock probes were introduced about a decade ago to detect single base variations in FISH format (Christian et al., 2001; Larsson et al., 2004; Lohmann et al., 2007). This technique is based on the extremely high sequence specificity of the ligation reaction that can discriminate single mutations if they are located close to the ligation point. Therefore, the padlock probes are designed in such a way that their 5′- and 3′-ends are complementary to the target DNA site with the mutation in the middle. When the padlock probe is hybridized to ssDNA it circularizes and the ligase closes the gap in the event of perfect complementarity. If there is a mismatch in the target, the ligase does not ligate the padlock ends and the circle is not formed. The next step in the assay is rolling circle amplification (RCA) that allows signal amplification. The RCA product is then detected by hybridization. Several attempts have been made to detect short DNA sequences in the human genome based on padlock probe design. Target-primed RCA is an approach that was used to detect point mutations in human mitochondrial DNA (Larsson et al., 2004). This method involves treatment of the target DNA with a restriction enzyme and exonuclease, then the use of the 3′-end of the target as a primer for RCA, and detection of the amplification product with fluorescent probes. This method can be used to detect individual DNA molecules with great specificity, but the efficiency of the detection is about 10%. Lohmann et al. attempted to employ target-primed RCA for detection of DNA on metaphase chromosomes (Lohmann et al., 2007). However, this method was only able to detect 1–10% of the target sites, and the authors decided that this technique is best suited for the targets that exist in multiple copies. It was concluded that the involvement of numerous enzymatic steps was significantly decreasing the efficiency of the detection. Thus, there still remains a need for an effective cytogenetic method to detect short, unique sequences in genomic DNA.
Here, we describe a novel approach for in situ visualization of short, single-copy DNA sequences within the human genome. The key step in our approach is the local opening of the double-stranded DNA with PNA openers followed by the padlock probe hybridization and rolling circle amplification (RCA). The RCA product is then detected by hybridization with fluorescent probes. We present evidence that our technique can be applied for visualization of unique sites in mitochondrial DNA and in DNA within the metaphase and interphase chromatin. We also show the potential of the method in multi-target detection and discrimination between sequence variants differing by two nucleotides. Finally, we demonstrate specificity of target detection by genotyping unique sequences on human X and Y-chromosomes in normal male and female cell lines.
RESULTS
Principle of the method
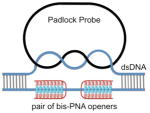
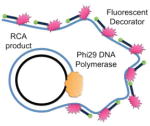
The first step in the procedure is the local opening of the target site in the duplex genomic DNA by a pair of peptide nucleic acid (PNA) oligomers. PNAs belong to a remarkable class of synthetic analogs of nucleic acids in which the negatively charged sugar–phosphate backbone has been replaced with a neutral peptide-like backbone. This feature gives PNAs the capacity to hybridize with high affinity and specificity to complementary RNA and DNA sequences, which has led to the development of a large variety of research and diagnostic assays (Blanco and Artero, 2010; Carbonari et al., 2011; Forrest, 2007; Singh et al., 2010). Here, we use two specially designed bis-PNAs (PNA openers) to spontaneously invade double-stranded DNA (dsDNA) at two closely located sites through simultaneous formation of Watson-Crick and Hoogsteen base pairs (Demidov and Frank-Kamenetskii, 2004; Egholm et al., 1995). The PNA openers bind to one of the two DNA strands of a target sequence, leaving the opposite strand displaced. The opened site is 20–30 nucleotides long, while the rest of DNA stays in the double stranded form. Once dsDNA is locally opened, the padlock probe is hybridized with the target sequence. After ligation of the padlock probe, the resulting circle forms about two turns of a double helix with the displaced DNA strand, creating a truly topological link (see Figure 1). The assembled circular DNA serves as a template for the RCA reaction, which yields a long, single-stranded amplicon that contains thousands of copies of the target sequence (Lizardi et al., 1998). The RCA step provides the required sensitivity to detect a single copy DNA site per genome (Zhang et al., 2006). Finally, fluorophore-tagged decorator probes are hybridized to the RCA product. The DNA amplicon remains firmly attached to its site of synthesis, so the fluorescent RCA product can be detected with a fluorescence microscope as a visible point source (Larsson et al., 2004).
Figure 1. Schematic depiction of the new method for sensitive and specific targeting of unique sequences on non-denatured human genomic DNA and images observed by fluorescent microscopy in experiments performed according to the scheme (with normal human male cells and probes specific to human autosomes).

Schematic: Our method consists of three major steps. I) The PNA openers specifically bind to two closely located homopurine DNA sites that are separated by several mixed purine-pyrimidine bases and locally open the double-stranded DNA. This opened region serves as a target for hybridization of an oligonucleotide probe to form a PD-loop. This binding is extremely sequence specific because nothing but the target location is accessible to the probe. II) The assembled circular DNA serves as a template for the RCA reaction, which yields a long, single-stranded amplicon that contains thousands of copies of the target sequence. III) For the detection step, fluorophore-tagged decorator probes are hybridized to the RCA product. The DNA amplicon remains firmly attached to its site of synthesis, so the multiply fluorescent labeled product can be imaged as a visible point source that can easily be detected with a fluorescence microscope. Images: (A) Multiple fluorescent spots in the vicinity of metaphase chromosomes and nuclei indicate the detection of short, human-specific DNA sequences within the mitochondrial genome in the cytoplasm. Data for target site MT-ND3 (AAGAAGAATTTTATGGAGAAAGG) designed for the mitochondrial-encoded NADH dehydrogenase 3 gene are shown. (B) Two fluorescent spots are clearly seen on human chromosomes 13 and 11, identified cytogenetically, when probe (GAGGGAGGTAGCCAGAGGAAG) specific to chromosome 13 or probe ALDH3B1 (GAGGGAAGACCCAGGAGGGAGG) designed for the aldehyde dehydrogenase 3 gene on chromosome 11 is applied. Insert shows the location of these spots correlates to the position of the target sequence determined by Human Genome BLAT (http://genome.ucsc.edu/cgi-bin/hgBlat) (C) Typical results observed in the large majority of nuclei (pair fluorescent spots) for several independently chosen target sites. Data for probe ALDH3B1 specific to chromosome 11 are shown. The fluorescent signals were acquired separately using two filter sets (DAPI for DNA and Cy3 for labelled RCA product). Each image is a superimposition of two separate images, with DAPI and Cy3 signals pseudo-colored in blue and red, respectively. Bright blue regions reflect chromatin-rich areas of the nuclei. See also Figures S2 and S3.
We have previously shown that the PNA-RCA approach can be used for fluorescent in situ detection of short single-copy DNA sequences in bacterial genomes (Smolina et al., 2007; Smolina et al., 2010; Smolina et al., 2008). Here we aimed at extending this approach to the human genome. The extension of the method to eukaryotic genomes is far from obvious. Human DNA, in contrast to bacterial DNA, is in a very condensed and packed form due to its interactions with histones. Although the fixation procedure somewhat stabilizes cell morphology and makes the cell membrane more permeable, the access of various reagents and probes to target DNA sites within the intact nucleosomes remain challenging.
Detection of specific sequences in human mitochondrial genome
We chose the first target sequences in mitochondrial DNA because it is known to exhibit prokaryotic features. Especially important is the fact that mtDNA lacks the nucleosome structure of metaphase chromosomes. The selected target site MT-ND3 within the NADH dehydrogenase 3 gene (Table 1) was opened with the corresponding PNAs and the padlock probe was added to hybridize the displaced single-stranded DNA. After ligation of the padlock probe the target sequence was amplified with RCA and simultaneously labelled with the fluorophore-tagged (Cy3) decorator probe. By counterstaining chromosomes and cell nuclei with DAPI, we observed multiple fluorescent spots in the vicinity of the metaphase chromosomes and nuclei that indicated the presence of mitochondrial genomes in the cytoplasm (Figure 1A). These results showed feasibility of the PNA-RCA approach for detecting short, human-specific DNA sequences in the mitochondrial genome.
Table 1.
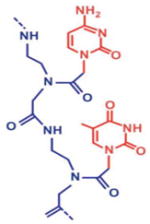
PD-sites, PNAs, and circularizable ODN probes
| I. Peptide Nucleic Acid (PNA) | II. Sequence-specific targeting of double-stranded DNA | III. Rolling circle amplification (RCA) | IV. Fluorescence in situ hybridization (FISH) |
|---|---|---|---|
|
|
|

|
binding sites are underlined;
eg1, Lys, and J denote the bis-PNA linker segment, the amino acid lysine, and the nucleobase pseudoisocytosine;
Detection of several target sites on human autosomes
To determine whether this approach can be applied to human nuclear DNA inside the chromatin structure, we targeted sequences within cell nuclei and metaphase chromosomes. Detection of DNA in metaphase chromosomes is especially challenging due to its highly condensed nucleosome structure. Specific probes were designed to target four unique sites on human autosomes (Table 1 and Supp. Fig. 1). The slides typically contained about 50 cell nuclei and about 10 metaphase chromosomal spreads. Since cell nuclei have pairs of autosomes, we expected to see double-spot signals for each of the chosen target sites. Figure 1C show typical results. Approximately 90% of nuclei produced the expected double-spot signals for each of the six target sites on autosomal chromosomes (Figure 3D).
Figure 3. Fluorescent images of metaphase chromosomes observed by fluorescent microscopy.

Normal human male cells and sites specific to human chromosomes were used. (A) Simultaneous detection of multiple unique sites in the human genome. Fluorescent spots are seen on chromosomes 8, 11 and 19. The location of the spots corresponds with the positions of the target genes on the chromosomes (insert - GPIHBP1, chr8 q24.3 – GAGGGAAGGGGATTTGGAGGGAGG; ALDH3B1, chr11 q13.2 – GAGGGAAGACCCAGGAGGGAGG; and CABP5, chr19 q13.33 - GAGGGAGGGCAGGCGGGAGGGAGG). (B–C) Sequence specificity of observed signals. The target sequence CABP5 designed for the calcium-binding protein 5 located on chromosome 19 is very similar to numerous other sites in the human genome. Using Human BLAST (http://www.ncbi.nlm.nih.gov/blast/) we found at least two sequence elements that differ from the target site by only two SNPs (Suppl. Table 1). Images observed by fluorescent microscopy in experiments performed according to the scheme with normal human male cells and probes specific to CABP5 gene located on chromosome 19 and mismatched probe 19_mm are shown. (B) No detectable fluorescent signals were observed on the metaphase chromosomes for sequences with mismatched sites that differ from the target sequences by only two SNPs. Insert -the position of the correct target sequence - CABP5 determined by BLAT. Potential false-positives for chr19 target site: GAGGGAGGGaAGGaGGGAGGGAGG (chr19_mm-probe) and GAGGGAGGGCAGGaGGaAGGGAGG. (C) Fluorescent signals observed on metaphase chromosomes when the chr19_mm probe was used. Mismatched probe chr19_mm has more than 80 hits located on various chromosomes. As expected, numerous signals were observed. The fluorescent signals were acquired separately using two filter sets (DAPI for DNA and Cy3 for labelled RCA product). Each image is a superimposition of two separate images, with DAPI and Cy3 signals pseudo-coloured in blue and red, respectively. See also Figure S1.
A metaphase cell has four copies of the genome, so we expected to see four signals per cell or two per metaphase chromosome pair. We were able to observe a detection signal on the majority of the metaphase spreads. Importantly, the localization of the signals agreed with the predicted location of the target on the chromosome as determined by BLAT (Suppl. Table 1). However, it proved to be difficult to distinguish signals on the sister chromatids and only one signal per metaphase chromosome was visually detected in most of our spreads. We believe that the chromatids were not sufficiently separated and the lack of signal resolution is likely due to the overlap of the RCA products. In approximately 30% of the cases, detailed analysis with ImageJ allowed us to distinguish the two signals on each of the metaphase chromosomes (Suppl. Fig. 3).
Reproducibility and irregularity of the signal are the major concerns in FISH assays (Levsky and Singer, 2003). To improve reproducibility of the method we tested several parameters: slide preparation, cell treatment and reaction conditions. We found that it is critical to perform the reactions under elevated coverslips as opposed to standard ones. This reduced variation across the slide, most likely by preventing local depletion of reagents. Figure 1B shows the highly localized signal obtained from the detection of the target sequences on chromosomes 11 and 13. No detectable fluorescent signal was observed on the nuclei or the metaphase chromosomes in the negative control experiments where at least one of the protocol steps was excluded.
Multi-target detection
In order to demonstrate that our method can detect multiple targets simultaneously we designed three probes for different genes with varied localization on chromosomes: glycosylphosphatidyl-inositol anchored high density lipoprotein binding protein 1 (GPIHBP1) at chr8 - distal end of the long arm; aldehyde dehydrogenase 3 family member B1 (ALDH3B1) at chr11 - near centromeric region; and calcium binding protein 5 (CABP5) at chr19 - middle of the q-arm. The three sequences were chosen in such a way that they had the same 7-bp poly-purine flanking sequences and unique random sequences in between. These unique sites were detected in parallel using one pair of PNA openers, three target-specific padlock probes, and the same fluorescently labelled decorator. Figure 3A shows the result of the detection; two fluorescent spots can be seen on each pair of targeted chromosomes. The locations of the spots correlate with the position of the target sequences determined by BLAT (Suppl. Fig 1).
Specificity of observed signals and efficiency of the method
Our extensive in vitro study (Demidov et al., 2001) and the data on bacterial cells (Smolina et al., 2010) provide strong evidence that the PNA-padlock probe-RCA approach has single-base sensitivity (Supp. Fig. 4). Our persistent observation of the double-spot signal on nuclei for each of the six target sites on autosomal chromosomes (Figure 2A) and the absence of spurious signals also support the high sequence specificity of this approach. In order to get additional proof of the specificity of our method, we designed probes for the X and Y-chromosomes and tested them in male and female cell lines. If these probes are sequence-specific, the X-probe should give two signals in the female cell line and one signal in the male. The Y-specific probe should give no signal in the female cell line and one signal in the male. Figure 2B shows the results for the detection with X- and Y-probes. For the male cell lines, each probe gave one signal per cell. For the female cell lines, the Y-probe produced no signals and the X-probe produced two.
Figure 2. Fluorescent images of interphase nuclei observed by fluorescent microscopy.

(A) Typical results (double-spot signals) were observed on human nuclei with every one of the unique target sites randomly chosen for autosomal parts of the human genome. All nuclei display two very bright fluorescent spots when probes corresponding to chromosome16 (AGAGGAAGCTACACTGAGAGGAAG) or to chromosome 21 (GAGGGAGGTAATTGAGGGAGG) were applied. (B) Special probes were designed for sex chromosomes (X and Y) in order to confirm specificity of the developed method. In experiments with male cell lines (XY) both probes, X chromosome - probe (AGAGGAAGAGTCGGAGAGGAAG) and Y chromosome - probe (GGAGGAGAATGGGTCTGAGGGAAG), give only one signal. Two signals for the X-probe were observed in nuclei of female cell lines (XX) and no signal was observed for the Y-probe. (C) Simultaneous, two-color detection of sequences on the X- and the Y-chromosomes in male cells. Both X and Y-probes were applied, ligated and amplified with RCA. The amplification product from the Y-probe is hybridized with the green fluorescent decorator, while the product from the X-probe is hybridized with the red decorator. (D) Number of detection signals on cell nuclei obtained per probe. A total of 72 reactions were analyzed, and 65 resulted in the expected 2 detection signals per probe (corresponding to 90%). Five of the cells had one or no detection signals, while two of the cells displayed three detection signals per probe. See also Figures S4.
One of the most attractive features of FISH is the ability to visualize multiple targets in different colors. In order to perform two-colored sex genotyping, special probes were designed for unique sequences on the X- and Y-chromosomes. When both X and Y-probes were applied simultaneously the RCA product specific for the X- probe would hybridize with the red fluorescent decorator, while the RCA product specific for the Y-probe would hybridize with the green fluorescent decorator. Figure 2C shows simultaneous, two-color detection of target sites on the X- and the Y-chromosomes in male cells, demonstrating the multiplicity of our approach.
In order to evaluate the PNA-RCA method for the detection of SNPs, we targeted various sequences within the human genome that could be opened by the same set of PNAs but required different padlock probes. For example, the target sequence specific for the calcium-binding protein 5 located on chromosome 19 (CABP5) is very similar to numerous other sites in the human genome. Two padlock probes were designed: one with the correct target sequence (chr19) and the other with two mismatched bases (chr19_mm). This mismatched probe would target about 80 other sites in the human genome that differ from the sequence on chromosome 19 by 2 SNPs (Figure 3B and C and Suppl. Table 1). As expected, the probe designed for CABP5 gene resulted in one positive signal on chromosome 19 and did not yield any false positives (Figure 3B). The fact that none of the mismatched sites yield a detection signal proves that our method is sensitive enough to discriminate between 2 mismatches in short sequences of genomic DNA. When a mismatched chromosome 19 probe was used, multiple signals were observed at various locations (Figure 3C).
DISCUSSION
Chemistry-based approaches have been successfully used for decades for sequence-specific recognition of DNA through base-pairing, and peptide nucleic acids (PNA) played an important role among other chemically modified analogues of nucleotides in different applications in biotechnology (Kaihatsu et al., 2004; Nielsen, 2010). Several research groups, including ours, have shown that various PNA modifications recognize duplex DNA with high sequence specificity and affinity. PNA-DNA interactions in duplex, triplex, and double-duplex invasive modes resulted in the rapid establishment of a new branch of research focussed on DNA diagnostics.
Here, we use a pair of pyrimidine bis-PNAs that sequence-specifically invade the DNA duplex to develop a FISH assay with a single nucleotide resolution power. PNA openers locally denature DNA within otherwise highly packed nucleosome structure and provide access to the displaced single-stranded DNA fragment for padlock probe hybridization. This critical step followed by padlock probe ligation, rolling circle amplification, and hybridization with decorator probe made it possible to visualize short unique DNA sequences in the interphase nuclei of human cells and in metaphase spreads. We have shown the potential of the method in multi-target detection and extremely high sequence specificity close to a single base resolution.
This extreme sequence specificity is achieved because of the combination of two molecular recognition events: simultaneous binding of both PNA openers to dsDNA and the padlock probe hybridization to the open target site followed by ligation. Statistical prediction indicates that the human genome has about 250 binding sites per each PNA pair (see experimental procedure for calculations). This implies that the specificity of the PNA-DNA interaction would not be enough for unique sequence detection. However, the addition of the next step, the assembly and ligation of the circular padlock probe makes this entire assay extremely sequence specific. The stringent strand-matching requirement for the padlock probe ligation has been previously reported (Nilsson, 2006; Zhang et al., 2006).
PNA-directed padlock probe’s application in biotechnology has certain limitations because the PNA binding site must consist of purines in one strand and pyrimidines in the other. However, these sequence limitations are relatively mild because the PNA binding sites can be very short and separated by up to 10 nucleotides with an arbitrary sequence (Bukanov et al., 1998). Our studies suggest that the PNA openers can be as short as hexamers. Statistically, such PD-loop sites are met every 200bp (see experimental procedure for calculations), which implies that every eukaryotic gene can be labelled at a unique target site. Unfortunately, the sequence restrictions posed by PNAs do limit the usefulness of this technique for the detection of SNPs. We envision that this method can be adapted to additional PNA variants, which would reduce the target sequence constraints (Demidov et al., 2002; Rapireddy et al., 2007; Zhang et al., 2000). Especially promising is the recently developed helical gamma-PNAs (γ-PNA) that contain the modified G-clamp nucleobases (Kuhn et al., 2010). Unlike bis-PNA openers, which sequence-specifically target duplex DNA only through triplex mode of binding, γ-PNAs can recognize dsDNA through direct Watson-Crick base-pairings via a strand invasion mechanism. Therefore, γ-PNAs have great potential for specific targeting of chosen duplex DNA sites in a sequence-unrestricted fashion. The substitution of one of the two bis-PNA openers by γ-PNA in the developed technique may overcome the sequence limitations of the original PD-loop design. Of course, it remains to be demonstrated that γ-PNA are as efficient as bis-PNA openers.
Fluorescent in situ hybridization plays an important role in cytogenetic analyses and gene mapping for several decades and such applications as prenatal diagnosis, tumour biology and genome analysis are difficult to imagine without FISH (Levsky and Singer, 2003). The resolution power of FISH is measured by 10s and 100s of kb, and even the best resolution of fiber-FISH is about few kilobases (Ersfeld, 2004). It should be also noted that as with any FISH technique, fiber-FISH requires DNA denaturation, and the resulting fragmented fluorescence signals prevent analysis at high resolution. This new method does not require any specific preliminary treatment of the cells or DNA. At the same time, it increases the resolution of FISH to a single base level and makes it possible to analyse chromosomes for allelic mutations. Recessive autosomal diseases and genetic crosses can now be diagnosed on the level of interphase nuclei and in metaphase spreads.
In summary, we demonstrated that the combination of PNAs, RCA, and FISH can be used to target human DNA in mitochondria, within the nucleus, and on metaphase spreads without preliminary denaturation. In the present study, we showed multi-target detection and discriminated between single-copy sequences that differ by two nucleotides at different loci. Further work will be focused on the detection of genetic variations and SNPs known to be linked to various diseases.
SIGNIFICANCE
We report a novel approach that can be used to visualize specific DNA segments that are about 20- nucleotides long within the human genome under DNA non-denaturing conditions. Our method expands the current applications of peptide nucleic acid probes to human cytogenetic. The sensitivity and specificity of the PNA-RCA-FISH assay was demonstrated by the detection of eight short DNA sequences in situ on human metaphase chromosomes and interphase nuclei. For the first time, it is possible to observe a unique, short target site in the whole genome of human beings with high sequence selectivity. Our approach has several advantages compared to standard FISH: it enables one to target and label a unique sequence element equivalent to the size of a PCR primer, the small size of the probes reduces any effects that probe incorporation has on the genome domain spatial arrangement, and denaturation of genomic DNA is omitted. The topological link between the target DNA and the ODN probe ensures that the precisely positioned label has to occupy a fixed site along the double helix. It may serve as a new method for highly localized detection of various marker sites within genomes. This method may open new perspectives in the investigation of structure dependent genome function or dysfunction; it could potentially be used for specific chromatin architecture studies. This technique might also be applied to detect genes of interest in non-denatured DNA, which is particularly useful for colocalization studies of DNA, RNA and proteins.
EXPERIMENTAL PROCEDURE
Selection of the target sites
The PD-loop sites were selected using human genome sequences available from the Genomes Database (ftp://ftp.ncbi.nih.gov/genbank/genomes/). Signature sites with different linker sequences between PNA-binding sites were selected (see Table 1). Specifically, we searched for sites RkNnRl (where R is a purine base and N is any base) choosing k and l between 7 and 8 and n between 2 and 10. PNA oligomers used in the project were purchased from PANAGENE or were from the laboratory collection. These PNAs recognize 6–10-bp-long sites on DNA. The collection of all PD-loop sites has been tested to select those sites that are unique and present in only one copy in a haploid genome according to human BLAST (http://www.ncbi.nlm.nih.gov/blast/). The locations of the target sites on human chromosomes are listed in Suppl. Table 1 and shown in Suppl. Figure 1.
Statistical prediction of PD-loop site availability within the human genome
The following calculation was used to determine the expected occurrence of PNA binding sites (RkNnRl) assuming a random sequence:
Consequently, for PNA openers that are 7 bp long and are interrupted by up to 10 bp of an arbitrary sequence, such sites are met statistically about every 800 bases. Our preliminary data indicate that, to form the PD-loop, the PNA openers can be as short as hexamers. If they again can be separated by up to 10 bp of an arbitrary sequence, we expect to have one such site per 200 bp, on average.
The sequence selectivity requirements for simultaneous PNA opening and ODN probe hybridization are stringent enough to eliminate the possibility of a false positive signal. Since the target sequence of the PNA openers is only 7–8 bp long, there are approximately 400,000 binding sites within the human genome for every PNA opener designed for a specific, 7 nucleotide-long target sequence. Within a random DNA sequence that has length of the human genome (3×109 bp), there are about 250 binding sites for two such PNA openers separated by 2 to 10 nucleotides of a random DNA sequence. Therefore, the specificity of the detection signal depends mostly on the circular probe assembly, which can only hybridize onto a unique, about 20-nt-long sequence that has been made accessible by the two PNAs and the stringent strand-matching requirement for ODN ligation.
Preparation of metaphase chromosomes
The normal male (GM10851; 46,XY) and normal female (GM15510; 46,XX) cell-lines (Coriell, Camden, NJ, USA) were used. Culture of cells and preparation of chromosomes were performed according to standard techniques. Lymphoblastoid cells were cultured at 37°C in RPMI 1640 medium supplemented with 10% Fetal Bovine Serum and 1% L-glutamine (200 mM), exposed for 25 minutes to Colcemid (final concentration 0.05ug/ml) and harvested according to standard cytogenetic protocol, using hypotonic 0.075M KCl and 1:3 acetic acid- methanol fixative. The fixation procedure was repeated 2–3 times. The fixed cell suspension was dropped onto clean microscope slides.
Slide pretreatment
The number and brightness of signals observed per metaphase spread varied with slide pretreatment conditions. In this study, the optimal and most reproducible results were obtained from the following set of pretreatments: treatment with 200 μg/ml RNaseA in 2×SSC at 37 °C for 1 h, followed by washes in the same buffer; treatment with 0.01% pepsin (Sigma) in 0.1 M HCl for 90 s at 37°C, followed by washes in PBS; then incubation in a 10mM Na phosphate buffer (pH 7.0) at 37°C for 30 minutes, followed by passing the slides through a series of ethanol baths (70%, 90%, 100%) for dehydration just before PD-loop assembly.
PD-loop assembly, oligonucleotide ligation, and RCA
In order to bind the PNA openers to the chromosomal DNA, 50μl of 10mM Na phosphate buffer (pH 7.0) with 0.8μM of each PNA was dropped on each slide, covered by a 24 × 50 mm slip, and sealed with rubber cement. The slides were incubated at 45–55°C for 4 hours, then the rubber cement was gently peeled off and the cover slip was removed. The slides were washed twice in PBS buffer to remove non-bound PNAs. The oligonucleotide probe was ligated to form the PD-loop by dropping 50μl of ligation mixture on each slide, which contained 1X T4 ligase buffer with 5u T4 DNA ligase (NEBiolabs) and 2μM of the padlock probe. The slides were incubated under a coverslip at room temperature for 1 hour. Then they were washed twice in 2X SSC buffer and once in buffer A (100mM TrisHCl, 150mM NaCl, and 0.05% Tween20) to remove excess non-ligated oligonucleotide probe. Then, 50μl of RCA reaction mixture was dropped onto each slide. The RCA reaction mixture contained 2μM primer, 10u of phi29 DNA polymerase, 200μM dNTPs, 1X phi29 DNA polymerase buffer (NEBiolabs), and 2μM fluorescently labelled decorator probe. The slides were covered by a slip and sealed with rubber cement, and the RCA reaction was carried out at 37°C for 2 hours in the humidifying chamber. We found that reaction performed under ‘chamber coverslips’ (1mm thick LifterSlip coverslips, Fisher) gave the best results because it reduced the variation across the slide by preventing local depletion in reagents. To enhance fluorescent signal, RCA reaction can be performed overnight (~16 hours). After RCA the slides were washed twice in buffer A and once in 4XT buffer. Subsequently, slides were air-dried and mounted with a drop of Vectashield antifade containing 5 μg/ml DAPI (4′,6-diamidino-2-phenylindole) for counterstaining of DNA. A cover slip was applied and the slides were allowed to stand at room temperature for 5 minutes. The coverslips were then secured with nail polish.
Fluorescent signal detection
The detection output was observed with a fluorescence microscope Olympus BX-70 equipped with a cooled charge-coupled device camera. Images were acquired using exposure times ranging from 0.2 to 1 second (depending on duration of RCA). Assuming a replication rate of phi29 DNA polymerase of 1400–1500 bases min−1 (Melin et al., 2007) and a circle size of about 80 nucleotides, each rolling-circle product increases in length by about 80,000 nucleotides h−1, with total circle amplification as high as 1000-fold per hour. Each repeat of the circular probe carries two sites for decorator probe hybridization, resulting in accumulation of fluorescent product directly on the target site with a density of one fluorescent label per 40 bases. Depending on duration of RCA, the RCA amplicon length varied between 160kb (2 hour) and more than 1Mb (for overnight RCA). Thus, the sensitivity of the approach was expected to be compatible with conventional FISH probes, which have 2–4 fluorescent labels per 100 bases (Cox and Singer, 2004). Fluorescent signals were captured separately using appropriate filter sets for DAPI and CY3. Images were pseudo-colored, processed, and merged using the CytoVision image software. To ensure that the detection signal is powerful enough to be distinguished from the background, the signal to background ratio was quantified with the Genus Package imaging software (Applied Imaging, Inc.). Suppl. Figure 2 demonstrates the quantification analysis.
Supplementary Material
Highlights.
PNA-directed assembly of the labelling complex on duplex DNA
Padlock probes combined with Rolling Circle Amplification for signal amplification
Multi-target visualization within the human genome with high sequence selectivity
Acknowledgments
We thank Prof. Maxim Frank-Kamenetskii for helpful discussions, Prof. Natalia Broude for critical reading and valuable comments on the manuscript, Dr. Peter Nielsen for providing us with PNA oligomers, and Dr. Charles Lee for slide preparation and technical assistance.
FUNDING
This work was supported by the National Institutes of Health [NIH/NGRR: 1R21RR025371–01 to I.S.]; Funding for open access charge: National Institutes of Health.
Footnotes
Supplementary Data are available online: Supplementary Table 1 and Supplementary Figures 1–4.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albertson DG, Pinkel D. Genomic microarrays in human genetic disease and cancer. Human molecular genetics. 2003;12(Spec No 2):R145–152. doi: 10.1093/hmg/ddg261. [DOI] [PubMed] [Google Scholar]
- Blanco AM, Artero R. A practical approach to FRET-based PNA fluorescence in situ hybridization. Methods. 2010;52:343–351. doi: 10.1016/j.ymeth.2010.07.010. [DOI] [PubMed] [Google Scholar]
- Bukanov NO, Demidov VV, Nielsen PE, Frank-Kamenetskii MD. PD-loop: a complex of duplex DNA with an oligonucleotide. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:5516–5520. doi: 10.1073/pnas.95.10.5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonari M, Cibati M, Sette N, Catizone A, Fiorilli M. Measurement of telomere length using PNA probe by cytometry. Methods in cell biology. 2011;103:189–202. doi: 10.1016/B978-0-12-385493-3.00008-5. [DOI] [PubMed] [Google Scholar]
- Christian AT, Pattee MS, Attix CM, Reed BE, Sorensen KJ, Tucker JD. Detection of DNA point mutations and mRNA expression levels by rolling circle amplification in individual cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:14238–14243. doi: 10.1073/pnas.251383598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox WG, Singer VL. Fluorescent DNA hybridization probe preparation using amine modification and reactive dye coupling. BioTechniques. 2004;36:114–122. doi: 10.2144/04361RR02. [DOI] [PubMed] [Google Scholar]
- Demidov VV, Frank-Kamenetskii MD. Two sides of the coin: affinity and specificity of nucleic acid interactions. Trends in biochemical sciences. 2004;29:62–71. doi: 10.1016/j.tibs.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Demidov VV, Kuhn H, Lavrentieva-Smolina IV, Frank-Kamenetskii MD. Peptide nucleic acid-assisted topological labeling of duplex dna. Methods. 2001;23:123–131. doi: 10.1006/meth.2000.1113. [DOI] [PubMed] [Google Scholar]
- Demidov VV, Protozanova E, Izvolsky KI, Price C, Nielsen PE, Frank-Kamenetskii MD. Kinetics and mechanism of the DNA double helix invasion by pseudocomplementary peptide nucleic acids. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:5953–5958. doi: 10.1073/pnas.092127999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egholm M, Christensen L, Dueholm KL, Buchardt O, Coull J, Nielsen PE. Efficient pH-independent sequence-specific DNA binding by pseudoisocytosine-containing bis-PNA. Nucleic acids research. 1995;23:217–222. doi: 10.1093/nar/23.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ersfeld K. Fiber-FISH: fluorescence in situ hybridization on stretched DNA. Methods Mol Biol. 2004;270:395–402. doi: 10.1385/1-59259-793-9:395. [DOI] [PubMed] [Google Scholar]
- Forrest GN. PNA FISH: present and future impact on patient management. Expert review of molecular diagnostics. 2007;7:231–236. doi: 10.1586/14737159.7.3.231. [DOI] [PubMed] [Google Scholar]
- Halling KC, Kipp BR. Fluorescence in situ hybridization in diagnostic cytology. Human pathology. 2007;38:1137–1144. doi: 10.1016/j.humpath.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Hell SW. Far-field optical nanoscopy. Science. 2007;316:1153–1158. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- Kaihatsu K, Janowski BA, Corey DR. Recognition of chromosomal DNA by PNAs. Chemistry & biology. 2004;11:749–758. doi: 10.1016/j.chembiol.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Kuhn H, Sahu B, Rapireddy S, Ly DH, Frank-Kamenetskii MD. Sequence specificity at targeting double-stranded DNA with a gamma-PNA oligomer modified with guanidinium G-clamp nucleobases. Artificial DNA, PNA & XNA. 2010;1:45–53. doi: 10.4161/adna.1.1.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson C, Koch J, Nygren A, Janssen G, Raap AK, Landegren U, Nilsson M. In situ genotyping individual DNA molecules by target-primed rolling-circle amplification of padlock probes. Nature methods. 2004;1:227–232. doi: 10.1038/nmeth723. [DOI] [PubMed] [Google Scholar]
- Levsky JM, Singer RH. Fluorescence in situ hybridization: past, present and future. Journal of cell science. 2003;116:2833–2838. doi: 10.1242/jcs.00633. [DOI] [PubMed] [Google Scholar]
- Lizardi PM, Huang X, Zhu Z, Bray-Ward P, Thomas DC, Ward DC. Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nature genetics. 1998;19:225–232. doi: 10.1038/898. [DOI] [PubMed] [Google Scholar]
- Lohmann JS, Stougaard M, Koch J. Detection of short repeated genomic sequences on metaphase chromosomes using padlock probes and target primed rolling circle DNA synthesis. BMC molecular biology. 2007;8:103. doi: 10.1186/1471-2199-8-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melin J, Jarvius J, Goransson J, Nilsson M. Homogeneous amplified single-molecule detection: Characterization of key parameters. Analytical biochemistry. 2007;368:230–238. doi: 10.1016/j.ab.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Nielsen PE. Sequence-selective targeting of duplex DNA by peptide nucleic acids. Current opinion in molecular therapeutics. 2010;12:184–191. [PubMed] [Google Scholar]
- Nilsson M. Lock and roll: single-molecule genotyping in situ using padlock probes and rolling-circle amplification. Histochemistry and cell biology. 2006;126:159–164. doi: 10.1007/s00418-006-0213-2. [DOI] [PubMed] [Google Scholar]
- Rapireddy S, He G, Roy S, Armitage BA, Ly DH. Strand invasion of mixed-sequence B-DNA by acridine-linked, gamma-peptide nucleic acid (gamma-PNA) Journal of the American Chemical Society. 2007;129:15596–15600. doi: 10.1021/ja074886j. [DOI] [PubMed] [Google Scholar]
- Singh RP, Oh BK, Choi JW. Application of peptide nucleic acid towards development of nanobiosensor arrays. Bioelectrochemistry. 2010;79:153–161. doi: 10.1016/j.bioelechem.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Smolina I, Lee C, Frank-Kamenetskii M. Detection of low-copy-number genomic DNA sequences in individual bacterial cells by using peptide nucleic acid-assisted rolling-circle amplification and fluorescence in situ hybridization. Applied and environmental microbiology. 2007;73:2324–2328. doi: 10.1128/AEM.02038-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolina I, Miller NS, Frank-Kamenetskii MD. PNA-based microbial pathogen identification and resistance marker detection: An accurate, isothermal rapid assay based on genome-specific features. Artificial DNA, PNA & XNA. 2010;1:76–82. doi: 10.4161/adna.1.2.13256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolina IV, Kuhn H, Lee C, Frank-Kamenetskii MD. Fluorescence-based detection of short DNA sequences under non-denaturing conditions. Bioorganic & medicinal chemistry. 2008;16:84–93. doi: 10.1016/j.bmc.2007.04.063. [DOI] [PubMed] [Google Scholar]
- Volpi EV, Bridger JM. FISH glossary: an overview of the fluorescence in situ hybridization technique. BioTechniques. 2008;45:385–386. 388, 390. doi: 10.2144/000112811. passim. [DOI] [PubMed] [Google Scholar]
- Zhang D, Wu J, Ye F, Feng T, Lee I, Yin B. Amplification of circularizable probes for the detection of target nucleic acids and proteins. Clinica chimica acta; international journal of clinical chemistry. 2006;363:61–70. doi: 10.1016/j.cccn.2005.05.039. [DOI] [PubMed] [Google Scholar]
- Zhang X, Ishihara T, Corey DR. Strand invasion by mixed base PNAs and a PNA-peptide chimera. Nucleic acids research. 2000;28:3332–3338. doi: 10.1093/nar/28.17.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.