

Abstract
2-arachidonylglycerol (2-AG) is the most abundant endocannabinoid in the central nervous system and is elevated after brain injury. Because of its rapid hydrolysis, however, the compensatory and neuroprotective effect of 2-AG is short-lived. Although inhibition of monoacylglycerol lipase, a principal enzyme for 2-AG degradation, causes a robust increase of brain levels of 2-AG, it also leads to cannabinoid receptor desensitization and behavioral tolerance. Alpha/beta hydrolase domain 6 (ABHD6) is a novel 2-AG hydrolytic enzyme that accounts for a small portion of 2-AG hydrolysis, but its inhibition is believed to elevate the levels of 2-AG within the therapeutic window without causing side effect. Using a mouse model of traumatic brain injury (TBI), we found that post-insult chronic treatment with a selective ABHD6 inhibitor WWL70 improved motor coordination and working memory performance. WWL70 treatment reduced lesion volume in the cortex and neurodegeneration in the dendate gyrus. It also suppressed the expression of inducible nitric oxide synthase and cyclooxygenase-2 and enhanced the expression of arginase-1 in the ipsilateral cortex at 3 and 7 days post-TBI, suggesting microglia/macrophages shifted from M1 to M2 phenotypes after treatment. The blood-brain barrier dysfunction at 3 and 7 days post-TBI was dramatically reduced. Furthermore, the beneficial effects of WWL70 involved up-regulation and activation of cannabinoid type 1 and type 2 receptors and were attributable to the phosphorylation of the extracellular signal regulated kinase and the serine/threonine protein kinase AKT. This study indicates that the fine-tuning of 2-AG signaling by modulating ABHD6 activity can exert anti-inflammatory and neuroprotective effects in TBI.
Key words: ABHD6, neuroinflammation, neuroprotection, traumatic brain injury, 2-arachidonylglycerol
Introduction
Traumatic brain injury (TBI) is an important worldwide public health problem and a leading cause of mortality and disability among young adults. Currently, approximately 2% of Americans are living with TBI-related disability with an annual cost of treating, rehabilitating, and caring for victims valued at $60.0 billion.1–3 TBI is characterized by the initial insult followed by the secondary injury associated with excitotoxicity, inflammation, oxidative stress, and neuronal death.4,5 Although the secondary injury provides a window of opportunity for treatment, there is still no Food and Drug Administration-approved therapeutic agent available. Because of the complexity of the TBI pathogenesis, an effective therapeutic agent should have multipotent properties capable of reducing excitotoxicity, oxidative stress, neuroinflammation, brain edema, and neuronal death.2,4–6
Accumulating evidence suggests that endocannabinoids 2-arachidonoylglycerol (2-AG) and anandamide (AEA) are two candidates to possess these properties.7–9 Several studies have reported that cannabinoids can alleviate blood-brain barrier dysfunction, brain edema, lesion volume, neuronal death, and improve the behavioral performance in the rodent models of TBI. The protective effects are attributable to their roles as anti-oxidants, anti-inflammatory and anti-excitotoxic agents and are likely mediated by activation of cannabinoid receptors 1 and 2 (CB1 and CB2) expressed in neurons and inflammatory cells.9–13 The use of cannabinoids as therapeutic agents, however, is still limited by the potential psychotropic effects caused by CB1 receptor activation. It is known that endocannabinoids are produced where they are needed; this “on demand” synthesis is believed to enhance the endocannabinoid tone in a site- and an event- specific manner, and therefore can avoid the undesirable side effects elicited by the non-selective, global CB1 receptor activation in neurons.14,15
2-AG is the most abundant endocannabinoid in the brain, and its level increases immediately after brain injury.9,16 Although this can be a compensatory mechanism, the protective effect of endogenous 2-AG is insufficient because of its rapid hydrolysis. Therefore, inhibition of 2-AG degradation should be able to sustain the brain levels of 2-AG and extend its therapeutic efficacy.17 Among the enzymes that catalyze 2-AG degradation, the monoacylglycerol lipase (MAGL) is responsible for approximately 85% and alpha/beta hydrolase domain 6 (ABHD6) accounts for only 4% of 2-AG hydrolysis in brain homogenates.18 Interestingly, it has been shown recently that inhibition of ABHD6 in neurons blocks 40–50% of 2-AG hydrolysis, suggesting it can compete with MAGL in efficacy and increase the endogenous levels of 2-AG.17,19 The selective inhibition of AHBD6 in vivo has so far not been reported, however.
Recent studies have shown that pharmacological inhibition or genetic deletion of MAGL causes 2-AG overload in the brain and results in CB1 receptor desensitization and behavioral tolerance.17,20,21 These results suggest that partial inhibition of 2-AG degradation might be more therapeutically useful. Therefore, inhibition of ABHD6, which is shown to lead to a moderate increase of 2-AG, may provide a better therapeutic potential by operating within the therapeutic window of 2-AG signaling and avoid the undesirable side effects, such as hypomobility observed with exogenous cannabinoids or chronic use of MAGL inhibitors.22–24
In this study, we aimed to investigate the protective effects of the selective ABHD6 inhibitor WWL70 on TBI-induced impairments in behavioral performance, neuroinflammation, and neurodegeneration. The involvement of CB1 and CB2 receptors and the potential therapeutic mechanisms of WWL70 were also investigated.
Methods
Reagents
ABHD6 inhibitor WWL70, CB1 antagonist AM 281, and CB2 antagonist AM 630 were purchased from Tocris Bioscience (Ellisville, MO). All other chemicals and reagents were purchased from Sigma (St. Louis, MO), unless stated otherwise.
Animals
Seven-week-old, male C57BL/6 mice weighing 25–30 g (Jackson Laboratory, Bar Harbor, ME) were used in this study. Animals were maintained under a controlled environment with a temperature of 23±2°C, a 12 h light/dark cycle, and access to food and water ad libitum. The study was performed in accordance to the guidelines of the Animal Use and Care Committee of the Uniformed Services University of the Health Sciences. Each experimental group contained 8 to 13 mice.
TBI model
Animals were subjected to TBI elicited by controlled cortical impact (CCI). Surgical procedures were performed under general anesthesia with isoflurane (2%) while the body temperature was maintained at 37°C using a heating pad coupled to a rectal probe. CCI was performed after craniotomy over the left parietal cortex using a stereotaxically positioned 3 mm diameter stainless steel tipped piston centered at 2 mm posterior and 2.5 mm lateral to bregma.25 Impact was induced using a stereotaxic impactor (Leica, Wetzlar, Germany) with piston velocity set at 5 m/sec and a depth penetration of 1.5 mm. Sham-operated mice were anesthetized with isoflurane, followed by craniotomy without trauma induction.
Drug administration
WWL70 (5 mg/kg or 10 mg/kg) dissolved in 1% dimethyl sulfoxide (DMSO) in physiologic saline or an equal volume of 1% DMSO in saline (10 mL/kg) was injected intraperitoneally 30 min after TBI, and then once a day for 3, 7, or 21 days depending on the experimental design (Fig. 1). To determine the cannabinoid receptor dependency, CB1 receptor antagonist AM 281 (3 mg/kg) and CB2 receptor antagonist AM 630 (3 mg/kg) were administered to additional groups of animals 30 min before WWL70 injection for 7 days.26 During the 21-day treatment regimen, animals were subjected to a battery of behavioral tests at different time points (Fig. 1). Two hours after the last injection on day 21 post-injury, animals were sacrificed and brain tissues were collected for histological analysis. Animals in the 3- and 7-days treatment groups were only used for histological and Western blot analyses (Fig 1).
FIG. 1.
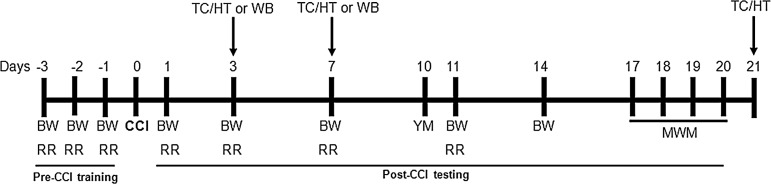
Schematic description of experimental procedures. Detailed timeline of all behavioral tests and experimental procedures starting at 3 days before the surgery (day −3). TC, tissue collection; HT, histology; WB, Western blot; BW, beam walk test; RR, rotarod test; CCI, controlled cortical impact; YM, Y maze test; MWM, Morris water maze test.
Assessment of functional recovery
Motor coordination was evaluated using beam-walk balance test and rotarod test. Spatial learning and memory task was assessed by Morris water maze test, and spontaneous alternation Y maze test was used to measure working memory.
1. Beam-walk balance test
Beam-walk balance test was performed to assess fine motor movements as described previously.1,27 The beam-walk apparatus consists of a wooden beam measuring 6 cm in width, 120 cm in length, and suspended 30 cm above a table. Animals were trained to walk on the beam before surgery, and those exhibiting less than 5 foot faults out of 50 steps were selected and randomly distributed in different treatment groups. The beam-walk balance test was performed on days 1, 3, 7, 11, and 14 post-injury, and the number of foot faults per 50 steps was recorded.
2. Rotarod test
Rotarod test was used to evaluate motor coordination. Animals were trained before surgery to coordinate their movement on an accelerating rotarod device (Ugo Basile, Collegeville, PA). The rotarod device was set to accelerate from 4 to 40 rpm within 2 min.28 Latency to fall from the device or to cling and rotate for two full rotations was recorded. After surgery, rotarod performance was evaluated on days 1, 3, 7, and 11 post-injury with three trials at 15 min intervals each day. The average of the three trials was recorded.
3. Morris water maze test
The Morris water maze test is commonly used to measure spatial learning and memory in several neurological disorders including TBI.29,30 The test was performed as described previously.1,31 The testing apparatus is comprised of a circular water tank (120 cm diameter) and an invisible acrylic glass platform (12 cm diameter), submerged 1 cm below the water surface. All animals were trained for 4 consecutive days (days 17–20 post-injury), followed by a probe trial 1 h after the last training session. Each training session was composed of four trials separated by a 5 min pause between trials.
During each trial, the platform was placed in one quadrant (western quadrant) of the pool, and each animal was randomly released in one of the remaining quadrants facing the wall of the tank and given 60 sec to locate the platform where they were left to spend 15 additional seconds to familiarize with the visual cue. If the animal failed to locate the platform within 60 sec on any given trial, he was led to the platform by the experimenter and allowed to remain there for 15 sec. For the probe trial, the platform was removed from the water and each mouse was released in the pool at a point diagonally opposite to the previous location of the platform (eastern quadrant), and allowed to swim for 60 sec to determine quadrant preference. Movement within the maze was monitored using a video camera and analyzed using the ANY-Maze software.
4. Spontaneous alternation Y maze test
Spontaneous alternation Y maze test is a hippocampus-based task that measures working memory,29,30,32 which was used to evaluate the effect WWL70 treatment on TBI- induced deficits in working memory. The Y maze device has a symmetrical wooden Y shape with arms measuring 25 cm long, 8 cm wide, and 15 cm high. The test was performed on day 10 post-TBI as reported previously.31,33 Each animal was placed in the central zone of the maze and allowed to explore the three arms. The sequence and number of entries to each arm were tracked with a video camera and recorded over a period of 5 min. An arm visit was referred to a mouse moving all four paws into the arm, and each alternation was defined as a consecutive entry in three different arms.34–36 The percentage of alternation was calculated based on the formula: total number of alternations/(total number of arm entries - 2)* 100 as reported previously.37,38
Histology
Histological analysis was performed on frozen brain sections to measure the lesion volume using hematoxylin and eosin (H&E) staining, and the number of degenerative cells was determined using Fluoro-Jade B (FJ-B) staining.
1. Evaluation of lesion volume
Lesion volume was measured on days 3, 7, and 21 post-injury. Animals were deeply anesthetized and transcardially perfused with physiologic saline followed by 4% formaldehyde. Brains were collected and fixed in the same fixative for 24 h and then transferred to 30% sucrose for 48 h. Serial coronal sections, 30 μm thick, were cut using a cryostat (Leica, Buffalo Grove, IL) starting at 600 μm anterior to the bregma. One of every six serial sections was stained with H&E and scanned with an Epson scanner. Each section was measured for the area of normal H&E staining with ImageJ software from the National Institutes of Health. Cavitation, hemorrhage, or loss of normal H&E staining were considered as lesions. The lesion area was calculated as the area of the contralateral side minus that of the ipsilateral side. The lesion volume for each brain was calculated as reported previously,39 using the formula {0.5A1+0.5(A1+A2)+……+ 0.5(An - 1+An)+0.5An}X; where A is the lesion area (mm2) for each slice and X is the distance (mm) between two sequential slices.
2. FJ-B staining
FJ-B staining was performed as described previously.40 In brief, one of every six serial sections was immersed in a solution containing 1% sodium hydroxide and 80% ethanol for 5 min followed by 2 min in 70% ethanol. Brain sections were transferred to 0.06% potassium permanganate for 10 min, followed by immersion in 0.0004% FJ-B solution containing 0.01% glacial acetic acid and then transferred to a 1 μg/mL of 4,6-diamino-2-phenylindole (DAPI) solution for 5 min. Sections were air-dried, cleared in xylene for 1 min before mounting with DPX. Slides were examined under a Nikon Eclipse TE 2000 U epifluorescence microscope at 488 nm wavelength using a 20× magnification objective. Degenerative cells in the hippocampal dentate gyrus were counted using the NIS-Elements and AR 3.0 software from Nikon Instruments (Melville, N.Y).
Immunohistochemistry
To assess the expression of intracellular adhesion molecule 1 (ICAM-1), F4/80 (a marker for microglia/macrophages), inducible nitric oxide synthase (iNOS) and arginase-1 (Arg-1), 30 μm thick frozen brain sections were immunostained with respective antibodies. In brief, sections were blocked with 5% normal donkey serum, then incubated overnight at 4°C with a polyclonal goat anti–ICAM-1 antibody (1:200; Santa Cruz Inc. CA), a mixture containing monoclonal rat anti-mouse F4/80 antibody (1:200; eBioscience, CA) and a polyclonal rabbit anti-iNOS antibody (1:300; Millipore, Temecula, CA) or a mixture containing monoclonal rat anti-mouse F4/80 antibody (1:200; eBioscience, CA) and a polyclonal goat anti-Arg-1 antibody (1:200; Santa Cruz, CA). Sections were rinsed and incubated for 1 h at room temperature with Texas Red-conjugated rabbit anti-goat antibody (1:500; Jackson ImmunoResearch Laboratories, West Grove, PA) to detect ICAM-1 and Arg-1, a Texas Red-conjugated goat anti-rabbit antibody to detect iNOS (1:500; Jackson ImmunoResearch Laboratories), or a FITC-conjugated goat anti-rat antibody (1:500; Jackson ImmunoResearch Laboratories) to detect F4/80. The sections were then transferred into a 1 μg/mL DAPI solution for 5 min. After DAPI staining, all sections were air-dried, mounted with Fluorogel mount (Electron Microscopy Sciences, Hatfield, PA), and visualized under a Nikon Eclipse TE 2000 U epifluorescence microscope using a 20× magnification objective. The 488 nm, 594 nm, and 350 nm wavelengths filters were used to visualize FITC, Texas Red, and DAPI staining, respectively.
Western blotting analysis
Western blotting was performed to determine the expression of CB1, CB2, cyclooxygenase (COX)-2, iNOS, extracellular-signal-regulated kinase (ERK)1/2, phosphorylated ERK1/2 (pERK1/2), AKT, pAKT, and beta actin. The ipsilateral cortices from the experimental animals were manually homogenized in ice-cold lysis buffer containing 50 mM HEPES, pH 7.5, 6 mM MgCl2, 1 mM EDTA, 75 mM sucrose, 2.5 mM benzamidine, 1 mM dithiothreitol, 1% Triton X-100, and the protease inhibitor cocktail,41 centrifuged at 10,000 rpm for 25 min. Proteins (30–40 μg) from each sample supernatant were separated by electrophoresis on 4–12% SDS-polyacrylamide gels (Invitrogen, Carlsbad, CA), and then transferred to a polyvinylidene difluoride membrane. The membranes were blocked with 5% fat-free milk for 1 h and incubated overnight at 4°C with rabbit polyclonal antibodies against CB1 (1:400; Santa Cruz, CA), CB2 (1:500; Santa Cruz, CA or 1:500; Cayman Chemical, MI), iNOS (1:1000; Millipore, Tenecula, CA), COX-2 (1:500), pERK1/2 (1:1000), ERK1/2 (1:1000), pAKT (1:1000), AKT (1:1000) (all from Cell Signaling Technology Inc., Danvers, MA) or a mouse anti-β-actin monoclonal antibody (1:5000; Sigma). The blots were washed in TBS-Tween and further incubated for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibodies (1: 5000; Bio-Rad Life Sciences, Hercules, CA). Reactive proteins were visualized by enhanced chemiluminescence according to the manufacturer's protocol (Thermo Scientific, Rockford, IL).
Evans blue extravasation
Quantification of Evans blue dye in brain tissues was performed as described previously,42 with minor modifications. Three and 7 days post-TBI, animals were injected with 2% Evans blue in saline at 5 mL/kg via the jugular veins. Four hours later, mice were transcardially perfused with heparin saline to clear Evans blue from blood vessels. Each cerebral hemisphere was weighed, homogenized in a three-fold volume of 50% trichloroacetic acid, and centrifuged at 10,000 rpm for 25 min. The level of Evans blue in each sample supernatant was measured using a spectrofluorometer microplate reader (Molecular Devices, Sunnyvale, CA) with excitation wavelength at 585 nm and emission wavelength at 610 nm, and then calculated based on a standard curve. Results were expressed in micrograms of Evans blue dye per milligram of tissue.
Statistical analysis
All data analyses were performed using GraphPad InStat 3 software (GraphPad Software, Inc., La Jolla, CA). The analysis of variance together with the Bonferroni multiple comparison post-test was used to compare differences among the multiple groups, and the Student t test was used to compare between two groups. Results were quantified and expressed as mean±standard error of the mean. Statistical significance was defined as p≤0.05.
Results
WWL70 alleviates TBI-induced deficits in fine motor movement and motor coordination
To evaluate the effect of WWL70 on TBI-induced deficits in fine motor movements, beam-walk balance test was performed, and the number of foot faults over a total of 50 steps was determined. The missteps in vehicle-TBI animals were 49±1, 34±3, and 27±4 on days 7, 11, and 14, respectively. Although post-treatment with WWL70 at 5 mg/kg did not have any effect, treatment with WWL70 at 10 mg/kg improved the performance significantly. In these treatment groups, only 18±3, 13±2, and 8±2 missteps were observed at 7, 11, and 14 days, respectively (Fig. 2A).
FIG. 2.

WWL70 improved TBI-induced alterations in motor coordination. Mice were treated with WWL70 (5 mg/kg or 10 mg/kg) 30 min after injury and the behavioral tests were performed at different time points. (A) Effect of WWL70 on fine motor movement was assessed by a beam-walk test, and the deficits in fine motor movements were recorded as foot faults. The number of foot faults dramatically increased at various time points after injury, although a partial recovery was shown at 11 and 14 days post-TBI. Treatment with WWL70 at 10 mg/kg but not at 5 mg/kg significantly reduced the number of foot faults at 7, 11 and 14 days post-TBI (**p<0.01; mean±standard error of the mean; n=8–13). (B) Effect of WWL70 on motor coordination was evaluated by rotarod test, and the deficits in motor coordination were reflected by the latency to fall (in sec) from the rotarod. Controlled cortical impact (CCI) decreased the latency to fall, which was significantly increased by WWL70 treatment. #p<0.05 and ##p<0.01 were obtained when the WWL70 (5 mg/kg, n=8) treated group was compared with the vehicle-TBI group (n=12) at the corresponding time points. *p<0.05 and ***p<0.001 were obtained when the WWL70 (10 mg/kg, n=12) treated group was compared with the vehicle-TBI group (n=12) on days 1, 3, and 7 post-TBI.
The rotarod test was used to assess the effect of WWL70 on TBI-induced impairment in motor coordination. The latency of animals to fall from the rotarod was recorded and expressed in seconds. There was a dramatic difference in the latency to fall when vehicle-treated TBI mice were compared with the sham-injured animals at 1, 3, and 7 days post-injury. The time was 57.23±4.22 versus 107.7±3.71 on day 1, 74.92±4.8 versus 111.63±2.83 on day 3, and 87.32±4.42 versus 114±2.65 on day 7 (p<0.01; Fig. 2B). WWL70 treatment improved motor coordination of TBI mice in a concentration dependent manner. The latency to fall in animals treated with WWL70 at 5 mg/kg increased from 74.92±4.8 to 99.57±5.21 on day 3 (p<0.01) and from 87.32±4.42 to 100.14±3.56 on day 7 (p<0.05) post-injury when compared with the vehicle-TBI groups. At 10 mg/kg, WWL70 treatment improved motor coordination starting on day 1 post-injury. The latency to fall extended from 57.23±4.22 to 70.83±5.21 on day 1 (p<0.05) and further increased to 105.33±4.5 on day 3 and 109.41±3.54 on day 7, which are all significantly higher than those of the vehicle-TBI groups. Notably, a time-dependent recovery was observed in motor coordination in both the vehicle- and the drug-treated groups (Fig. 2A and B), and there is no significant difference in rotarod performance on day 11 post-injury among sham-injured, vehicle and WWL70 treated groups (Fig. 2B).
These results demonstrated that WWL70 treatment substantially alleviated TBI-induced deficits in motor coordination. Because better effects were achieved with WWL70 at 10 mg/kg, this concentration was used in the subsequent experiments to assess the role of WWL70 on TBI-induced alterations in learning/memory, histological, and molecular changes.
WWL70 has no effect on spatial learning and memory but improves TBI-induced deficits in working memory performance
Spatial learning and memory were evaluated on days 17–20 after injury. Sham-injured animals reached to the platform with an average escape latency of 39.7±1.93 sec on day 18, and 31.66±2.16 sec on day 19 post-injury. Compared with sham-injured animals, the escape latency for vehicle- (51.59±2.01 sec; 44.4±2.6 sec) and WWL70- (51.59±2.01 sec; 44.4±2.6 sec) treated TBI animals was significantly greater on days 18 (p<0.01) and 19 (p<0.05), respectively (Fig. 3A). There was no significant difference in escape time among sham, vehicle-treated, and WWL70-treated TBI mice on day 20 post-TBI (Fig. 3A). In the probe trial performed to measure spatial memory 1 h after the end of the last training session, sham-injured animals spent significantly more time (20.59±1.12 sec) in the western quadrant where the platform was previously located than the vehicle-treated mice (15.1±1.31; p<0.05; Fig. 3B). Although there was a slight increase in the time spent in the western quadrant in WWL70-treated animals, there was no significant difference between these animals and the vehicle-treated mice (p>0.05; Fig. 3B).
FIG. 3.

WWL70 improved traumatic brain injury (TBI)-induced deficits in working memory performance. Mice were treated with WWL70 (10 mg/kg) 30 min after injury, and behavioral tests to determine the role of WWL70 on TBI-induced deficits in spatial learning and memory and on working memory were performed at different time points. The Morris water maze test was conducted 17–20 days after TBI. (A) During the training period of the test, sham-injured animals spent significantly less time to locate the platform compared with controlled cortical impact (CCI)-vehicle treated animals on training days 2 (p<0.01) and 3 (p<0.05) post-injury, indicating there is a spatial learning deficit in TBI animals. There is no significant difference between the WWL70 and vehicle-treated TBI mice (p>0.05); (B) In the probe trial performed on day 20 after injury, vehicle-treated TBI mice spent significantly less time in the western quadrant than the sham-injured mice. The time spent by WWL70-treated animals was not significantly different from that spent by sham-injured and vehicle-treated TBI animals (n=8–13). (C) Spontaneous alternation Y maze test was performed on day 10 post-injury, and deficits in hippocampus performance were determined by the number of alternations achieved during 5 min exploration in the maze. CCI reduced the number of alternations, which was significantly increased by WWL70 treatment. *p<0.05 and **p<0.01 were obtained when the sham and WWL70-treated groups were compared with the TBI-vehicle group (mean±standard error of the mean; n=8–13).
Then we assessed whether WWL70 could affect the working memory performance using Y-maze. TBI mice performed a significantly lower percent Y maze arms alternation compared with the sham control animals (49.3±5.07% versus 64.2±7.22%; p<0.05). WWL70 treatment completely restored the ability of TBI mice to continuously alternate arms during Y maze exploration (69.67±4.98 %; Fig. 3C). These results indicate that the WWL70 treatment restores TBI-induced deficits in working memory but has no significant effect on spatial memory.
WWL70 reduces brain lesion in TBI mice and the effect is blocked by CB1 receptor antagonist
To investigate the effect of WWL70 on TBI-induced brain injury, H&E staining was performed to estimate the brain lesion volume at 3, 7, and 21 days post-injury.39,43 Although there was no significant improvement on day 3 post-injury, WWL70 treatment at 10 mg/kg significantly reduced the lesion volume from 14.23±0.32 mm3 to 10.88±0.57 mm3 on day 7, and from 17.59±0.95 mm3 to 13.32±0.95 mm3 on day 21 post-TBI (Fig. 4A, B). To determine which cannabinoid receptor was involved in the protective effect of WWL70 on lesion volume, TBI mice were administered CB1 or CB2 antagonist together with WWL70 for 7 consecutive days. The lesion volume in the WWL70- (10 mg/kg) and AM281- (3 mg/kg) treated group is 15.25±1.19 mm3, which is similar to that of the vehicle-TBI group (14.23±0.32 mm3), suggesting the protective effect of WWL70 is blocked by CB1 receptor antagonist. On the other hand, the use of CB2 antagonist AM 630 (3 mg/kg) did not affect the protective effect of WWL70, indicating the reduction of lesion volume by WWL70 is independent of CB2 receptor activation (Fig. 4C).
FIG. 4.

WWL70 reduced lesion volume in TBI mouse brain and the effect was blocked by CB1 receptor antagonist. Brains were collected at 3, 7, and 21 days post-injury, sectioned and stained with hematoxylin and eosin (H&E) to assess the lesion volume. (A) Representative H&E stained brain sections showing noticeable lesions on the ipsilateral hemispheres of both vehicle- and WWL70-treated mice. (B) Quantification of lesion volume by National Institutes of Health ImageJ software indicated that the lesion volume increased over time in vehicle-treated animals and was significantly reduced by WWL70 treatment at 7 and 21 days post-injury (*p<0.05; mean±standard error of the mean; n=4). (C) The reduction of lesion volume by WWL70 on day 7 post-injury was completely blocked by co-administration of CB1 antagonist AM 281, but not the CB2 antagonist AM 630 (*p<0.05; mean±standard error of the mean; n=4). CCI, controlled cortical impact. Color image is available online at www.liebertpub.com/neu
WWL70 attenuates neuronal degeneration in the dentate gyrus of TBI mice and the effect is partially reversed by CB1 and CB2 antagonists
FJ-B staining is a commonly used method to detect neuronal degeneration after TBI.43,44 Three and 7 days after TBI, the number of FJ-B positive cells in the ipsilateral dentate gyrus was significantly reduced by WWL70 when compared with the vehicle treated-animals. The average number of FJ-B positive cells per dentate gyrus without and with WWL70 treatment was 972±175 and 558±70 (p<0.05) on day 3, 1590±150 and 870±105 (p<0.01) on day 7 (Fig. 5A-B). No FJ-B positive cells were found in the dentate gyrus of the sham control animals, as well as in the contralateral dendate gyrus of the TBI animals (data not shown). To investigate whether cannabinoid receptors are involved in the neuroprotective effect of WWL70, we administered TBI mice with CB1 or CB2 antagonist together with WWL70 for 7 consecutive days. As illustrated in Figure 5C, co-treatment of WWL70 with CB1 antagonist or CB2 antagonist significantly reduced the neuroprotective effect of WWL70. The average numbers of FJ-B positive cells per dentate gyrus in WWL70 plus CB1 antagonist and WWL70 plus CB2 antagonist-treated TBI groups were 912±73 and 1021±102; both were significantly higher than the WWL70-treated TBI group (602±84). These results indicate that the effect of WWL70 on neurodegeneration is mediated, at least in part, by activation of CB1 and CB2 receptors.
FIG. 5.

WWL70 attenuated neuronal degeneration in the dentate gyrus of TBI mice and the effect was partially blocked by CB1 and CB2 receptor antagonists. Brain sections were stained with FJ-B to determine the degenerating cells in the dentate gyrus. (A) Representative micrographs showing FJ-B positive cells in the hippocampal dentate gyrus of both vehicle-and drug-treated groups at 3 and 7 days post-TBI (degenerating cells shown in green and 4,6-diamino-2-phenylindole positive cells in blue). Scale bar=50 μm. (B) Treatment with WWL70 significantly reduced the number of degenerating cells in dentate gyrus at 3 and 7 days post-TBI (*p<0.05 and **p<0.01; mean±standard error of the mean; n=4). (C) The reduction of WWL70 on FJ-B positive cells/dentate gyrus on day 7 post-TBI was significantly blocked by co-administration of AM281 and AM630, the respective CB1 and CB2 receptor antagonists (**p<0.01; mean±standard error of the mean; n=4). CCI, controlled cortical impact. Color image is available online at www.liebertpub.com/neu
WWL70 attenuates blood-brain barrier breakdown
Blood-brain barrier (BBB) breakdown is an important hallmark of TBI pathology usually observed in inflamed tissues.45 To investigate the effect of WWL70 on TBI-induced BBB dysfunction, we examined two factors that are associated with increased BBB permeability. First, we determined the expression of ICAM-1, a marker of leukocyte transmigration,46–49 which is shown to be up-regulated by inflammatory molecules such as iNOS and tumor necrosis factor (TNF)-α.45,50 The enhanced immunoreactivity of ICAM-1 was mainly observed in the cortex adjacent to the lesion site at 3 and 7 days post-TBI and was suppressed by WWL70 treatment (Fig. 6A). Next, we measured the levels of Evans blue extravasation in brain tissues as an index of BBB permeability using a fluorometric analysis. The levels of Evans blue in the ipsilateral cerebral hemisphere were 3.51±0.43 μg/g and 5.46±1.22 μg/g on days 3 and 7 post-TBI, which were higher than those in the sham control group (2.04±0.1 μg/g). The increased Evans blue extravasation was significantly attenuated by WWL70 treatment (Fig. 6B). In the contralateral hemispheres, there was no significant difference in Evans blue levels among sham control, vehicle-, and WWL70-treated TBI groups.
FIG. 6.

WWL70 attenuated blood-brain barrier breakdown. (A) Immunoreactivity of ICAM-1 in the ipsilateral cerebral cortices increased at 3 and 7 days post-TBI and decreased by WWL70 treatment. Representative microphotographs from both vehicle- and WWL70-treated groups are shown (ICAM-1 shown in red, and 4,6-diamino-2-phenylindole shown in blue). Scale bar=50 μm. (B) Evans blue extravasation in the ipsilateral brain tissue was increased at 3 and 7 days post-TBI and significantly reduced by WWL70 treatment (*p<0.05; mean±standard error of the mean; n=4). CCI, controlled cortical impact. Color image is available online at www.liebertpub.com/neu
WWL70 suppresses iNOS and COX-2 expression in TBI mice brain
The pro-inflammatory mediators such as COX-2 and iNOS are known to be up-regulated and contribute to the progression of the secondary brain injury.51–54 To test whether the therapeutic effect of WWL70 is associated with the inhibition of these pro-inflammatory mediators, their expression in the ipsilateral cerebral cortex was assessed by Western blot. Compared with the sham control group, the expression of COX-2 and iNOS was dramatically increased on days 3 and 7 after injury, and the increased expression was almost completely blocked by the WWL70 treatment (Fig. 7A-C).
FIG. 7.

WWL70 suppressed inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX)-2 expression in TBI mice brain. Brain homogenates were electrophoresed and analyzed by Western blot for the expression of iNOS, COX-2, and beta actin. (A) Representative immunoblots showing the expression of iNOS and COX-2 in the ipsilateral cortices of both vehicle- and WWL70-treated animals at 3 and 7 days post-TBI; actin was used as a loading control. (B, C) Quantification of iNOS and COX-2 signal intensity indicating that the expression of iNOS and COX-2 was increased post-TBI and significantly reduced by WWL70 treatment (*p<0.05 and **p<0.01; mean±standard error of the mean; n=6). CCI, controlled cortical impact.
WWL 70 suppresses the expression of iNOS and increases the expression of Arg-1
iNOS is expressed in microglia and macrophages under inflammatory conditions.55,56 Using Western blot analysis, we found that WWL70 suppressed iNOS induction in the ipsilateral cerebral cortex on days 3 and 7 post-TBI. The density of microglia near the lesion site, however, was not altered by the treatment. We hypothesized that the microglia phenotype might be altered after the treatment. To test this possibility, the expression of iNOS and Arg-1, the pro- and anti-inflammatory molecules induced in reactive microglia,57,58 was examined using monoclonal antibodies against iNOS and Arg-1 and polyclonal antibody against F4/80, a marker of reactive microglia/macrophages. Consistent with our hypothesis, we found that more that 70% of F4/80 positive cells expressed iNOS in vehicle-treated TBI mice cortices on days 3 and 7 post-injury, whereas in the WWL70-treated group, the expression of iNOS was only found in 20% of F4/80 positive microglia/macrophages (Figs. 8A, B). By contrast, about 25–30% of F4/80 positive cells express Arg-1 in WWL 70-treated groups on days 3 and 7 post-injury, when compared with the vehicle-treated TBI group, in which only 2% F4/80 positive cells were shown to express Arg-1 (Figs. 9A, B). These results suggest that treatment with WWL70 shifted microglia/macrophages from M1 to M2 phenotypes. None to very few microglia/macrophages were found in the contralateral hemispheres of the TBI and control animals (data not shown).
FIG. 8.

WWL70 decreased the number of inducible nitric oxide synthase (iNOS) expressing microglia cells. (A) Representative microphotographs of double immunostaining of iNOS and F4/80 in ipsilateral cerebral cortices from both vehicle- and WWL70-treated groups on days 3 and 7 post-TBI are shown. The merged image showed yellow fluorescence, meaning that iNOS staining (red) is co-localized with F4/80 (green). Nuclei were stained with 4,6-diamino-2-phenylindole as shown with blue fluorescence. Scale bar=50 μm. The inserts in the merged micrographs are higher magnification photographs of the arrow-pointed boxes. (B) Quantification of the overlap fraction of iNOS immunopositive/F4/80 positive microglia/macrophages indicating that WWL70 treatment significantly suppressed iNOS expression in inflammatory cells on days 3 and 7 post-surgery (***p<0.001; mean±standard error of the mean; n=4). CCI, controlled cortical impact. Color image is available online at www.liebertpub.com/neu
FIG. 9.

WWL70 enhanced arginase (Arg)-1 expression. (A) Representative microphotographs of double immunostaining of Arg-1 and F4/80 in ipsilateral cerebral cortices from both vehicle- and WWL70-treated groups on days 3 and 7 post-TBI are shown. The merged image showed yellow fluorescence, meaning that Arg-1 staining (red) is co-localized with F4/80 (green). Nuclei were stained with 4,6-diamino-2-phenylindole (blue). Scale bar=50 μm. The inserts in the merged micrographs are higher magnification photographs of the arrow-pointed boxes. (B) Quantification of the overlap fraction of Arg-1 positive/F4/80 positive cortical cells indicating that WWL70 treatment significantly enhanced Arg-1 expression on days 3 and 7 post-injury (***p<0.001; mean±standard error of the mean; n=4). CCI, controlled cortical impact. Color image is available online at www.liebertpub.com/neu
Treatment with WWL70 enhances CB1 and CB2 expression in TBI mice brain
Given the fact that the protective effect of WWL70 on TBI-induced lesion and neurodegeneration was mediated by CB1 and CB2 receptors, we sought to explore the impact of WWL70 treatment on their levels of expression at 3 and 7 days after TBI using Western blot analysis. Although there was no difference in the expression levels of both CB1 and CB2 receptors between the TBI and sham control animals, treatment with WWL70 significantly increased the expression of both CB1 and CB2 receptors. There was a 40% increase in the expression of CB1 at 3 and 7 days post-treatment, and the expression of CB2 increased by 60% when compared with the vehicle-treated TBI mice and the sham control animals (Fig. 10A–C).
FIG. 10.

WWL70 up-regulated the expression of CB1 and CB2 receptors in TBI mouse brain. Brain homogenates from experimental animals were electrophoresed by SDS-PAGE and analyzed by Western blot for the expression of CB1, CB2, and beta actin. (A) Representative immunoblots showing the expression of CB1 and CB2 in the ipsilateral cortices of both vehicle and WWL70-treated animals at 3 and 7 days post-TBI. Actin was used as a loading control. (B, C) Quantification of CB1 and CB2 signal intensity indicating both CB1 and CB2 expression was up-regulated by WWL70 treatment on days 3 and 7 post-injury (*p<0.05 and **p<0.01; mean±standard error of the mean; n=6). CCI, controlled cortical impact.
WWL70 restores TBI-induced decrease in ERK phosphorylation and increases AKT phosphorylation
To determine the downstream signaling mechanisms involved in the neuroprotective effects of WWL70, we assessed the effect of WWL70 on phosphorylation of two pro-survival kinases, ERK1/2 and the serine/threonine protein kinase AKT. There was a significant decrease in the level of ERK1/2 phosphorylation in the ipsilateral cerebral cortex at 3 and 7 days post-TBI when compared with the sham controls. After WWL70 treatment, the phosphorylation of ERK1/2 was restored (Figs. 11A, B). Although there were no significant changes in the levels of phophorylated AKT between sham control and vehicle-treated TBI mice, treatment with WWL70 substantially increased AKT phosphorylation by 17±9% and 38±4% on days 3 and 7 post-TBI, respectively (Figs. 11A, C).
FIG. 11.

WWL70 restored extracellular-signal-regulated kinase (ERK) and increased serine/threonine protein kinase AKT phosphorylation in TBI mouse brain. Brain homogenates were electrophoresed and analyzed by Western blot for ERK and AKT phosphorylation. (A) Representative immunoblots showing the expression of ERK, pERK, AKT, and pAKT in the ipsilateral cortices of both vehicle- and WWL70-treated groups at 3 and 7 days post-TBI. Actin was used as a loading control. (B) Quantification of pERK/ERK ratio showing a significant increase in ERK phosphorylation by WWL70 when compared with the vehicle-TBI group at 3 and 7 days post-injury. (*p<0.05; mean±standard error of the mean; n=6). (C) Quantification of pAKT/AKT ratio showing a significant increase in AKT phosphorylation by WWL70 compared with the vehicle-TBI group at 3 and 7 days post-injury (*p<0.05 and **p<0.01; mean±standard error of the mean; n=6). CCI, controlled cortical impact.
Discussion
This is the first study to document the beneficial effect of ABHD6 inhibition in a mouse model of TBI. Our results provide clear evidence that selective inhibition of ABHD6 by WWL70 produced a broad-spectrum therapeutic effect in mice with TBI. WWL70 reduced lesion volume in the cortex and neurodegeneration in the dentate gyrus; suppressed the expression of COX-2 and iNOS and increased the expression of Arg-1; attenuated BBB breakdown; and improved the functional recovery including motor coordination, fine motor movement, and working memory. Further, we demonstrated that the beneficial effects of WWL70 are mediated by the activation of CB1 and CB2 receptors and the enhanced phosphorylation of cell survival kinases ERK1/2 and AKT.
The animal model of TBI induced by CCI in this study reproduced several behavioral deficits as shown by impairments in motor coordination, fine motor movement, and memory performance. These deficits were reversed by post-injury treatment with WWL70. Notably, WWL70 at 10 mg/kg had a better effect for the improvement of TBI-induced deficits in motor coordination and fine motor movement and restored the working memory performance. These results are in harmony with the previous reports that administration of exogenous 2-AG reduces TBI-induced neurological deficits associated with the fine motor movements from 24 h to weeks or even months after closed head injury.9,12 A recent study indicates that administration of a single dose of N-arachidonoyl-L-serine, a cannabinoid-like compound, also ablates TBI-induced behavioral deficits over 14 days.10 Prolonged oral administration of a cannabinoid is also shown to improve cognitive performance in a mouse model of Alzheimer's disease.51 All these studies demonstrate that exogenous as well as endogenous cannabinoids can improve behavioral performance after brain injury.
CCI-induced TBI produces lesions at the site of impact, and the lesion volume is parallel to the injury severity.43,52,53 Consistent with the findings by others,52,54 our results also indicate that the lesion volume in the vehicle-treated TBI mice increased over time after injury. Chronic administration of WWL70 reversed that progress and produced a substantial reduction of the lesion volume. It is known that the hippocampal dentate gyrus is the epicenter for cognitive and memory function and, therefore, neuronal death in the dentate gyrus could contribute to TBI-induced cognitive impairment.59,60 In the present study, we found that WWL70 significantly reduced neuronal degeneration in the dentate gyrus, as well as the working memory deficits after brain injury.
It has been demonstrated that ABHD6 is located in both cerebral cortex and hippocampal neurons.19,61 Therefore, inhibition of ABHD6 by WWL70 may lead to an increase of 2-AG in these brain areas. Unlike MAGL, the principal 2-AG inactive enzyme that is located in the presynaptic nerve terminals, ABHD6 resides in the post-synaptic neurons.19 Therefore, ABHD6 and MAGL may control the different pools of 2-AG degradation. In addition, ABHD6 appears to be a membrane protein with its active site facing cytosol and is therefore well suited to guard the intracellular pools of 2-AG at the site of its production.17–19,24 Further, unlike the selective MAGL inhibitor, which causes a robust 2-AG increase, CB1 receptor desensitization in the brain, and the behavioral tolerance, chronic inactivation of ABHD6 is shown to cause a moderate increase of 2-AG, which can maintain the cannabinoid receptor efficacy and avoid the undesirable side effects.21,24
TBI-induced neuronal cell death is often accompanied by microglial activation and the release of pro-inflammatory mediators.62,63 The pro-inflammatory mediators such as COX-2 and iNOS are inducible and markedly up-regulated after TBI and believed to play an important role in the secondary process of injury.64–67 Our findings showed that WWL70 suppressed the expression of COX-2 and iNOS in brain homogenates and inflammatory cells at 3 and 7 days post-TBI. It has been shown that 2-AG protects neurons from insults by acting either as a COX-2 inhibitor,68 or via a signaling mechanism downstream of CB1 receptor activation.69 The inhibition of COX-2 and iNOS expression may improve the functional outcome in rodent model of TBI as suggested previously.66,70
Despite the fact that WWL70 treatment did not affect the density of microglia cells near the lesion site in the TBI mice brain, the iNOS expression in these cells was significantly reduced. In contrast, the expression of Arg-1 was dramatically increased in microglia. These results suggest that WWL70 treatment can switch microglia from a pro-inflammatory phenotype (M1) to an anti-inflammatory phenotype (M2).57,71,72 It has been reported that Arg-1 is upregulated in M2 phenotype microglia/macrophages that possess anti-inflammatory function and are also implicated in the repair process after injury.59,71,73 Our finding is consistent with a very recent report showing that administration of exogenous 2-AG in a mouse model of experimental autoimmune encephalomyelitis shifted microglia/macrophages from M1 to M2 phenotypes, as demonstrated by an increased expression of Arg-1 in the spinal cord white matter.73
BBB disruption that occurs after TBI may contribute to the inflammatory response and is reflected by the increased expression of ICAM-1 produced by the endothelial and immune cells.66 Previous reports have shown that exogenous 2-AG administration reduces BBB leakage and attenuates ICAM-1 expression after TBI.11,66 Consistently, we found that the inhibition of ABHD6 attenuated ICAM-1 expression and Evans blue extravasation in the ipsilateral mouse brain at 3 and 7 days post-injury. It has been reported that the expression of ICAM-1 is up-regulated by inflammatory molecules such as TNF-α,45 and the selective inhibition of iNOS results in a substantial decrease in the expression levels of TNF-α and ICAM-1.50
It is still debatable whether the action of cannabinoids is coupled to the CB1 and CB2 receptors, and the relative contribution of these receptors is likely dependent on the model systems applied and the cell types involved.74,75 In the current study, we found that the protective effect of WWL70 on lesion volume was blocked by CB1 receptor antagonist whereas its effect on hippocampal neurodegeneration was partially mediated by CB1 and CB2 receptors. These results are consistent with the findings by the use of exogenous 2-AG in the mouse model of close head injury.12 Although the therapeutic effect of 2-AG is thought to be mediated by CB1 receptor activation, the involvement of CB2 is not excluded.9,11,12
Indeed, the same group has recently reported that N-arachidonoyl-L-serine, a cannabinoid-like compound structurally related to the endocannabinoid family, exerts neuroprotective effects in closed head injury via CB2 receptor- and transient receptor potential vanilloid 1- dependent mechanisms.10 In contrast to the results obtained by the chronic use of MAGL inhibitor, which causes CB1 receptor desensitization and down-regulation,21 we found that sustained inhibition of ABHD6 by WWL70 significantly increased the expression of both CB1 and CB2 receptors in the ipsilateral cerebral cortex at 3 and 7 days post-TBI. The up-regulation of cannabinoid receptors has also been demonstrated in a mouse model of intestinal inflammation,76 alcoholism, and other pathological conditions,77–80 in which the 2-AG levels are dramatically elevated.77,78 All these findings suggest that a moderate increase of 2-AG can up-regulate the expression of CB1 and CB2 receptors and enhance the cannabinergic signaling under TBI and other pathological conditions.
Phosphorylation of ERK1/2 and PI3K/AKT is thought to be the common signaling event that occurs downstream of cannabinoid receptor activation81 and is involved in the control of the inflammatory response and neuronal survival.81–83 In this study, we found that CB1 and CB2 up-regulation by WWL70 is also associated with the activation of AKT and the restoration of ERK phosphorylation in TBI mice brain. Similarly, the administration of N-arachidonoyl-L-serine to mice subjected to close head injury was also found to reverse the reduction of phosphorylated ERK1/2 and lead to the increase of AKT phosphorylation in both the ipsilateral and contralateral cortices at the early stage of brain injury.10
Conclusion
This study suggests that chronic inactivation of ABHD6 may allow more precise fine-tuning of cannabinoid receptor signaling and provide therapeutic relief without causing undesirable side effects. It is noteworthy that inhibition of ABHD6 has recently been shown to suppress seizure activity in a mouse model of Huntington's disease.84 Because inhibition of ABHD6 does not down-regulate but actually enhances the expression of the cannabinoid receptors without causing behavioral tolerance, it is reasonable to believe that selective inhibition of ABHD6 can be used as an attractive strategy for the treatment of patients with TBI and other neurological and neurodegenerative diseases.
Acknowledgments
This work was supported by grants from the Blast Lethality Injury and Research Program (R600-070-00000-00-106109) and the Defense Medical Research and Development Program (0130-10-00003-00002). The authors are grateful to Drs. De-Maw Chuang and Fengshan Yu for careful reading and critical comments on the manuscript. We also acknowledge Ms. Laura Tucker and Dr. Amanda Fu from the Center for Neuroscience and Regenerative Medicine for their help during behavioral tests and CCI surgery.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Loane D.J. Pocivavsek A. Moussa C.E. Thompson R. Matsuoka Y. Faden A.I. Rebeck G.W. Burns M.P. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat. Med. 2009;15:377–379. doi: 10.1038/nm.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pitkänen A. Longhi L. Marklund N. Morales D.M. McIntosh T.K. Neurodegeneration and neuroprotective strategies after traumatic brain injury. Drug Discov. Today: Disease Mechanisms. 2005;2:409–418. [Google Scholar]
- 3.Faul M. Xu L. Wald M. Coronado V. Centers for Disease Control and Prevention. National Center for Injury Prevention and Control; Atlanta, GA: 2010. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. [Google Scholar]
- 4.Jain K.K. Neuroprotection in traumatic brain injury. Drug Discov. Today. 2008;13:1082–1089. doi: 10.1016/j.drudis.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Loane D.J. Faden A.I. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol. Sci. 2010;31:596–604. doi: 10.1016/j.tips.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Werner C. Engelhard K. Pathophysiology of traumatic brain injury. Br. J. Anaesth. 2007;99:4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- 7.Di Marzo V. Bifulco M. De Petrocellis L. The endocannabinoid system and its therapeutic exploitation. Nat. Rev. Drug Discov. 2004;3:771–784. doi: 10.1038/nrd1495. [DOI] [PubMed] [Google Scholar]
- 8.Mackie K. Cannabinoid receptors as therapeutic targets. Annu. Rev. Pharmacol. Toxicol. 2006;46:101–122. doi: 10.1146/annurev.pharmtox.46.120604.141254. [DOI] [PubMed] [Google Scholar]
- 9.Shohami E. Cohen-Yeshurun A. Magid L. Algali M. Mechoulam R. Endocannabinoids and traumatic brain injury. Br. J. Pharmacol. 2011;163:1402–1410. doi: 10.1111/j.1476-5381.2011.01343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen-Yeshurun A. Trembovler V. Alexandrovich A. Ryberg E. Greasley P.J. Mechoulam R. Shohami E. Leker R.R. N-arachidonoyl-L-serine is neuroprotective after traumatic brain injury by reducing apoptosis. J. Cereb. Blood Flow Metab. 2011;31:1768–1777. doi: 10.1038/jcbfm.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Panikashvili D. Shein N.A. Mechoulam R. Trembovler V. Kohen R. Alexandrovich A. Shohami E. The endocannabinoid 2-AG protects the blood-brain barrier after closed head injury and inhibits mRNA expression of proinflammatory cytokines. Neurobiol. Dis. 2006;22:257–264. doi: 10.1016/j.nbd.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 12.Panikashvili D. Simeonidou C. Ben-Shabat S. Hanus L. Breuer A. Mechoulam R. Shohami E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413:527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- 13.van der Stelt M. Veldhuis W.B. van Haaften G.W. Fezza F. Bisogno T. Bar P.R. Veldink G.A. Vliegenthart J.F. Di Marzo V. Nicolay K. Exogenous anandamide protects rat brain against acute neuronal injury in vivo. J. Neurosci. 2001;21:8765–8771. doi: 10.1523/JNEUROSCI.21-22-08765.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hwang J. Adamson C. Butler D. Janero D.R. Makriyannis A. Bahr B.A. Enhancement of endocannabinoid signaling by fatty acid amide hydrolase inhibition: a neuroprotective therapeutic modality. Life Sci. 2010;86:615–623. doi: 10.1016/j.lfs.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naidoo V. Karanian D.A. Vadivel S.K. Locklear J.R. Wood J.T. Nasr M. Quizon P.M. Graves E.E. Shukla V. Makriyannis A. Bahr B.A. Equipotent inhibition of fatty acid amide hydrolase and monoacylglycerol lipase—dual targets of the endocannabinoid system to protect against seizure pathology. Neurotherapeutics. 2012;9:801–813. doi: 10.1007/s13311-011-0100-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mechoulam R. Panikashvili D. Shohami E. Cannabinoids and brain injury: therapeutic implications. Trends Mol. Med. 2002;8:58–61. doi: 10.1016/s1471-4914(02)02276-1. [DOI] [PubMed] [Google Scholar]
- 17.Savinainen J.R. Saario S.M. Laitinen J.T. The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2-arachidonoylglycerol signalling through cannabinoid receptors. Acta Physiol. (Oxf) 2012;204:267–276. doi: 10.1111/j.1748-1716.2011.02280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blankman J.L. Simon G.M. Cravatt B.F. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marrs W.R. Blankman J.L. Horne E.A. Thomazeau A. Lin Y.H. Coy J. Bodor A.L. Muccioli G.G. Hu S.S. Woodruff G. Fung S. Lafourcade M. Alexander J.P. Long J.Z. Li W. Xu C. Möller T. Mackie K. Manzoni O.J. Cravatt B.F. Stella N. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat. Neurosci. 2010;13:951–957. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chanda P.K. Gao Y. Mark L. Btesh J. Strassle B.W. Lu P. Piesla M.J. Zhang M.Y. Bingham B. Uveges A. Kowal D. Garbe D. Kouranova E.V. Ring R.H. Bates B. Pangalos M.N. Kennedy J.D. Whiteside G.T. Samad T.A. Monoacylglycerol lipase activity is a critical modulator of the tone and integrity of the endocannabinoid system. Mol. Pharmacol. 2010;78:996–1003. doi: 10.1124/mol.110.068304. [DOI] [PubMed] [Google Scholar]
- 21.Schlosburg J.E. Blankman J.L. Long J.Z. Nomura D.K. Pan B. Kinsey S.G. Nguyen P.T. Ramesh D. Booker L. Burston J.J. Thomas E.A. Selley D.E. Sim-Selley L.J. Liu Q.S. Lichtman A.H. Cravatt B.F. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat. Neurosci. 2010;13:1113–1119. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mangieri R.A. Piomelli D. Enhancement of endocannabinoid signaling and the pharmacotherapy of depression. Pharmacol. Res. 2007;56:360–366. doi: 10.1016/j.phrs.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scotter E.L. Abood M.E. Glass M. The endocannabinoid system as a target for the treatment of neurodegenerative disease. Br. J. Pharmacol. 2010;160:480–498. doi: 10.1111/j.1476-5381.2010.00735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marrs W.R. Horne E.A. Ortega-Gutierrez S. Cisneros J.A. Xu C. Lin Y.H. Muccioli G.G. Lopez-Rodriguez M.L. Stella N. Dual inhibition of alpha/beta-hydrolase domain 6 and fatty acid amide hydrolase increases endocannabinoid levels in neurons. J. Biol. Chem. 2011;286:28723–28728. doi: 10.1074/jbc.M110.202853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Myer D.J. Gurkoff G.G. Lee S.M. Hovda D.A. Sofroniew M.V. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761–2772. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- 26.Di Filippo C. Rossi F. Rossi S. D'Amico M. Cannabinoid CB2 receptor activation reduces mouse myocardial ischemia-reperfusion injury: involvement of cytokine/chemokines and PMN. J. Leukoc. Biol. 2004;75:453–459. doi: 10.1189/jlb.0703303. [DOI] [PubMed] [Google Scholar]
- 27.Sell S.L. Avila M.A. Yu G. Vergara L. Prough D.S. Grady J.J. DeWitt D.S. Hypertonic resuscitation improves neuronal and behavioral outcomes after traumatic brain injury plus hemorrhage. Anesthesiology. 2008;108:873–881. doi: 10.1097/ALN.0b013e31816c8a15. [DOI] [PubMed] [Google Scholar]
- 28.Galante M. Jani H. Vanes L. Daniel H. Fisher E.M. Tybulewicz V.L. Bliss T.V. Morice E. Impairments in motor coordination without major changes in cerebellar plasticity in the Tc1 mouse model of Down syndrome. Hum. Mol. Genet. 2009;18:1449–1463. doi: 10.1093/hmg/ddp055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bryan K.J. Lee H. Perry G. Smith M.A. Casadesus G. Transgenic mouse models of alzheimer's disease: behavioral testing and considerations. In: Buccafusco J.J., editor. Methods of Behavior Analysis in Neuroscience. CRC Press; Boca Raton (FL): 2009. [PubMed] [Google Scholar]
- 30.Chauhan N.B. Gatto R. Chauhan M.B. Neuroanatomical correlation of behavioral deficits in the CCI model of TBI. J. Neurosci. Methods. 2010;190:1–9. doi: 10.1016/j.jneumeth.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 31.Yu F. Zhang Y. Chuang D.M. Lithium reduces BACE1 overexpression, beta amyloid accumulation, and spatial learning deficits in mice with traumatic brain injury. J. Neurotrauma. 2012;29:2342–2351. doi: 10.1089/neu.2012.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka K. Yagi T. Shimakoshi R. Azuma K. Nanba T. Ogo H. Tamura A. Asanuma M. Effects of galantamine on L-NAME-induced behavioral impairment in Y-maze task in mice. Neurosci. Lett. 2009;462:235–238. doi: 10.1016/j.neulet.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 33.Lloyd E. Somera-Molina K. Van Eldik L.J. Watterson D.M. Wainwright M.S. Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. J. Neuroinflammation. 2008;5:28. doi: 10.1186/1742-2094-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Canas P.M. Porciuncula L.O. Cunha G.M. Silva C.G. Machado N.J. Oliveira J.M. Oliveira C.R. Cunha R.A. Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by beta-amyloid peptides via p38 mitogen-activated protein kinase pathway. J. Neurosci. 2009;29:14741–14751. doi: 10.1523/JNEUROSCI.3728-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dauge V. Sebret A. Beslot F. Matsui T. Roques B.P. Behavioral profile of CCK2 receptor-deficient mice. Neuropsychopharmacology. 2001;25:690–698. doi: 10.1016/S0893-133X(01)00291-3. [DOI] [PubMed] [Google Scholar]
- 36.Tchantchou F. Graves M. Ortiz D. Rogers E. Shea T.B. Dietary supplementation with 3-deaza adenosine, N-acetyl cysteine, and S-adenosyl methionine provide neuroprotection against multiple consequences of vitamin deficiency and oxidative challenge: relevance to age-related neurodegeneration. Neuromolecular Med. 2004;6:93–103. doi: 10.1385/NMM:6:2-3:093. [DOI] [PubMed] [Google Scholar]
- 37.Dall'Igna O.P. Fett P. Gomes M.W. Souza D.O. Cunha R.A. Lara D.R. Caffeine and adenosine A(2a) receptor antagonists prevent beta-amyloid (25-35)-induced cognitive deficits in mice. Exp. Neurol. 2007;203:241–245. doi: 10.1016/j.expneurol.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 38.Hughes R.N. The value of spontaneous alternation behavior (SAB) as a test of retention in pharmacological investigations of memory. Neurosci. Biobehav. Rev. 2004;28:497–505. doi: 10.1016/j.neubiorev.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 39.Dash P.K. Orsi S.A. Zhang M. Grill R.J. Pati S. Zhao J. Moore A.N. Valproate administered after traumatic brain injury provides neuroprotection and improves cognitive function in rats. PLoS One. 2010;5:e11383. doi: 10.1371/journal.pone.0011383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmued L.C. Hopkins K.J. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- 41.Tchantchou F. Xu Y. Wu Y. Christen Y. Luo Y. EGb 761 enhances adult hippocampal neurogenesis and phosphorylation of CREB in transgenic mouse model of Alzheimer's disease. FASEB J. 2007;21:2400–2408. doi: 10.1096/fj.06-7649com. [DOI] [PubMed] [Google Scholar]
- 42.Ay I. Francis J.W. Brown R.H., Jr. VEGF increases blood-brain barrier permeability to Evans blue dye and tetanus toxin fragment C but not adeno-associated virus in ALS mice. Brain Res. 2008;1234:198–205. doi: 10.1016/j.brainres.2008.07.121. [DOI] [PubMed] [Google Scholar]
- 43.Yu F. Wang Z. Tchantchou F. Chiu C.T. Zhang Y. Chuang D.M. Lithium ameliorates neurodegeneration, suppresses neuroinflammation, and improves behavioral performance in a mouse model of traumatic brain injury. J. Neurotrauma. 2012;29:362–374. doi: 10.1089/neu.2011.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hall E.D. Bryant Y.D. Cho W. Sullivan P.G. Evolution of post-traumatic neurodegeneration after controlled cortical impact traumatic brain injury in mice and rats as assessed by the de Olmos silver and fluorojade staining methods. J. Neurotrauma. 2008;25:235–247. doi: 10.1089/neu.2007.0383. [DOI] [PubMed] [Google Scholar]
- 45.Frank P.G. Lisanti M.P. ICAM-1: role in inflammation and in the regulation of vascular permeability. Am. J. Physiol. Heart Circ. Physiol. 2008;295:H926–H927. doi: 10.1152/ajpheart.00779.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Di Lorenzo A. Manes T.D. Davalos A. Wright P.L. Sessa W.C. Endothelial reticulon-4B (Nogo-B) regulates ICAM-1-mediated leukocyte transmigration and acute inflammation. Blood. 2011;117:2284–2295. doi: 10.1182/blood-2010-04-281956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y. Shaw S.K. Ma S. Yang L. Luscinskas F.W. Parkos C.A. Regulation of leukocyte transmigration: cell surface interactions and signaling events. J. Immunol. 2004;172:7–13. doi: 10.4049/jimmunol.172.1.7. [DOI] [PubMed] [Google Scholar]
- 48.Schneider C. Schuetz G. Zollner T.M. Acute neuroinflammation in Lewis rats—a model for acute multiple sclerosis relapses. J. Neuroimmunol. 2009;213:84–90. doi: 10.1016/j.jneuroim.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 49.Soetikno V. Sari F.R. Veeraveedu P.T. Thandavarayan R.A. Harima M. Sukumaran V. Lakshmanan A.P. Suzuki K. Kawachi H. Watanabe K. Curcumin ameliorates macrophage infiltration by inhibiting NF-kappaB activation and proinflammatory cytokines in streptozotocin induced-diabetic nephropathy. Nutr. Metab. (Lond) 2011;8:35. doi: 10.1186/1743-7075-8-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kan W.H. Hsu J.T. Schwacha M.G. Choudhry M.A. Raju R. Bland K.I. Chaudry I.H. Selective inhibition of iNOS attenuates trauma-hemorrhage/resuscitation-induced hepatic injury. J. Appl. Physiol. 2008;105:1076–1082. doi: 10.1152/japplphysiol.90495.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martin-Moreno A.M. Brera B. Spuch C. Carro E. Garcia-Garcia L. Delgado M. Pozo M.A. Innamorato N.G. Cuadrado A. de Ceballos M.L. Prolonged oral cannabinoid administration prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in Tg APP 2576 mice. J. Neuroinflammation. 2012;9:8. doi: 10.1186/1742-2094-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Elliott M.B. Jallo J.J. Tuma R.F. An investigation of cerebral edema and injury volume assessments for controlled cortical impact injury. J. Neurosci. Methods. 2008;168:320–324. doi: 10.1016/j.jneumeth.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 53.Taylor A.N. Rahman S.U. Sanders N.C. Tio D.L. Prolo P. Sutton R.L. Injury severity differentially affects short- and long-term neuroendocrine outcomes of traumatic brain injury. J. Neurotrauma. 2008;25:311–323. doi: 10.1089/neu.2007.0486. [DOI] [PubMed] [Google Scholar]
- 54.Hall E.D. Sullivan P.G. Gibson T.R. Pavel K.M. Thompson B.M. Scheff S.W. Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: more than a focal brain injury. J. Neurotrauma. 2005;22:252–265. doi: 10.1089/neu.2005.22.252. [DOI] [PubMed] [Google Scholar]
- 55.Iravani M.M. Kashefi K. Mander P. Rose S. Jenner P. Involvement of inducible nitric oxide synthase in inflammation-induced dopaminergic neurodegeneration. Neuroscience. 2002;110:49–58. doi: 10.1016/s0306-4522(01)00562-0. [DOI] [PubMed] [Google Scholar]
- 56.Arimoto T. Bing G. Up-regulation of inducible nitric oxide synthase in the substantia nigra by lipopolysaccharide causes microglial activation and neurodegeneration. Neurobiol. Dis. 2003;12:35–45. doi: 10.1016/s0969-9961(02)00017-7. [DOI] [PubMed] [Google Scholar]
- 57.Loane D.J. Byrnes K.R. Role of microglia in neurotrauma. Neurotherapeutics. 2010;7:366–377. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rojo A.I. Innamorato N.G. Martin-Moreno A.M. De Ceballos M.L. Yamamoto M. Cuadrado A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson's disease. Glia. 2010;58:588–598. doi: 10.1002/glia.20947. [DOI] [PubMed] [Google Scholar]
- 59.Gao X. Deng-Bryant Y. Cho W. Carrico K.M. Hall E.D. Chen J. Selective death of newborn neurons in hippocampal dentate gyrus following moderate experimental traumatic brain injury. J. Neurosci. Res. 2008;86:2258–2270. doi: 10.1002/jnr.21677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hilton G.D. Stoica B.A. Byrnes K.R. Faden A.I. Roscovitine reduces neuronal loss, glial activation, and neurologic deficits after brain trauma. J. Cereb. Blood Flow Metab. 2008;28:1845–1859. doi: 10.1038/jcbfm.2008.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Straiker A. Hu S.S. Long J.Z. Arnold A. Wager-Miller J. Cravatt B.F. Mackie K. Monoacylglycerol lipase limits the duration of endocannabinoid-mediated depolarization-induced suppression of excitation in autaptic hippocampal neurons. Mol. Pharmacol. 2009;76:1220–1227. doi: 10.1124/mol.109.059030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lenzlinger P.M. Morganti-Kossmann M.C. Laurer H.L. McIntosh T.K. The duality of the inflammatory response to traumatic brain injury. Mol. Neurobiol. 2001;24:169–181. doi: 10.1385/MN:24:1-3:169. [DOI] [PubMed] [Google Scholar]
- 63.Li B. Mahmood A. Lu D. Wu H. Xiong Y. Qu C. Chopp M. Simvastatin attenuates microglial cells and astrocyte activation and decreases interleukin-1beta level after traumatic brain injury. Neurosurgery. 2009;65:179–186. doi: 10.1227/01.NEU.0000346272.76537.DC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ahmad M. Rose M.E. Vagni V. Griffith R.P. Dixon C.E. Kochanek P.M. Hickey R.W. Graham S.H. Genetic disruption of cyclooxygenase-2 does not improve histological or behavioral outcome after traumatic brain injury in mice. J. Neurosci. Res. 2008;86:3605–3612. doi: 10.1002/jnr.21809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clark R.S. Kochanek P.M. Schwarz M.A. Schiding J.K. Turner D.S. Chen M. Carlos T.M. Watkins S.C. Inducible nitric oxide synthase expression in cerebrovascular smooth muscle and neutrophils after traumatic brain injury in immature rats. Pediatr. Res. 1996;39:784–790. doi: 10.1203/00006450-199605000-00007. [DOI] [PubMed] [Google Scholar]
- 66.Khan M. Im Y.B. Shunmugavel A. Gilg A.G. Dhindsa R.K. Singh A.K. Singh I. Administration of S-nitrosoglutathione after traumatic brain injury protects the neurovascular unit and reduces secondary injury in a rat model of controlled cortical impact. J. Neuroinflammation. 2009;6:32. doi: 10.1186/1742-2094-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kunz A. Park L. Abe T. Gallo E.F. Anrather J. Zhou P. Iadecola C. Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J. Neurosci. 2007;27:7083–7093. doi: 10.1523/JNEUROSCI.1645-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang J. Chen C. Endocannabinoid 2-arachidonoylglycerol protects neurons by limiting COX-2 elevation. J. Biol. Chem. 2008;283:22601–22611. doi: 10.1074/jbc.M800524200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Du H. Chen X. Zhang J. Chen C. Inhibition of COX-2 expression by endocannabinoid 2-arachidonoylglycerol is mediated via PPAR-gamma. Br. J. Pharmacol. 2011;163:1533–1549. doi: 10.1111/j.1476-5381.2011.01444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thau-Zuchman O. Shohami E. Alexandrovich A.G. Trembovler V. Leker R.R. The anti-inflammatory drug carprofen improves long-term outcome and induces gliogenesis after traumatic brain injury. J. Neurotrauma. 2012;29:375–384. doi: 10.1089/neu.2010.1673. [DOI] [PubMed] [Google Scholar]
- 71.Kuo H.S. Tsai M.J. Huang M.C. Chiu C.W. Tsai C.Y. Lee M.J. Huang W.C. Lin Y.L. Kuo W.C. Cheng H. Acid fibroblast growth factor and peripheral nerve grafts regulate Th2 cytokine expression, macrophage activation, polyamine synthesis, and neurotrophin expression in transected rat spinal cords. J. Neurosci. 2011;31:4137–4147. doi: 10.1523/JNEUROSCI.2592-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee S. Huen S. Nishio H. Nishio S. Lee H.K. Choi B.S. Ruhrberg C. Cantley L.G. Distinct macrophage phenotypes contribute to kidney injury and repair. J. Am. Soc. Nephrol. 2011;22:317–326. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lourbopoulos A. Grigoriadis N. Lagoudaki R. Touloumi O. Polyzoidou E. Mavromatis I. Tascos N. Breuer A. Ovadia H. Karussis D. Shohami E. Mechoulam R. Simeonidou C. Administration of 2-arachidonoylglycerol ameliorates both acute and chronic experimental autoimmune encephalomyelitis. Brain Res. 2011;1390:126–141. doi: 10.1016/j.brainres.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 74.Nomura D.K. Morrison B.E. Blankman J.L. Long J.Z. Kinsey S.G. Marcondes M.C. Ward A.M. Hahn Y.K. Lichtman A.H. Conti B. Cravatt B.F. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334:809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oz M. Receptor-independent actions of cannabinoids on cell membranes: focus on endocannabinoids. Pharmacol. Ther. 2006;111:114–144. doi: 10.1016/j.pharmthera.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 76.Izzo A.A. Fezza F. Capasso R. Bisogno T. Pinto L. Iuvone T. Esposito G. Mascolo N. Di Marzo V. Capasso F. Cannabinoid CB1-receptor mediated regulation of gastrointestinal motility in mice in a model of intestinal inflammation. Br. J. Pharmacol. 2001;134:563–570. doi: 10.1038/sj.bjp.0704293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jeong W.I. Osei-Hyiaman D. Park O. Liu J. Batkai S. Mukhopadhyay P. Horiguchi N. Harvey-White J. Marsicano G. Lutz B. Gao B. Kunos G. Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metab. 2008;7:227–235. doi: 10.1016/j.cmet.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 78.Julien B. Grenard P. Teixeira-Clerc F. Van Nhieu J.T. Li L. Karsak M. Zimmer A. Mallat A. Lotersztajn S. Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology. 2005;128:742–755. doi: 10.1053/j.gastro.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 79.Teixeira-Clerc F. Julien B. Grenard P. Tran Van Nhieu J. Deveaux V. Li L. Serriere-Lanneau V. Ledent C. Mallat A. Lotersztajn S. CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Nat. Med. 2006;12:671–676. doi: 10.1038/nm1421. [DOI] [PubMed] [Google Scholar]
- 80.van der Poorten D. Shahidi M. Tay E. Sesha J. Tran K. McLeod D. Milliken J.S. Ho V. Hebbard L.W. Douglas M.W. George J. Hepatitis C virus induces the cannabinoid receptor 1. PLoS One. 2010:5. doi: 10.1371/journal.pone.0012841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Galve-Roperh I. Aguado T. Palazuelos J. Guzman M. Mechanisms of control of neuron survival by the endocannabinoid system. Curr. Pharm. Des. 2008;14:2279–2288. doi: 10.2174/138161208785740117. [DOI] [PubMed] [Google Scholar]
- 82.Diaz-Laviada I. Ruiz-Llorente L. Signal transduction activated by cannabinoid receptors. Mini. Rev. Med. Chem. 2005;5:619–630. doi: 10.2174/1389557054368808. [DOI] [PubMed] [Google Scholar]
- 83.Molina-Holgado F. Rubio-Araiz A. Garcia-Ovejero D. Williams R.J. Moore J.D. Arevalo-Martin A. Gomez-Torres O. Molina-Holgado E. CB2 cannabinoid receptors promote mouse neural stem cell proliferation. Eur. J. Neurosci. 2007;25:629–634. doi: 10.1111/j.1460-9568.2007.05322.x. [DOI] [PubMed] [Google Scholar]
- 84.Horne E.A. Marrs W. Kalume F. Blankman J. Li W. Coy J. Swinney K. Cherry A. Fung S. Cravatt B. Oakley J.C. Stella N. CB1 regulates epileptic activity in a mouse model of early-onset Huntington's disease. Presented at the Society for Neuroscience Annual Meeting; 2011. pp p.668.606/Q662. [Google Scholar]