

Abstract
The use of combination antiretroviral nanoparticles (cART NPs) was investigated as a novel treatment approach for the inhibition of HIV-1 replication. We developed nanoparticles of biodegradable polymer, poly-(dl-lactide-co-glycolic acid; PLGA) containing efavirenz (EFV) and boosted lopinavir (lopinavir/ritonavir; LPV/r) by a high-pressure homogenization method. The method resulted in >79% drug entrapment efficiency for each of the three drugs. The average size of cART NPs was 138.3±55.4 nm as measured by dynamic light scanning, confirmed by scanning electron microscopy (SEM) with an average surface charge of −13.7±4.5. Lissamine-rhodamine-labeled fluorescent PLGA NPs exhibited efficient uptake in nonimmune (HeLa cells) and immune (H9 T cells) cells as measured by confocal microscopy. Cells treated with cART NPs resulted in minimal loss of cell viability over 28 days. Subcellular fractionation studies demonstrated that HIV-1-infected H9 monocytic cells treated with cART NPs contained significantly (p<0.05) higher nuclear, cytoskeleton, and membrane antiretroviral drug levels compared to cells treated with drug solutions alone. Finally, cART NPs efficiently inhibited HIV-1 infection and transduction. The IC50 for each of the three drugs in the cART NPs was <31 nM. These experiments demonstrate the efficacy of a novel PLGA NPs formulation for the delivery of cART to inhibit HIV-1 replication.
Introduction
In 2009, approximately 34 million people were infected with human immunodeficiency virus-1 (HIV-1) worldwide according to the World Health Organization.1 Despite over 30 years of research, HIV-1 remains incurable and a preventive vaccine is not available. While recent advances may eventually lead to vaccine therapy for HIV, current therapies depend on effective antiretroviral drug delivery.2 The development of combination antiviral therapy has reduced HIV morbidity and mortality, but not significantly enough to provide hope for the eradication of the disease. Several recent clinical trials provide hope that additional prevention measures may substantially reduce the rate of new HIV infections. Both the iPREX and tenofovir 1% microbicide gel prophylaxis trials demonstrated antiretroviral drugs administered before sexual encounter prevented HIV-1 infection.3,4 However, both trials demonstrated that efficacy of protection from contracting HIV correlated with patient adherence to the prescribed regimens. Indeed, adherence remains a major cause of drug treatment failure in HIV-positive patients.
Sustained treatment of HIV-1 infection requires using a combination of antiretroviral agents acting on different stages of viral replication. However, combination antiretroviral therapy has several significant disadvantages such as multiple drug–drug interactions, additive toxicity from the combination therapy leading to poor adherence, and the potential to develop multidrug resistance. Emerging data show that even when plasma viral loads are nondetectable, there may still be detectable amounts of replicating virus in other areas of the body, namely CD4+ T-lymphocytes, macrophages, gut-associated lymphatic tissue (GALT), genital tissue, and brain.5–8 The persistent, stable reservoirs of HIV that are not efficiently eradicated even with combination antiretroviral therapy have significant implications. Poor bioavailability and low residence time of orally administered antiretroviral drugs are the primary reasons for the reservoirs. Many antiretroviral drugs have poor aqueous solubility and permeability properties. These drugs are also substrates for efflux transporter systems (i.e., P-glycoprotein), which exist in cells of the gastrointestinal tract, lymphocytes, and brain capillaries.9 This leads to poor gastrointestinal bioavailability and reduced drug concentrations in anatomical sites where HIV-1 replicates. The development of drug delivery systems that provide sustained release of antiretroviral drugs at these sites may improve long-term treatment success rates and retard the development of drug-resistant viruses.
Nanotechnology has emerged as a promising approach for augmenting delivery of antiretroviral drugs. Previously, we demonstrated that a single intraperitoneal (ip) injection of nanoparticles containing a combination of antiretroviral drugs (cART NPs) results in sustained in vivo release of antiretroviral drugs in mice. Interestingly, we observed high levels of antiretroviral drugs in HIV reservoirs such as brain and other organs such as liver, spleen, kidney, serum, and testes for a period of 28 days whereas antiretroviral drug solutions showed detectable drug levels for only 48–72 h.10 The antiretroviral drug levels in the tissue and serum from the nanoparticle formulation were all greater than the IC90 for wild-type virus at day 28. Thus, cART NPs clearly showed potential as a sustained therapeutic modality for HIV treatment. In the present study, we investigated cell uptake, long-term cytotoxicity of cART NPs, and intracellular distribution of antiretroviral drugs after uptake of cART NPs into nonimmune HeLa and immune H9 T cells. Furthermore, the functionality of cART NPs was also assessed by examining their effect on virus production in immune T cells. Treatment of infected cells with cART NPs significantly reduced virus production. These data provide further evidence of the potential of cART NPs as a sustained-release treatment strategy.
Materials and Methods
Materials
Efavirenz and lopinavir/ritonavir were purchased from United States Pharmacopeia. Poly-lactide-co-glycolide (average MW 52,000 Da, inherent viscosity: 0.59 dl/g in hexafluoroisopropanol) was purchased from Birmingham Polymers (Birmingham, AL). Lissamine-rhodamine DHPE was purchased from Invitrogen, (Carlsbad, CA). H9 cells and TZM-bl reporter cells were obtained from the NIH AIDS Research and Reference Reagent Program.11 HeLa and SupT1 cell lines were purchased from the American Tissue Culture Collection (ATCC, Manassas, VA).12,13 Cell media (DMEM or RPMI-1640) with antibiotics, 10% fetal bovine serum (FBS), and l-glutamine were purchased from Fisher Scientific (St. Louis, MO). The CellTiter Glo kit was purchased from Promega (Promega, Madison, WI). The protein fractionation kit that was used was the Pierce subcellular protein fractionation kit (Thermo, Thermo Scientific, Logan, UT). Western blotting primary antibody was a mouse monoclonal anti-p55 antibody (1:1,000, Abcam, Cambridge, MA). The secondary antibody was an antimouse HRP (1:5,000, Applied Biosystems, Inc., Life Technology, Carlsbad, CA).
Nanoparticle preparation
Antiretroviral (AR) drugs (efavirenz, lopinavir/ritonavir) loaded poly-lactide-co-glycolide (PLGA) NPs were prepared using the emulsion-solvent evaporation method.14–17 Briefly, AR drug powder (15 mg of each AR drug) and 150 mg of PLGA were dissolved in 30 ml methylene chloride by heating in an incubating shaker at 37°C with concomitant slow stirring for a minimum of 45 min. After the PLGA and drugs were dissolved, the methylene chloride phase was added to a solution of 0.5% polyvinyl alcohol (PVA) and 2% Poloxamer 407 (Pluronic F127). The crude emulsion was placed into the solvent container for a high-pressure homogenizer (model MP-120, Microfluidics, Inc., Walton, MA). The homogenizer was set at 15,000 psi and the emulsion was circulated through the high-pressure homogenizer for five cycles. The resultant submicronic emulsion was collected and the organic phase was evaporated overnight. The emulsion was ultracentrifuged (23,000×gfor 20 min each) to remove unencapsulated drug and PVA, then lyophilized (−55°C for 4 h and then ranging from −20°C to 20°C over 40 h with vacuum (model LD85, Millrock Technologies, Kingston, NY) to obtain a dry powder. These cART NPs were used in the various cell-based experiments. To fabricate fluorescent NPs for cellular uptake studies, Lissamine-rhodamine DHPE (1 mg/ml, Invitrogen) was added to the methylene chloride solution in place of the antiretroviral drugs and the procedure was repeated to make fluorescent NPs.
Nanoparticle characterization
Nanoparticles were evaluated for size using dynamic light scattering and surface charge by a zeta potential analyzer (ZetaPlus, Brookhaven Instruments, Holtsville, NY). The nanoparticle size was also confirmed by imaging using a Hitachi S-4700 field-emission scanning electron microscopy (SEM).
HeLa, H9, U937, and SupT1 cell cultivation
H9 cells are a human CD4 cell line. HeLa cells are an epithelial cell line and U937 are a monocytic cell line. HeLa cells were maintained in Dulbecco's modified Eagle's media (DMEM) supplemented with 10% FBS, 4 mM l-glutamine, and 1% penicillin/streptomycin. U937, SupT1, and H9 cell lines were grown as a suspension in RPMI-1640 media (Hyclone) with 10% FBS and 1% penicillin/streptomycin and maintained in a logarithmic growth phase. All cells were grown at 37°C and 5% CO2.
Cell viability assay
For cell viability assays HeLa, H9, and U937 cells were grown in 96-well plates at 37°C in 5% CO2 at a density of 4,000 cells/well. Viability was assessed on the days indicated using the CellTiter Glo protocol according to the manufacturer's instructions. Chemiluminescence was read on a BioTek Instrument ELISA plate reader (Winooski, VT). Triplicate values were averaged and analyzed comparing cART NPs, blank NP, respectively, and control cells. Blank NPs and cART NPs were added at 0.5 mg/ml as this concentration in preliminary experiments demonstrated no cytotoxicity at 48 h (data not shown). The goal was to extend the cytotoxicity assays for a total of 28 days. The results were based on triplicate experiments and presented as mean±standard error of the mean (SEM).
Infection of H9 cells with HIV-1NL4-3 and treatment with cART NPs
H9 cells were seeded at 0.5×106 cells/ml and infected with HIV-1NL4-3 (moi=0.05) at 37°C in 5% CO2 for 24 h. The cells were then centrifuged (1000 RPM×10 min at RT) and medium was removed and replaced with fresh RPMI-1640 medium with 10% FBS and 1% penicillin/streptomycin. Antiretroviral drugs were weighed individually and combined into a single microfuge tube and 30 μl of ethanol was added to dissolve the drugs. The ethanol drug solution was diluted with phosphate-buffered saline (470 μl; PBS) such that the concentration of antiretroviral drugs was 1 mg/ml. H9 cells were treated postinfection (1 dpi) with no drug, antiretroviral drug solution (1 mg/ml), or cART NPs (equivalent to 1 mg/ml of antiretroviral drugs). After incubation with drug solutions or NP formulation for 24 h, cells were centrifuged, washed with PBS, and incubated with fresh media and maintained for 7 days.
Subcellular fractionation
H9 cells were centrifuged and washed with PBS on day 7 postinfection (dpi). The protein fractionation procedure followed the manufacturer's protocol to fractionate H9 cells. The PBS was removed and cytoplasmic extract buffer was added to the cell pellet. Cells were pelleted by centrifugation, and the cytoplasmic extract was removed from the pellet. Membrane extract buffer was added to the new pellet and centrifuged. The supernatant was removed and kept as the membrane extract buffer. This procedure was repeated to obtain the nuclear extract, chromatin bound nuclear extract, and cytoskeletal extract using nuclear extract buffer, nuclear extract buffer with micrococcal nuclease and calcium chloride, and pellet extraction buffer, respectively. All samples were placed on ice during this procedure and then frozen (−80°C) until analyzed.
Samples of the subcellular fraction extracts were prepared for high-pressure liquid chromatography (HPLC) as previously reported.10,14 A solid-phase extraction cartridge (Strata-X SPE, Phenomenex, Torrance, CA) was used to prepare the samples. After initial mobilization of the solid phase with methanol, the cartridge was rinsed with double distilled water. Samples (500 μl) were added to the solid phase in 5% methanol. Antiretroviral agents were eluted off the column with 100% methanol, refrigerated for 30 min, and centrifuged at 13,500 rpm at 4°C for 15 min. An aliquot of the supernatant was placed in autosampler vials with glass inserts and injected into the HPLC. Solid-phase extraction of spiked control samples was performed in parallel for each assay and recovered at >97% efficiency. Triplicate samples were injected and the peak area was compared with the peak area of the standard curves (2.25–50 μg/ml) for the three drugs. Intraday and interday variability of the HPLC assay was <10%.
Evaluation of cART NPs for inhibition of HIV-1 replication and transduction
To determine the 50% inhibitory concentration (IC50) values for cART NPs, TZM-bl cells were seeded in 24-well plates at a density of 0.4×105 cells per well. After 24 h, the cells were treated with six 10-fold dilutions of cART NPs starting with 0.05 mg/ml [equivalent to 0.0405, 0.0399, and 0.0399 mg/ml of ritonavir (RTV), lopinavir (LPV), and efavirenz (EFV), respectively] based on the entrapment efficiency. After 4 h, the NPs were removed and the cells were washed with media. The following day, the cells were inoculated with HIV-1NLX virus (25 μl) for 4 h, washed, and incubated for 48 h. Cells were washed with PBS, lysed with 150 μl M-PER solution (Thermo Scientific, Rockford, IL), and clarified by centrifugation. Luciferase assays were performed using a GloMax-Multi+Microplate luminometer with Instinct software (Promega, Madison, WI) and read immediately. The data obtained were normalized from relative luminescence units (RLU), plotted, and IC50 obtained using GraphPad Prism software.
For virus transduction experiments, TZM-bl cells were seeded at 0.4×105 per well in 24-well plates, incubated overnight, and inoculated with 25 μl of HIV-1 vector pseudotyped with vesicular stomatitis virus glycoprotein (VSVg) for 4 h and washed. After an additional 48 h, the cells were harvested and assayed for luciferase activity as described above. Data represent eight total replicates.
For the SupT1 infections, 5×106 cells were inoculated with HIV-1NL4-3 virus (500 μl) for 4 h, washed with media, and incubated overnight. The cells were then treated at 1 dpi with 0.05 mg/ml NPs (blank or cART) for 24 h and washed twice. After an additional 48 h, the supernatants and cells were collected separately. The cells were washed with PBS, lysed with 400 μl M-PER solution, clarified by centrifugation (20,000×g for 5 min), concentrated by ultracentrifugation through a 20% sucrose cushion, and the pellet resuspended in 1× sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) sample buffer. Cell or concentrated supernatants were resolved by SDS–PAGE and transferred to PVDF membranes for western blot analysis. Proteins were detected by western blot using anti-HIV-1 Gag or GAPDH primary antibodies (1:1,000) followed by species-specific secondary antibodies conjugated with HRP (1:5,000). Bands were detected by chemiluminescence exposed to film. Images were obtained by scanning films and cropped using Adobe Photoshop.
Confocal laser scanning microscopy
H9 and HeLa cells were cultured on 12-well tissue culture slides in their supplemented DMEM media as described. Cells were plated at 105 cells/well and incubated with and without Lissamine-rhodamine DHPE NPs at 1 and 2 mg/ml for 2, 4, and 24 h in vitro. These concentrations match human drug levels within a dosing interval. At each time point, cells were collected and centrifuged at 850 rpm for 5 min and reconstituted in 200 μl of media. Cells were cytospun onto glass slides at 850 rpm for 4 min. Spun cells were fixed with 3.7% formaldehyde at 37°C for 15 min. Cells were rinsed with PBS three times and incubated with DAPI at 300 ng/ml for 30 min at room temperature. Following mounting, cells were imaged on an LSM 510 META NLO confocal microscope (Carl Zeiss inc., Thornwood, NY) at the Integrative Biological Imaging Facility at Creighton University. Images were taken without software enhancement.
Statistical analysis
Statistical analysis was performed using SPSS-PC (ver. 18.0, SPSS, Chicago, IL). The results are presented as mean±standard error of the mean. Analysis of variance (ANOVA) was performed on cell viability data. Student's t test was performed on subcellular fractionation and viral infection data. Nonlinear regression was used in GraphPad Prism (ver. 5; GraphPad, Inc., San Diego, CA) software to determine the IC50 of cART NPs.
Results
Prior to using cART NPs for cellular toxicity, cell viability, and subcellular fractionation experiments, the biophysical properties of the PLGA NPs were assessed. PLGA NPs were fabricated by the emulsion-solvent evaporation method with high-pressure homogenizer and lyophilized under vacuum. The use of the high-pressure homogenizer resulted in a smaller size compared to the previous method using a probe homogenizer.14 The size of the cART NPs was assessed in triplicate by dynamic light scattering and confirmed by scanning electron microscopy (Fig. 1). The average diameter of the cART NPs was 138.3±55.4 nm whereas the average surface charge of three batches of cART NPs was −13.7±4.5. The drug loading and entrapment efficiency of the cART NPs were measured by dissolving a known quantity of the nanoparticle powder in acetonitrile and analyzing it by HPLC. The drug loading efficiency (DL) is defined as entrapped efavirenz, lopinavir, and ritonavir content in the NP cores calculated from the mass of the incorporated drugs using the following equation: 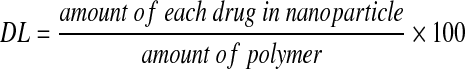 . The encapsulation efficiency (EE) is defined as the ratio of the mass of each encapsulated drug to the mass of the drug used for nanoparticle preparation using the following equation:
. The encapsulation efficiency (EE) is defined as the ratio of the mass of each encapsulated drug to the mass of the drug used for nanoparticle preparation using the following equation: 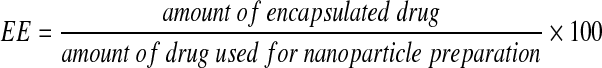 . The results are summarized in Table 1. The drug loading of all three drugs was similar (28.7–35.1%) and their incorporation efficiencies were >79%. These data demonstrate efficient entrapment for all three drugs. From these measurements, the μg/mg equivalent of EFV and LPV/r contained in the cART NPs was calculated. These data were used for all subsequent experiments as well as concentration-response determinations.
. The results are summarized in Table 1. The drug loading of all three drugs was similar (28.7–35.1%) and their incorporation efficiencies were >79%. These data demonstrate efficient entrapment for all three drugs. From these measurements, the μg/mg equivalent of EFV and LPV/r contained in the cART NPs was calculated. These data were used for all subsequent experiments as well as concentration-response determinations.
FIG. 1.
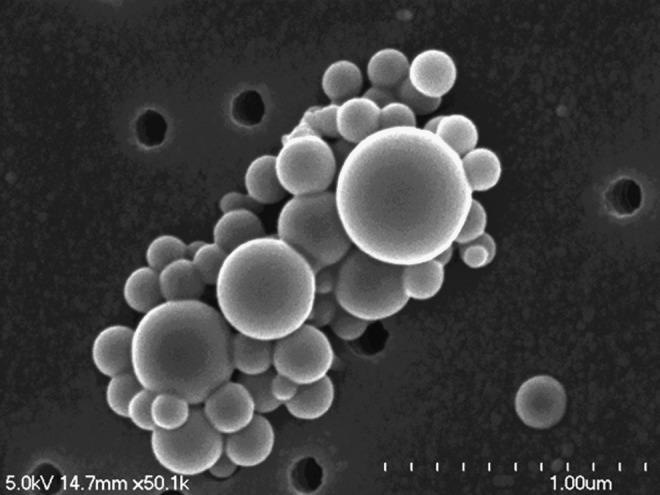
Scanning electron microscopic micrograph of combination antiretroviral nanoparticles (cART NPs).
Table 1.
Drug Loading and Entrapment Efficacy for Combination Antiretroviral Nanoparticles (n=3)
| |
Drug |
||
|---|---|---|---|
| RTV | LPV | EFV | |
| Mean drug loading (%) (±SD) | 28.7 (18.1) | 32.5 (27.1) | 35.1 (34.7) |
| Mean entrapment efficiency (%) (±SD) | 81.0 (21.4) | 79.8 (13.6) | 79.5 (12.9) |
RTV, ritonavir; LPV, lopinavir; EFV, efavirenz.
To determine the cellular toxicity of the cART NPs, cell viability experiments were performed with nonimmune (HeLa) and immune (H9 and U937) cells. Cells were treated with blank or cART NPs and cell viability was monitored by measuring intracellular ATP levels. No significant reduction in cellular viability was observed in any of the cell lines treated with cART NPs compared to blank NPs or untreated (control) cells for up to 28 days (Fig. 2). These results demonstrate that the cART NP formulation treatment was not significantly cytotoxic when compared to untreated cells or cells treated with blank NPs in vitro.
FIG. 2.

Chemiluminescence of cell viability for HeLa (A), H9 (B), and U937 (C) cell lines over time. All cells received no drugs (control), blank NPs (0.5 mg/ml), and cART NPs at 0.5 mg/ml.
Efficient uptake of PLGA NPs in monocyte-derived macrophages was demonstrated previously.14 To extend those studies, the uptake of NPs in both the human T cells (H9) and fibroblasts (HeLa) was assessed using fluorescent NPs and immunocytochemical analysis. Cells were treated with either 1 or 2 mg/ml of Lissamine-rhodamine DHPE NPs, fixed at 2, 4, and 24 h, and imaged by fluorescent confocal microscopy (Fig. 3A and B, HeLa and H9 cells, respectively). Abundant levels of rhodamine (red fluorescence) were observed within both the HeLa and H9 cells at all time points, although it appeared to reach the highest intensity at 4 h. As expected, there was a noticeable increase in intensity between cells treated with 1 and 2 mg/ml of Lissamine-rhodamine NPs at all time points suggesting that a saturating concentration of NPs was not a factor influencing uptake at these concentrations.
FIG. 3.
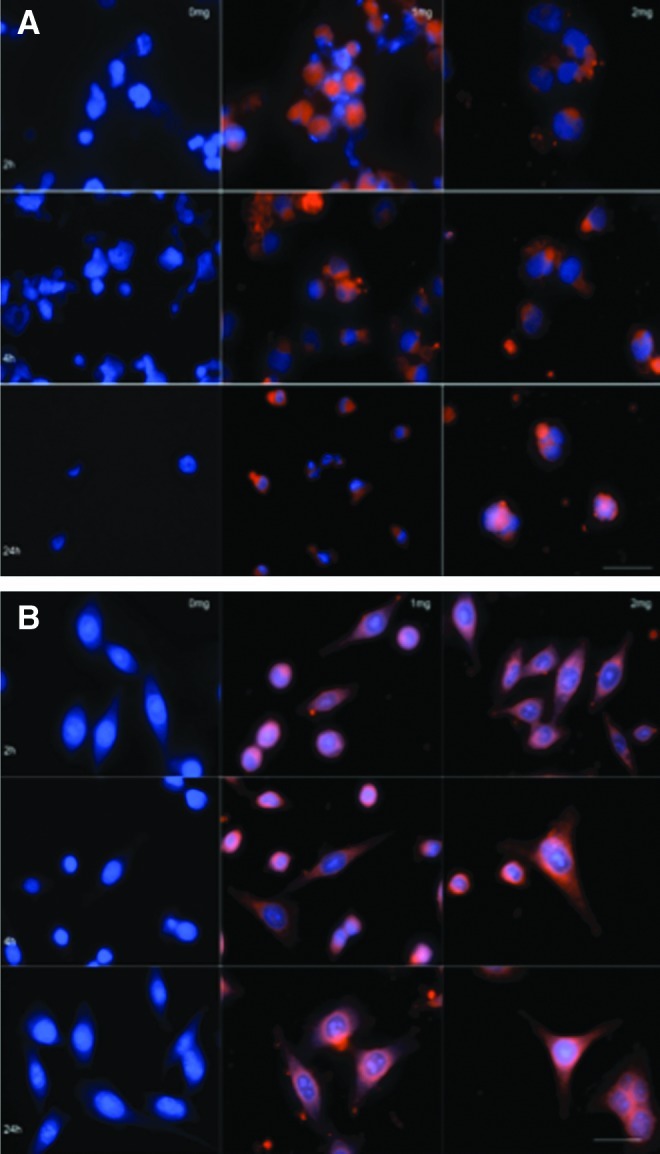
Confocal composite of NP uptake by HeLa (A) and H9 (B) cells. Cells were incubated with 1 and 2 mg Lissamine NPs, cells were fixed, DAPI (300 ng/ml) was added, and images were taken using an LSM 510 META NLO confocal microscope (Carol Zeiss, Inc., Thorwood, NT) (magnification 65×). Scale bar=100 μm (A) and 75 μm (B).
The specific intracellular concentration and location of antiretroviral drugs taken up by immune cells infected with HIV-1 were evaluated using subcellular fractionation and HPLC. HIV-1-infected H9 cells were treated with ART drug solutions or cART NPs for 7 days and separated into cytoplasmic, membrane, soluble nuclear, chromatin, and cytoskeletal protein fractions. The amount of each antiretroviral drug was measured from each fraction (Fig. 4). There were significantly higher amounts of all three antiretroviral drugs in the membrane, cytoskeleton, and nuclear fractions of cells treated with cART NPs compared to those fractions of cells treated with drug solutions. The overall average (±SEM) membrane concentrations of the antiretroviral drugs (per 106 H9 cells) in the cells treated with cART NPs were 15.0±1.0 (RTV), 15.6±1.6 (LPV), and 12.9±1.3 μg (EFV). Conversely, only ritonavir achieved a detectable level in the membrane fraction (0.2±0.1 μg) when cells were treated with drug solution.
FIG. 4.

Subcellular antiretroviral drug levels. Comparison of antiretroviral treatment (ART) levels in the subcellular compartments of H9 cells cultured with cART NPs (A) or free drug (B) after 7 days of culture with HIV-1NL4-3 (*p<0.05). Error bars denote standard error of the mean.
Next, the antiviral efficacy of the cART NP formulation was investigated. First to be examined was whether the cART NPs reduced HIV-1 viral protein expression in infected human T cells. To do this, SupT1 T cells were infected with HIV-1NL4-3 for 24 h and then treated with either 0.05 mg/ml cART NPs or control (blank) NPs. After an additional 48 h incubation, the cells were analyzed for viral protein expression by western blot analysis of p55-Gag protein in both supernatant (Fig. 5A) as well as cell lysates (Fig. 5B). Western blot analysis of GAPDH was used as a loading control (Fig. 5B bottom panel), demonstrating equivalent protein levels in all the SupT1 cells over the course of the experiment. The cART NP treatment resulted in a substantial reduction of p55-Gag levels in both the supernatants and lysates compared to the blank nanoparticle-treated cells. Next, the ability of the cART NPs to inhibit HIV-1 transduction was tested. For these studies, TZM-bl reporter cells were pretreated with cART or blank NPs 1 day prior to infection with HIV-1 pseudotyped with the vesicular stomatitis virus glycoprotein. At 48 hpi (hours postinfection) the cells were harvested and the amount of luciferase produced was measured by enzymatic assay. As shown in Fig. 5C, the cells treated with 0.05 mg/ml cART NPs showed a significant reduction (p<0.001) of infection compared to untreated cells or cells treated with blank NPs. Finally, a concentration-response (IC50) curve of each drug in the cART NPs was calculated using the TZM-bl cells. For these experiments, the cells were again treated with a series of 10-fold dilutions of NPs 1 day prior to infection with wild-type HIV-1NL4-3. The IC50 curve is shown in Fig. 5D and produced an r2 value of 0.884. Based on the amount of the three antiretroviral drugs (μg/mg) incorporated into the nanoparticles, the calculated IC50 values of the individual drugs in the cART NPs were 14.01, 16.54, and 30.73 nM for ritonavir, lopinavir, and efavirenz, respectively.
FIG. 5.

cART NPs efficacy in HIV-1-infected cells in vitro. Supernatants (A) and cell lysates (B) were analyzed by western blot with anti-Gag and anti-GAPDH antibodies as indicated. SupT1 T cells were infected with HIV-1 NLX and treated with 0.05 mg/ml at the same time for 24 h. After extensive washing the cells were incubated for an additional 48 h, and the cell supernatants and lysates were harvested. (C) TZM-bl reported cells were inoculated with HIV-1+VSVg and the next day treated with 0.05 mg/ml of either cART or blank NPs. After 48 h, the cells were lysed and assayed for luciferase expression using a luminometer and reported as relative light units (RLU). A significant (p<0.001) reduction for cART NPs was observed. Data represent the mean of eight individual infects and error bars represent standard error of the mean. (D) TZM-bl cells were treated with six 10-fold dilutions of cART and the next day inoculated with HIV-1 NLX. After 48 h, the cells were lysed and luciferase assays were performed. RLU values were obtained with a luminometer. Data are representative of triplicate samples. Error bars denote standard error of the mean.
Discussion
The use of nanotechnology as a drug delivery system has been reported for a number of years for chemotherapy agents.18–20 Nanoparticles have been reported to concentrate in tumor masses, inflammatory sites, and infection sites.21 Nanotechnology has been considered for the delivery of antimicrobial agents, including antiretroviral agents, for more than a decade. Combination antiretroviral therapy is currently the standard of care for HIV-1 treatment, but thus far only anti-HIV nanocarriers containing one antiretroviral drug have been reported. Consistent with the standard of care, we have developed PLGA nanoparticles that contain a combination of three antiretroviral drugs. In a recent study, we demonstrated that a single 20 mg/kg cART NP dose in mice results in sustained in vivo release of all three antiretroviral drugs for 28 days.10 Although we showed that single ip injection of cART NPs can result in sustained in vivo delivery of antiretroviral drugs, it was important to characterize the intracellular delivery of antiretroviral drugs from nanoparticles and demonstrate the release of functional antiretroviral drugs after intracellular uptake. In view of this, in the present investigation, we evaluated the ability of cART NPs to deliver antiretroviral drugs to nonimmune and immune cells, their cytotoxicity, and their functionality in HIV-infected cells in vitro.
Since the method of fabrication of the nanoparticles was different than previously reported14 the biophysical properties of the cART NPs were reinvestigated in this report. The size of the cART NPs was smaller than those produced by the previous method, likely due to the use of the high-pressure homogenizer in place of a probe homogenizer. We consider this an improvement since the size of the nanoparticles can affect the biodistribution and cellular uptake.22 The percent of drug entrapment efficiency was greater than 79% for all three ART drugs. Drug entrapment efficiency was improved using the high-pressure homogenizer. Previously we reported drug entrapment efficiencies of 38%, 45%, and 86%, respectively, for ritonavir, lopinavir, and efavirenz.14 These results suggest that all three drugs are efficiently encapsulated into the cART NPs.
The results of the cytotoxicity data in nonimmune and immune human cells show that the PLGA nanoparticles do not induce significant cytotoxicity out to 28 days in vitro. Importantly, the lack of cytotoxicity in the cell lines up to 28 days in vitro would suggest that the cART NP formulation offers some long-term advantages over the same drugs administered orally to patients with HIV-1 infection. Our previous study in mice demonstrated the presence of high levels of all three drugs in all tissues examined out to day 28 after a single 20 mg/kg dose.10 These mice showed no adverse effects from the single injection as measured by clinical evaluation. These data could offer an advance over the chronic oral dosing of antiretroviral agents, which leads to cellular toxicity and mitochondrial dysfunction over time.23,24
Confocal microscopy images show significant uptake of fluorescent Lissamine PLGA NPs in both HeLa and H9 cells. These data extend previous studies that showed substantial 6-hydroxycoumarin NP uptake by monocyte-derived macrophages14 and demonstrate that nonphagocytic cells can efficiently uptake the particles. Immunofluorescence and the concentration of Lissamine NP exposure followed by cellular up-take out to 24 h suggest that the concentration of NPs and fluorescence were not saturated. Thus both immune and nonimmune cells are able to engulf large numbers of these particles over extended periods of time. Additionally, the amount of antiretroviral drugs in the nuclear, cytoskeleton, and membrane fractions was significantly higher from cART NPs in HIV-1-infected cells at 7 dpi compared to the drug solutions. Cells treated with the antiretroviral drug solutions had undetectable levels of efavirenz and lopinavir in all subcellular fractions suggesting low retention of soluble drugs. One concern is that the drugs in solutions were more apt to be removed as medium was exchanged over the 7-day time course while cART NPs were phagocytosed by the cells and drugs were released from the nanoparticles. Ritonavir, lopinavir, and efavirenz levels in the membrane fraction averaged >12-fold more from cART NPs compared to cells treated with drug solutions. It has been previously reported that the drugs from the protease inhibitor class have poor permeability and are substrates for drug efflux transporter pumps such as P-glycoprotein and breast cancer resistance protein (BCRP).25,26 Furthermore, it has also been demonstrated that treatment of HIV-infected H9 cells can up-regulate expression of P-glycoprotein and BCRP resulting in reduced drug uptake in the cells. These factors could be responsible for negligible concentrations of antiretroviral drugs in the various subcellular fractions of the cells treated with drug solutions. However, encapsulation of antiretroviral drugs in nanoparticles would increase their cellular uptake and permeability and might prevent protease inhibitor-mediated P-glycoprotein up-regulation in the HIV-infected cells. PLGA nanoparticles have also been reported to undergo endolysosomal escape leading to cytoplasmic delivery of the encapsulated drug,27 which is clearly evident from the subcellular fractionation studies These results suggest that the antiviral efficacy of these drugs in the nanoparticle formulation could be superior to the drugs given in oral pill form for actively infected T cells. Our previous data have documented that cART NPs are ingested by monocyte-derived macrophages.14
Several trials have shown a significant correlation between intracellular antiretroviral drug levels and efficacy.28–32 These trials documented the intracellular nucleoside reverse transcriptase inhibitors zidovudine triphosphate and lamivudine triphosphate levels with efficacy parameters, mainly time to reach <50 copies/ml of HIV RNA at 24 and 52 weeks. Additional studies examined the pharmacodynamic relationships between protease inhibitor trough serum concentration and virologic success (defined as an HIV viral load of <50 copies/ml).33–35 Lamotte et al. found that intracellular and trough lopinavir levels in patients receiving boosted lopinavir therapy were significantly correlated with virologic success.33 Anderson et al. demonstrated that peak serum indinavir levels >7 μg/ml were significantly correlated with a median CD4 cell count rise of >350/μl in patients receiving zidovudine/lamivudine+indinavir combination therapy.35 There have been no reports documenting antiviral efficacy with nonnuclear reverse transcriptase inhibitor (NNRTI) serum or intracellular levels. Finally, a recent report in patients who take orally administered antiretroviral drugs has demonstrated limited concentrations in tissue leading to a lack of antiretroviral efficacy in lymph nodes, ileum, and rectal tissue that were studied.36 The subcellular results presented here show significantly higher lopinavir, ritonavir, and efavirenz intracellular levels from cART NPs compared to soluble drugs in HIV-infected monocytic cells after 24 h of incubation in vitro. The cART NPs could be a superior drug delivery system to deliver combination antiretroviral drugs to target tissues compared to the soluble drugs administered orally. Using the nanoparticle delivery system, antiretroviral intracellular levels will remain high for a prolonged period compared to soluble drugs administered orally. More research into the tissue penetration of antiretroviral drugs delivered by nanotechnology is necessary.
The utility of efavirenz and boosted lopinavir into a combination nanoparticle is not considered a standard, first-line regimen for treatment-naive HIV patients. However, previous studies have documented efficacy when these drugs were used in human trials.37,38 Riddler et al. showed boosted lopinavir+efavirenz as a nucleoside reverse transcriptase inhibitor (NRTI)-sparing option had efficacy similar to efavirenz+two NRTIs, respectively, after 96 weeks of follow-up.37 The ratios of the three drugs used for the cART NPs (1:1:1) are not typical for oral drug therapy with these treatments. Efavirenz is administered 600 mg once daily usually at bedtime. Lopinavir/ritonavir combination therapy is administered 500/125 mg twice daily with efavirenz. However, the area-under-the-serum-concentration time curves (AUC) are similar between efavirenz and lopinavir.
The cART NPs were effective against HIV-1 in vitro, inhibiting virus infection of T cells and VSVg-pseudotyped transduction of TZM-bl indicator cells. Pretreatment of TZM-bl cells inhibited virus infections over 500-fold. Posttreatment of infected SupT1 cells, measured by detection of the major core proteins, p55-Gag, demonstrated that nanoparticle delivery of antiretroviral drugs potently reduced viral production in both supernatants as well as cell lysates in a human T cell line. Additionally, the concentration-response (IC50) calculations indicated a similar level of potency for the three drugs incorporated into the nanoparticle formulation as compared to the IC50 of the free drugs alone. Combined, these results demonstrate that the nanoparticle formulation provides a safe and effective means of delivering combination antiretroviral therapy to reduce HIV-1 viral activity in vitro.
The sustained release of antiretroviral drugs from cART NPs is advantageous for a number of reasons. If HIV-infected patients were able to administer a subcutaneous injection of cART NPs on a monthly basis, their adherence could be significantly improved. Of interest, survey data from potential user groups of preexposure prophylaxis demonstrated that bimonthly or monthly injections were the top two preferred routes of administration for prophylactic regimens.39 Finally, sustained-release cART NPs could potentially result in a reduction in development of drug resistance. This could also contribute to an advantage for this treatment option.40,41 Further research is necessary to test these hypotheses.
Conclusions
This study demonstrates significant uptake of the cART NPs compared to the soluble antiretroviral drugs, the subsequent release of levels of antiretroviral drugs in the nuclear, cytoskeleton, and membrane fractions of cells with no toxicity for 28 days, and the antiviral potency with IC50 values in the nM range. The higher intracellular antiretroviral drug delivery by nanoparticles may significantly reduce HIV-1 infectivity by inhibiting HIV replication at lower doses.
Acknowledgments
The following reagent was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: H9 cell line from Dr. Robert Gallo, TZM-bl cells from Drs. John Kappes, Xiaoyun Wu, and Tranzyme, Inc. The authors would like to thank Dr. Han Chen of the Microscopy Core Research Facility at the University of Nebraska-Lincoln for his assistance in SEM and Dr. Heather C. Jensen-Smith of the Integrated Biomedical Imaging Facility at Creighton University for her assistance with the confocal microscopy.
The authors would like to acknowledge the Creighton University Integrated Biomedical Imaging Facility for providing the confocal images. This facility, supported by the C.U. Medical School, was constructed with support from the National Center for Research Resources (5P20RR016469) and the National Institute for General Medical Science (NIGMS) (8P20GM103427).
This work was supported by National Institutes of Health grants 1R15AI076039-01A1 (to C.J.D.), 5RO1AI080348 (to M.B.), and grants from the National Center for Research Resources (5P30RR031151-01) and the National Institute of General Medical Sciences (8 P30 GM103509-03) and Creighton University Graduate School Summer Faculty Research Fellowship (to A.S.). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.UNAIDS. AIDS epidemic update World Health Organization. 2009;30 [Google Scholar]
- 2.Barouch DH. Liu J. Li H. Maxfield LF. Lynch DM. Lampietro MJ. SanMiguel A. Seaman MS. Ferrari G. Forthal D. Ourmanov I. Hirsch VM. Carville A. Mansfield KG. Stablein D. Pau MG. Schuitemaker H. Sadoff JC. Billings EA. Rao M. Robb ML. Kim JH. Marovich MA. Goudsmit J. Michael NL. Vaccine protection against acquisition of neutralization-resistant SIV challenges in rhesus monkeys. Nature. 2012;482:89–93. doi: 10.1038/nature10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdool Karim Q. Abdool Karim SS. Frohlich JA. Grobler AC. Baxter C. Mansoor LE. Kharsany AB. Sibeko S. Mlisana KP. Omar Z. Gengiah TN. Maarschalk S. Arulappan N. Mlotshwa M. Morris L. Taylor D the CAPRISA 004 Trial Group. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide for the prevention of HIV infection in women. Science. 2010;329:1168–1174. doi: 10.1126/science.1193748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grant RM. Lama JR. Anderson PL. McMahan V. Lui AY. Vargas L. Goicochea P. Gasapia M. Guanira-Carranza JV. Ramirez-Cardich ME. Montoya-Herrera O. Fernandez T. Veloso VG. Buchbinder SP. Chariyalertsak S. Schechter M. Bekker LG. Mayer KH. Kallas EG. Amico KR. Mulligan K. Bushman LR. Hance RJ. Ganoza C. Defechereux P. Postle B. Wang F. McConnell JJ. Zheng JH. Lee J. Rooney JF. Jaffe HS. Martinez AI. Burns DN. Glidden DV the iPrEx Study Team. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363:2587–2599. doi: 10.1056/NEJMoa1011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chun TW. NIckle DC. Justement JS. Meyers JH. Roby G. Hallahan CW. Kottilil S. Moir S. Mican JM. Mullins JI. Ward DJ. Kovacs JA. Mannon PJ. Fauci AS. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J Infect Dis. 2008;197:714–720. doi: 10.1086/527324. [DOI] [PubMed] [Google Scholar]
- 6.Clements JE. Babas T. Mankowski JL. Suryanarayana K. Piatak M., Jr Tarwater PM. Lifson JD. Zink MC. The central nervous system as a reservoir for simian immunodeficiency virus (SIV): Steady-state levels of SIV DNA in brain from acute through asymptomatic infection. J Infect Dis. 2002;186:905–913. doi: 10.1086/343768. [DOI] [PubMed] [Google Scholar]
- 7.Gunthard HF. Havlir DV. Fiscus S. Zhang AQ. Eron J. Mellors J. Gulick R. Frost SD. Brown AJ. Schleif W. Valentine F. Jonas L. Meibohm A. Ignacio CC. Isaacs R. Gamagami R. Emini E. Haase A. Richman DD. Wong JK. Residual human immunodeficiency virus (HIV) type 1 RNA and DNA in lymph nodes and HIV RNA in genital secretions and in cerebrospinal fluid after suppression of viremia for 2 years. J Infect Dis. 2001;183:1318–1327. doi: 10.1086/319864. [DOI] [PubMed] [Google Scholar]
- 8.Yeh RF. Renk NL. Kashuba AD. Dumond JB. Tappouni HL. Tien HC. Chen YC. Vourvahis M. Horton Al. Fiscus SA. Patterson KB. Genital tract, cord blood, and amniotic fluid exposures of seven antiretroviral drugs during and after pregnancy in human immunodeficiency virus type1-infected women. Antimicrob Agents Chemother. 2009;53:2367–2374. doi: 10.1128/AAC.01523-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kay MS. Silent, but deadly–eliminating reservoirs of latent HIV. Trends Biotechnol. 2003;2:420–423. doi: 10.1016/j.tibtech.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 10.Destache CJ. Belgum T. Goede M. Shibata A. Belshan MA. Antiretroviral release from poly(DL-lactide-co-glycolide) nanoparticles in mice. J Antimicrob Chemother. 2010;65:2183–2187. doi: 10.1093/jac/dkq318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mann DL. O'Brien SJ. Gilbert DA. Reid Y. Popovic M. Read-Connole E. Gallo RC. Gazdar AF. Origin of the HIV-susceptible human CD4+ cell line H9. AIDS Res Hum Retroviruses. 1989;5:253–255. doi: 10.1089/aid.1989.5.253. [DOI] [PubMed] [Google Scholar]
- 12.Sundstrom C. Nilsson K. Establishment and characterization of a human histiocytic lymphoma cell line (U937) Int J Cancer. 1976;17:565–577. doi: 10.1002/ijc.2910170504. [DOI] [PubMed] [Google Scholar]
- 13.Maddon PJ. Dalgleish AG. McDougal JS. Clapham PR. Weiss RA. Axel R. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell. 1986;47:333–348. doi: 10.1016/0092-8674(86)90590-8. [DOI] [PubMed] [Google Scholar]
- 14.Destache CJ. Belgum R. Christensen K. Shibata A. Sharma A. Dash AK. Combination antiretroviral drugs in PLGA nanoparticles for HIV-1. BMC Infect Dis. 2009;9:198. doi: 10.1186/1471-2334-9-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee WK. Park JY. Yang EH. Suh H. Kim SH. Chung DS. Choi K. Yang CW. Park JS. Investigation of the factors influencing the release rates of cyclosporin A-loaded micro- and nanoparticles prepared by high-pressure homogenizer. J Control Release. 84(3):115–123. doi: 10.1016/s0168-3659(02)00239-0. 200. [DOI] [PubMed] [Google Scholar]
- 16.Mehta AK. Yadav KS. Sawant KK. Nimodipine loaded PLGA nanoparticles: Formulation optimization using factorial design, characterization and in vitro evaluation. Curr Drug Deliv. 2007;4(3):185–193. doi: 10.2174/156720107781023929. [DOI] [PubMed] [Google Scholar]
- 17.Dong Y. Feng SS. Poly(D,L-lactide-co-glycolide) (PLGA) nanoparticles prepared by high pressure homogenization for paclitaxel chemotherapy. Int J Pharm. 2007;342(1–2):208–814. doi: 10.1016/j.ijpharm.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 18.Couvreur P. Kante B. Crislain L. Roland M. Speiser P. Toxicity of polyalkcyanoacrylate nanoparticles II: Doxorubicin-loaded nanoparticles. J Pharm Sci. 1982;71:790–792. doi: 10.1002/jps.2600710717. [DOI] [PubMed] [Google Scholar]
- 19.Beck P. Kreuter J. Reszka R. Richtner I. Influence of polybutylcyanacrylate nanoparticles and liposomes on the efficacy and toxicity of anticancer drug mitoxantrone in murine tumour models. J Microencapsul. 1993;10:101–114. doi: 10.3109/02652049309015316. [DOI] [PubMed] [Google Scholar]
- 20.Conway MA. Madrigal-Estebas L. McClean S. Brayden DJ. Millis KH. Protection against Bordetella pertussis infection following parenteral or oral immunization with antigens entrapped in biodegradable particles: Effect of formulation and route of immunization on induction of Th1 and Th2 cells. Vaccine. 2001;19:1940–1950. doi: 10.1016/s0264-410x(00)00433-3. [DOI] [PubMed] [Google Scholar]
- 21.Shenoy D. Little S. Langer R. Amiji M. Poly(ethylene oxide)-modified poly(b-amino) ester nanoparticles as a pH-sensitive system for tumor-targeted delivery of hydrophobic drugs: Part 2. In vivo distribution and tumor localization studies. Pharm Res. 2005;22:2107–2114. doi: 10.1007/s11095-005-8343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahapatro A. Singh DK. Biodegradable nanoparticles are excellent vehicle for site directed in-vivo delivery of drugs and vaccines. J Nanobiotechnol. 2011;9:55. doi: 10.1186/1477-3155-9-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vidal F. Domingo JC. Guallar J. Saumoy M. Cordobilla B. Sanchez de la Rosa R. Giralt M. Alvarez ML. Lopez-Dupla M. Torres F. Villarroya F. Cihlar T. Domingo P. In vitro cytotoxicity and mitochondrial toxicity of tenofovir alone and in combination with other antiretrovirals in human renal proximal tubule cells. Antimicrob Agents Chemother. 2006;50:3824–3832. doi: 10.1128/AAC.00437-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Groener JB. Seybold U. Vollbrecht T. Bogner JR. Decrease in mitochondrial transmembrane potential in peripheral blood mononuclear cells of HIV-uninfected subjects undergoing HIV postexposure prophylaxis. AIDS Res Hum Retroviruses. 2011;27:969–972. doi: 10.1089/AID.2010.0348. [DOI] [PubMed] [Google Scholar]
- 25.Speck RR. Yu X-F. Hildreth J. Flexner C. Differential effects of P-glycoprotein and multidrug resistance protein-1 on productive human immunodeficiency virus infection. J Infect Dis. 2002;186(3):332–340. doi: 10.1086/341464. [DOI] [PubMed] [Google Scholar]
- 26.Peroni RN. Di Gennaro SS. Hocht C. Chiappetta DA. Rubio MC. Sosnik A. Bramuglia GF. Efavirenz is a substrate and in turn modulates the expression of the efflux transporter ABCG2/BCRP in the gastrointestinal tract of the rat. Biochem Pharmcol. 2011;82(9):1227–1233. doi: 10.1016/j.bcp.2011.07.081. [DOI] [PubMed] [Google Scholar]
- 27.Panyam J. Zhou WZ. Prabha S. Sahoo SK. Labhasetwar V. Rapid endo-lysosomal escape of poly(DL-lactide-co-glycolide) nanoparticles: Implications for drug and gene delivery. FASEB J. 2002;16(10):1217–1226. doi: 10.1096/fj.02-0088com. [DOI] [PubMed] [Google Scholar]
- 28.Moore JD. Acosta EP. Johnson VA. Bassett R. Eron JJ. Fischl MA. Long MC. Kuritzkes DR. Sommadossi JP. Intracellular nucleoside triphosphate concentrations in HIV-infected patients on duel nucleoside reverse transcriptase inhibitor therapy. Antivir Ther. 2007;12(6):981–986. [PubMed] [Google Scholar]
- 29.Fletcher CV. Kawie SP. Kakuda TN. Anderson PL. Bushman LR. Brundage RC. Remmel RP. Zidovudine triphosphate and lamivudine triphosphate concentration-response relationships in HIV-infected persons. AIDS. 2000;14(14):2137–2144. doi: 10.1097/00002030-200009290-00010. [DOI] [PubMed] [Google Scholar]
- 30.Anderson PL. Kakuda TN. Kawle S. Fletcher CV. Antiviral dynamics and sex differences of zidovudine and lamivudine triphosphate concentrations in HIV-infected individuals. AIDS. 2003;17(15):2159–2168. doi: 10.1097/00002030-200310170-00003. [DOI] [PubMed] [Google Scholar]
- 31.Fletcher CV. Anderson PL. Kakuda TN. Schacker TW. Henry K. Concentration-controlled compared with conventional antiretroviral therapy for HIV infection. AIDS. 2002;16(4):551–560. doi: 10.1097/00002030-200203080-00006. [DOI] [PubMed] [Google Scholar]
- 32.Aweeka FT. Kang M. Yu JY. Lizak P. Alston B. Chung RT the AIDS Clinical Trials Group 5092s Study Team. Pharmacokinetic evaluation of the effects of ribavirin on zidovudine triphosphate formation: ACTG 5092s Study Team. HIV Med. 2007;8(5):288–294. doi: 10.1111/j.1468-1293.2007.00472.x. [DOI] [PubMed] [Google Scholar]
- 33.Lamotte C. Landman R. Peytavin G. Mentre F. Gerbe J. Brun-Vezinet F. Boue F. Spiridon G. Valantin MA. Michelet C. Farinotti R. Yeno P. Once-daily dosing of saquinavir soft-gel capsules and ritonavir combination in HIV-infected patients. (IMEA015 Study) Antivir Ther. 2004;9(2):247–256. [PubMed] [Google Scholar]
- 34.Chaillou S. Durant J. Garraffo R. Georgenthum E. Roptin C. Levenbergh P. Dunais B. Bondain V. Roger PM. Dellamonica P. Intracellular concentration of protease inhibitors in HIV-1-infected patients: Correlation with MDR-1 gene expression and low dose of ritonavir. HIV Clin Trial. 2002;3(6):493–501. doi: 10.1310/0873-bvdp-akay-445u. [DOI] [PubMed] [Google Scholar]
- 35.Anderson PL. Brundage RC. Kakuda TN. Fletcher CV. CD4 response is correlated with peak plasma concentrations of indinavir in adults with undetectable human immunodeficiency virus ribonucleic acid. Clin Pharmacol Ther. 2002;71:280–285. doi: 10.1067/mcp.2002.121910. [DOI] [PubMed] [Google Scholar]
- 36.Cohen J. Tissue says blood is misleading, confusing HIV cure efforts. Science. 2011;334(6063):1614. doi: 10.1126/science.334.6063.1614. [DOI] [PubMed] [Google Scholar]
- 37.Riddler SA. Haubrich R. DiRenzo AG. Peeples L. Powderly WG. Klingman KL. Garren KW. George T. Rooney JF. Brizz B. Lalloo UG. Murphy RL. Swindells S. Havlir D. Mellors JW the AIDS Clinical Trials Groups Study A5142 Team. Class-sparing regimens for initial treatment of HIV-1 infection. N Engl J Med. 2008;358:2095–2106. doi: 10.1056/NEJMoa074609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Calmy A. Petoumenos K. Lewden C. Law M. Bocquentin F. Hesse K. Cooper D. Carr A. Bonnet F. Aquitaine Cohort Australian HIV Observational Database, and St Vincent's Hospital Cohort Study Groups. Combination antiretroviral therapy without a nucleoside reverse transcriptase inhibitor: Experience from 334 patients in three cohorts. HIV Med. 2007;8:171–180. doi: 10.1111/j.1468-1293.2007.00448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eisingerich AB. Wheelock A. Gomez GB. Garnett GP. Dybul MR. Piot PK. Attitudes and acceptance of oral and parenteral HIV preexposure prophylaxis among potential user groups: A multinational study. PLoS One. 2012;7:e282380. doi: 10.1371/journal.pone.0028238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farmer P. Leandre F. Mukherjee J. Gupta R. Tarter L. Kim JY. Community-based treatment of advanced HIV disease: Introducing DOT-HAART (directly observed therapy with highly active antiretroviral therapy) Bull World Health Org. 2001;79:1145–1151. [PMC free article] [PubMed] [Google Scholar]
- 41.Bangsberg DR. Ragland K. Monk A. Deeks SGA. Single tablet regimen is associated with higher adherence and viral suppression than multiple tablet regimens in HIV+ homeless and marginally housed people. AIDS. 2010;24:2835–2840. doi: 10.1097/QAD.0b013e328340a209. [DOI] [PMC free article] [PubMed] [Google Scholar]