

Abstract
Although diabetic cardiomyopathy is associated with enhanced intramyocardial triacylglycerol (TAG) levels, the role of TAG catabolizing enzymes in this process is unclear. Because the TAG hydrolase, adipose triglyceride lipase (ATGL), regulates baseline cardiac metabolism and function, we examined whether alterations in cardiomyocyte ATGL impact cardiac function during uncontrolled type 1 diabetes. In genetic (Akita) and pharmacological (streptozotocin) murine models of type 1 diabetes, cardiac ATGL protein expression and TAG content were significantly increased. To determine whether increased ATGL expression during diabetes is detrimental or beneficial to cardiac function, we studied streptozotocin-diabetic mice with heterozygous ATGL deficiency and cardiomyocyte-specific ATGL overexpression. After diabetes, streptozotocin-diabetic mice with heterozygous ATGL deficiency displayed increased TAG accumulation, lipotoxicity, and diastolic dysfunction comparable to wild-type mice. In contrast, myosin heavy chain promoter (MHC)-ATGL mice were resistant to diabetes-induced increases in intramyocardial TAG levels, lipotoxicity, and cardiac dysfunction. Moreover, hearts from diabetic MHC-ATGL mice exhibited decreased reliance on palmitate oxidation and blunted peroxisome proliferator--activated receptor-α activation. Collectively, this study shows that after diabetes, increased cardiac ATGL expression is an adaptive, albeit insufficient, response to compensate for the accumulation of myocardial TAG, and that overexpression of ATGL is sufficient to ameliorate diabetes-induced cardiomyopathy.
Cardiovascular disease is the leading cause of mortality in diabetic patients (1,2). Whereas coronary vessel disease and atherosclerosis have been identified as the primary reasons for the increased incidence of diabetic cardiovascular dysfunction (3), a significant number of diabetic patients continue to experience left ventricular dysfunction even in the absence of coronary artery disease or hypertension (4–6). This cardiac dysfunction is referred to as diabetic cardiomyopathy (4–6). Although the causes are multifactorial, the initiating event that contributes to the development of diabetic cardiomyopathy appears to be an early maladaptation in cardiac energy metabolism (7–9) attributable to early onset of intramyocardial lipid accumulation that occurs secondary to altered substrate supply and utilization (10,11). However, the involvement of intramyocardial lipid accumulation in the pathogenesis of diabetic cardiomyopathy has not been clearly defined.
Under normal aerobic conditions, cardiomyocyte ATP is obtained via oxidation of various substrates, including fatty acids (FAs), glucose, lactate, and ketone bodies (12), with FA being the primary energy substrates utilized by the heart. Because the heart has limited potential to synthesize FA, exogenous FAs are supplied to cardiomyocytes via three distinct pathways, all of which are sensitive to insulin inhibition. These pathways include triacylglycerol (TAG) hydrolysis in adipose tissue with an ensuing increase in albumin-bound FA in the plasma (12), hydrolysis of TAG in circulating lipoproteins by lipoprotein lipase (9,13,14), and hydrolysis of TAG stores within cardiomyocytes (15,16). During diabetes, the lack of insulin action results in inadequate myocardial glucose transport and oxidation, thereby relieving insulin's inhibition of FA supply and utilization (8,15). Furthermore, diabetes augments intramyocardial FA availability and oxidation, which promotes pathological TAG accumulation within the cardiomyocyte (17,18). The majority of previous studies have primarily investigated how FA uptake and oxidation influences the progression of diabetic cardiomyopathy (19). However, the role that TAG catabolism plays in influencing metabolism and function during the development of diabetic cardiomyopathy is currently unknown.
The rate-limiting step for cytosolic TAG hydrolysis to diacylglycerol is mediated via adipose triglyceride lipase (ATGL; PNPLA2) (20–23). Consistent with the importance of ATGL in TAG hydrolysis, mice and humans with loss of ATGL activity have development of severe myocardial steatosis and lipotrophic cardiomyopathy, resulting in premature mortality (20–23). Recently, we have shown that overexpression of myocardial ATGL is sufficient to reduce TAG content and to improve systolic function in the healthy heart (24). Because FAs derived from TAG hydrolysis plays a critical role during diabetes (15,16,25), we investigated whether myocardial ATGL-mediated TAG hydrolysis influences cardiac function during the development of type 1 diabetes.
RESEARCH DESIGN AND METHODS
Mice.
Male C57BL/6J wild-type (WT; Ins2WT/WT) and type 1 diabetic Akita (Ins2WT/C96Y) mice were purchased from Jackson Laboratory (stock numbers: WT, 000664; Ins2WT/C96Y, 003548). Mice with cardiomyocyte specific ATGL overexpression driven by myosin heavy chain promoter (MHC-ATGL) and heterozygous ATGL knockout (ATGL KO Het) mice were generated in the laboratory of R.Z. (20,21,23,24). Mice were housed on a 12-h light and12-h dark cycle with ad libitum access to chow diet (with 13.5% kcal from fat; #5001 from Laboratory Diet) and water. Nonfasted mice of mixed gender were used. All experiments were performed according to protocols approved by the University of Alberta Institutional Animal Care and Use Committee.
Induction of diabetes using streptozotocin.
Mice from the MHC-ATGL colony were injected with a single intraperitoneal dose of streptozotocin (STZ; Sigma) in saline at 185 mg/kg body weight. Mice from the ATGL KO Het colony were injected with a lower dose of STZ at 165 mg/kg body weight because of significant mortality associated with this strain in response to STZ.
Echocardiography.
Transthoracic echocardiography was performed as described previously (24).
Tissue lipid analysis.
Tissue TAG and sn-1,2-diacylglycerol content were determined as previously described (24). Quantification of palmitoyl CoA and ceramides was performed using Waters Acquity UPLC (26–28).
Serum analysis.
Using colorimetric assay kits, serum free FA (#999–34691; Wako) and TAG (#2780–400H; Infinity TAG reagent) were measured. Serum insulin was measured using an ELISA assay kit from Crystal Chem (#90080).
Gene expression analysis.
Gene expression analysis was performed using quantitative RT-PCR as previously described (29).
Immunoblot analysis.
Immunoblot analysis was performed as described previously (24). All densitometric data were corrected against total protein loading visualized via Ponceau S or Memcode (Pierce) staining.
Histology.
Hearts were fixed in 10% formalin and paraffin-embedded for subsequent Masson trichrome staining, followed by visualization as described previously (24).
Electron microscopy.
Transmission electron microscopy was performed using a Gatan Orius CCD camera (Morgagni 268 model operating at 80 kV; Philips-FEI). Left ventricular tissue was fixed in 2.5% glutaraldehyde and 2% paraformaldehyde, cut into small blocks (∼1 × 0.5 × 0.4 mm), and fixed for 24 h at room temperature. Tissue was postfixed with 1% osmium tetroxide and dehydrated using increasing concentrations of ethanol (50–100%). Blocks were embedded in Spurr resin and sectioned at 70–90 nm. Sections were stained with 4% uranyl acetate and Reynolds lead citrate.
Citrate synthase and β-hydroxyacyl-CoA dehydrogenase activity assays.
Spectrophotometric citrate synthase and β-hydroxyacyl-CoA dehydrogenase activity assays were performed similar to methods described previously (30,31).
Heart perfusions.
Hearts were aerobically perfused in the working mode at 11.5 mmHg preload and 50 mmHg afterload with Krebs-Henseleit buffer containing 1.2 mmol/L palmitate prebound to 3% delipidated bovine serum albumin, 5 mmol/L glucose, and 50 μU/mL insulin as described previously (24).
Statistical analysis.
Results are expressed as means ± SEM. Statistical analyses were performed using Graph Pad Prism software. Comparisons between two groups were made by unpaired two-tailed Student t test. For comparisons between more than two groups, one-way ANOVA followed by Newman-Keuls multiple comparison test was used. To compare the respective sources of variation (genotype and treatment), data were analyzed using two-way ANOVA followed by Bonferroni post hoc test. P < 0.05 was considered statistically significant.
RESULTS
Increased ATGL protein expression in the hearts of type 1 diabetic Akita mice.
Because diabetic mice exhibit elevated intramyocardial TAG levels (7,15), we investigated whether ATGL is altered in the heart during uncontrolled diabetes. We examined myocardial TAG content and ATGL protein expression in 12-week-old nondiabetic WT (Ins2WT/WT) mice and type 1 diabetic Akita (Ins2WT/C96Y) mice. Consistent with previous findings (32), Akita mice demonstrated characteristic features of uncontrolled diabetes, such as hyperglycemia (Ins2WT/WT: 9.2 ± 0.6 mmol/L glucose; Ins2WT/C96Y: 31.6 ± 1.7 mmol/L glucose; n = 5; P < 0.05) and decreased body weight (Ins2WT/WT: 28.5 ± 0.43 g; Ins2WT/C96Y: 24.4 ± 0.33 g; n = 5; P < 0.05) at an age when diastolic dysfunction also has been reported (32). Interestingly, despite increased myocardial TAG content (Fig. 1A), elevated ATGL protein expression also was observed in Akita mice (Fig. 1B, C).
FIG. 1.

ATGL deficiency exacerbates diabetes-induced lipotoxicity and systolic dysfunction. A: Cardiac TAG in 12-week-old fed WT (Ins2WT/WT) and Akita (Ins2WT/C96Y) mice. B and C: Immunoblots showing cardiac ATGL protein expression in fed WT (Ins2WT/WT) and Akita (Ins2WT/C96Y) hearts (n = 5; *P < 1 × 10−8). Cardiac ATGL protein expression (D, E) and cardiac TAG (F) in WT and ATGL haploinsufficient (ATGL KO Het) mice. Ejection fraction (G) and fractional shortening (H) by transthoracic echocardiography. I: Representative whole-heart sections stained with Masson trichrome for visualizing structural morphology in 13- to 15-week-old mice (n = 7–8). +P < 0.001 vs. WT-SAL; *P < 0.001 vs. WT-SAL; **P < 0.001 vs. ATGL KO Het-SAL; @P < 0.001 vs. WT STZ.
Partial loss of ATGL does not protect from STZ-induced cardiac dysfunction.
We originally speculated that elevated ATGL protein levels during diabetes would increase lipolysis to promote excess FA utilization, which would be detrimental to cardiac function. As a result, we hypothesized that a reduction in ATGL protein expression would be beneficial for the diabetic heart. To test this, we used saline (SAL)- and STZ-treated WT mice and mice with global heterozygous ATGL deficiency (ATGL KO Het). SAL-treated ATGL KO Het mice showed decreased ATGL protein expression (Fig. 1D, E) but unchanged cardiac TAG levels (Fig. 1F). Body weight (WT-SAL: 29.8 ± 1.5 g; ATGL KO Het-SAL: 28.4 ± 1.3 g; n = 7–8), serum glucose (WT-SAL: 7.7 ± 0.5 mmol/L; ATGL KO Het-SAL: 6.9 ± 0.7 mmol/L; n = 7–8), serum TAG (WT-SAL: 83 ± 10.1 mg/dL; ATGL KO Het-SAL: 69.7 ± 18.8 mg/dL; n = 7–8), systolic (Fig. 1G, H) and diastolic cardiac function (Table 1), and overall cardiac morphology (Fig. 1I) also were comparable between ATGL KO Het and WT mice. Thus, in the absence of diabetes, partial deficiency of ATGL does not significantly alter systemic metabolism or cardiac function.
TABLE 1.
In vivo heart function of WT and ATGL KO Het mice at 4 weeks after STZ
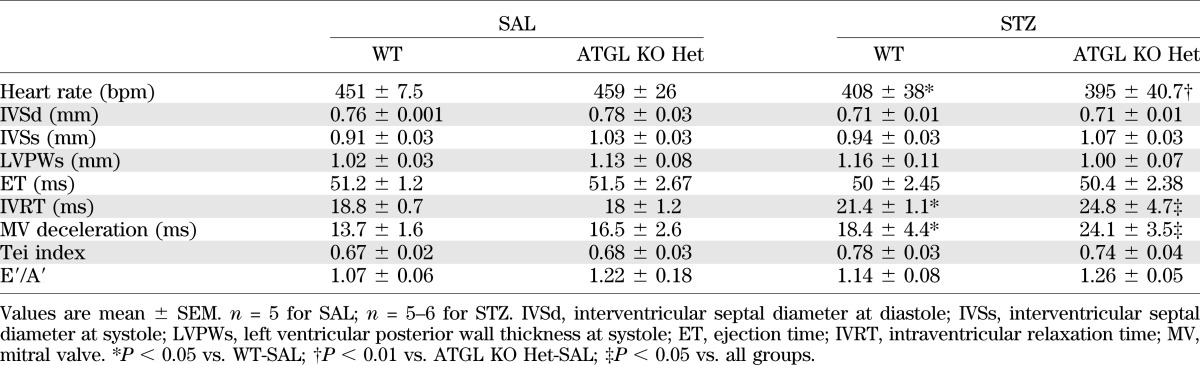
After 4 weeks of uncontrolled type 1 diabetes, the magnitude of induction of ATGL protein expression and cardiac TAG content was comparable between hearts from WT and ATGL KO Het mice (Fig. 1D–F). Moreover, diabetes-induced alterations in body weight (WT STZ: 21.5 ± 0.6 g; ATGL KO Het STZ: 23.4 ± 0.4 g; n = 7–8), serum glucose (WT STZ: 27.4 ± 1.9 mmol/L; ATGL KO Het STZ: 24.3 ± 1.6 mmol/L; n = 7–8), serum TAG (WT STZ: 432.7 ± 100.7 mg/dL; ATGL KO Het STZ: 345.5 ± 97.5 mg/dL; n = 7–8), heart rate (Table 1), systolic cardiac function (Fig. 1G, H), and cardiac dilatation (Fig. 1I) were comparable between genotypes. Furthermore, ventricular wall dimensions (Table 1) were similar between hearts from diabetic WT and ATGL KO Het mice, suggesting the absence of overt cardiac hypertrophy. However, in contrast to our original hypothesis, markers of diastolic function such as mitral valve deceleration rate and isovolumic relaxation time were significantly increased in diabetic ATGL KO Het mice compared with WT mice (Table 1), suggesting that this decrease in ATGL expression after uncontrolled diabetes was associated with diastolic dysfunction. Together, these findings demonstrate that reduction of ATGL protein expression does not protect against diabetes-induced functional impairment of the myocardium.
Partial loss of ATGL does not protect from myocardial peroxisome proliferator–activated receptor-α activation and lipotoxicity after diabetes.
Because peroxisome proliferator–activated receptor-α (PPARα) activation regulates myocardial lipid metabolism (33–35), we also measured protein expression of PPARα in hearts from WT and ATGL KO Het mice. In the absence of diabetes, PPARα expression tended to be higher in hearts from ATGL KO Het mice compared with WT mice (Fig. 2A, B, white bars). Because PPARα mediates its metabolic outcomes via increased gene expression of specific mitochondrial proteins involved in FA sensing (UCP3) (36,37) and storage (perilipin 5) (38), we also measured the expression of these proteins in both genotypes. Consistent with subtle changes in PPARα expression, protein expression of perilipin 5 and UCP3 was unchanged or decreased in ATGL KO Het hearts at baseline, respectively (Fig. 2A, C, D). However, diabetes significantly augmented cardiac PPARα protein expression in both WT and ATGL KO Het mice with concomitant increases in perilipin 5 and UCP3 protein expression (Fig. 2A–D). In agreement with the ability of PPARα to promote lipotoxicity, palmitoyl CoA (Fig. 2E) and ceramides (Fig. 2F) were similarly increased in both genotypes after diabetes.
FIG. 2.

ATGL haploinsufficiency does not protect hearts from diabetes-induced PPARα activation, lipotoxicity, and mitochondrial dysfunction. A–D: Cardiac PPARα, perilipin 5, and UCP3 protein expression in WT and ATGL KO Het mice. Palmitoyl-CoA (E), ceramides (F), and representative electron micrographs (G) of heart sections visualized at 14,000× magnification. Scale bars indicate 0.5 μm. Lipid droplets are marked by white arrows and mitochondrial cristae dysmorphosis is indicated by yellow arrows. Mice were 13–15 weeks old (n = 7–8). +P < 0.001 vs. WT-SAL; *P < 0.001 vs. WT-SAL; **P < 0.001 vs. ATGL KO Het-SAL.
Based on these findings, we next examined whether PPARα activation was associated with changes in mitochondrial ultrastructure and lipid droplet accumulation. At baseline, no morphological alterations in mitochondria or significant lipid droplet accumulation were observed in hearts from ATGL KO Het mice (Fig. 2G). After diabetes, hearts from both genotypes exhibited increased lipid droplet accumulation. However, lipid droplets were significantly enlarged in ATGL KO Het diabetic hearts (Ferret diameter expressed as nm represents the size of the lipid droplet in an average of two fields at 14,000× magnification; WT STZ 109.7 ± 8.7 nm versus ATGL KO Het STZ 176.2 ± 5.3 nm; P = 0.0001). ATGL KO Het hearts also displayed mitochondrial cristae dysmorphosis or mitochondrial “tear” compared with WT hearts after diabetes (Fig. 2G). These data suggest that partial deficiency of ATGL does not significantly alter the diabetic response of transactivating PPARα (39,40), promoting TAG accumulation and lipotoxic cardiomyopathy.
Diabetes-induced TAG accumulation is prevented in ATGL-overexpressing mice.
Because ATGL KO Het mice were susceptible to diabetes-induced TAG accumulation and cardiac dysfunction, we speculated that MHC-ATGL mice would be protected from excessive intramyocardial TAG accumulation and cardiomyopathy after diabetes. To test this, we injected WT and MHC-ATGL mice with either SAL or STZ and studied TAG accumulation and lipotoxic FA metabolic intermediates 4 weeks thereafter. Before and after diabetes, fed glucose (Fig. 3A), body weight (Fig. 3B), and serum TAG levels (Fig. 3C) were comparable between genotypes. Consistent with our previous findings (24), hearts from MHC-ATGL mice showed a significant increase in ATGL mRNA levels (Fig. 3D) and protein expression (Fig. 3E), as well as decreased TAG content (Fig. 3F). After diabetes, hearts from WT, but not MHC-ATGL, mice exhibited a two-fold increase in ATGL mRNA and protein expression (Fig. 3D, E) and TAG content (Fig. 3F), as well as significantly increased lipid droplet deposition (Fig. 3G). The reduction in TAG in hearts from diabetic MHC-ATGL mice also was accompanied by decreases in diacylglycerol content (Fig. 3H). Taken together, these data suggest that ATGL overexpression protects the heart from increased accumulation of TAG during diabetes.
FIG. 3.

ATGL overexpression protects against diabetes-induced lipotoxicity. Fed serum glucose (A), body weight (B), and serum TAG (C) in WT and MHC-ATGL mice. Cardiac Atgl mRNA expression (D) and ATGL protein expression (E). Cardiac TAG (F) and representative electron micrographs (G) of heart sections visualized at 14,000× magnification. Scale bars indicate 0.5 μm. Lipid droplets are marked by white arrows. H: DAG in WT and MHC-ATGL mice in the absence and presence of diabetes. Mice were 12–14 weeks old (n = 8–10). +P < 0.001 vs. WT-SAL; *P < 0.001 vs. WT-SAL; **P < 0.001 vs. MHC ATGL-SAL; @P < 0.001 vs. WT STZ.
MHC-ATGL mice are resistant to diabetes-induced metabolic remodeling and lipotoxicity.
Although diabetes impairs myocardial glucose utilization and increases FA oxidation (17), at similar heart rates (Fig. 4A) ex vivo perfusion of diabetic MHC-ATGL hearts revealed increased glucose oxidation (Fig. 4B) and decreased palmitate oxidation (Fig. 4C) compared with WT hearts, resulting in increased reliance on glucose oxidation for Krebs cycle acetyl-CoA production (Fig. 4D). Moreover, β-hydroxyacyl-CoA dehydrogenase enzyme activity (Fig. 4E) and PGC1-α mRNA expression (Fig. 4F), which correlates with muscle FA oxidative capacity (41,42), were augmented in STZ-treated WT but not in STZ-treated MHC-ATGL mice. Importantly, enhanced lipid utilization in STZ-treated WT mice translated into increased cardiac accumulation of the lipotoxic intermediates, palmitoyl CoA (Fig. 4G) and ceramides (Fig. 4H), an effect that was not observed in diabetic MHC-ATGL hearts.
FIG. 4.

ATGL-overexpressing hearts exhibit reduced palmitate oxidation after diabetes. Hearts from male 40- to 50-week-old mice were perfused for 60 min ex vivo with 1.2 mmol/L [9, 10-3H]palmitate, 5 mmol/L [U-14C]glucose, and 50 μU/mL insulin (n = 5–7; *P < 0.05). Heart rate (A), glucose oxidation (B), palmitate oxidation (C), and Krebs cycle acetyl-CoA production (D). β-hydroxyacyl-CoA dehydrogenase enzyme activity (HADH) (E), cardiac Pgc1α mRNA expression (F), palmitoyl CoA (G), and ceramides (H) in 12- to 14-week-old WT and MHC ATGL mice (n = 6–8) in the absence or presence of diabetes. +P < 0.001 vs. WT-SAL; *P < 0.001 vs. WT-SAL; @P < 0.001 vs. WT STZ.
MHC-ATGL mice are protected from diabetes-induced cardiac dysfunction.
We next investigated whether resistance to metabolic remodeling and lipotoxicity in hearts from diabetic MHC-ATGL mice translated into improved cardiac function. Diastolic function (Table 2) was comparable between SAL-injected WT and MHC-ATGL mice. However, as reported previously (24), systolic function at baseline was significantly enhanced in MHC-ATGL mice, as was evident from increased ejection fraction (Fig. 5A–D), fractional shortening (Fig. 5A, C), and circumferential fiber shortening velocity (Fig. 5D). Mitral valve E-wave velocity also was increased in hearts from MHC-ATGL mice (Table 2). Importantly, in diabetic WT mice, but not diabetic MHC-ATGL mice, a significant decrease in systolic function (Fig. 5A–D) was observed, which was associated with a concomitant decrease in mitral valve E-wave velocity, increased left ventricular volume at systole (Table 2), and augmented ejection and isovolumic contraction time.
TABLE 2.
In vivo heart function of WT and cardiac-restricted ATGL-overexpressing mice at 4 weeks after STZ
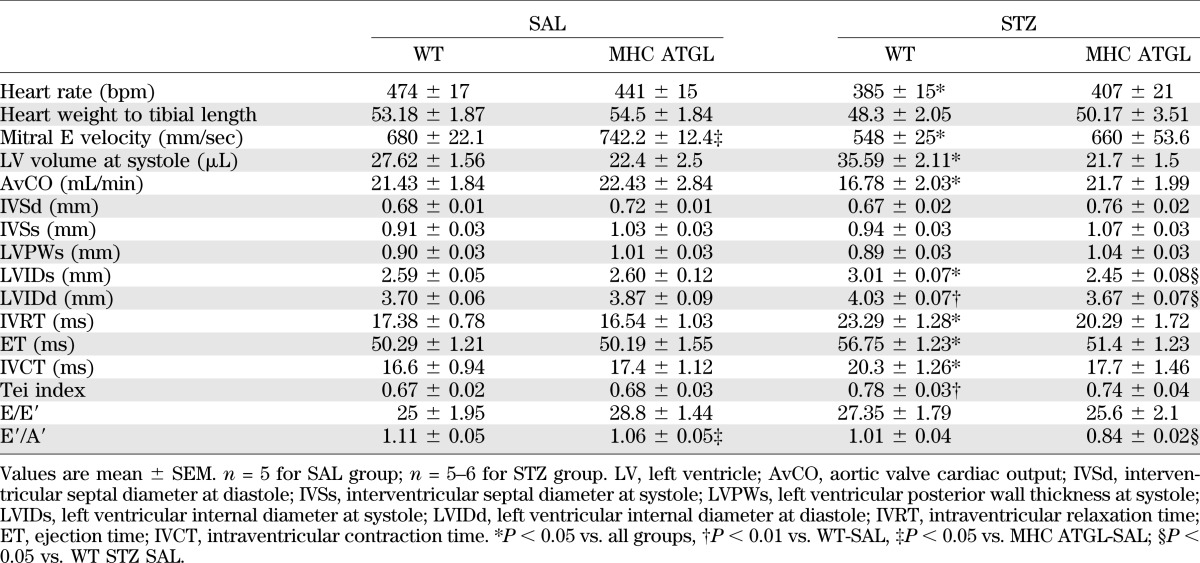
FIG. 5.

ATGL overexpression protects against diabetes-induced systolic dysfunction and cardiomyopathy. Transthoracic echocardiography images (A), ejection fraction (B), fractional shortening (C), velocity of circumferential fiber shortening (VcF) (D), and representative whole-heart sections stained with Masson trichrome for visualizing structural morphology (E). Mice were 13–15 weeks old (n = 10–12). +P < 0.001 vs. WT-SAL; *P < 0.001 vs. WT-SAL; @P < 0.001 vs. WT STZ SAL.
In addition to changes in systolic function, isovolumic relaxation time was increased in hearts from diabetic WT mice, but not diabetic MHC-ATGL mice, indicating diastolic dysfunction (Table 2). Moreover, diabetic WT mice exhibited significant cardiac dilatation, which was not observed in diabetic MHC-ATGL mice (Fig. 5E, Table 2). These changes between genotypes were not associated with overt cardiac hypertrophy (Table 2). Together, these data show that cardiomyocyte-specific ATGL overexpression protects mice from development of cardiac dysfunction induced by uncontrolled diabetes.
Altered metabolism in hearts from diabetic MHC-ATGL mice occurs independent of changes in markers of glucose utilization.
Because alterations in cardiac energy metabolism could underlie the limited susceptibility of MHC-ATGL hearts to diabetes-induced dysfunction, we examined markers of biochemical pathways influencing glucose and fat utilization. Enhanced glucose utilization in MHC-ATGL hearts at baseline (24) and after diabetes was independent of alterations in GLUT1 mRNA levels (Fig. 6A). GLUT4 protein levels were lower in MHC-ATGL hearts and decreased significantly after diabetes in both genotypes (Fig. 6B, C), which was not associated with changes in Akt serine 473 phosphorylation (Fig. 6B, D). Although PDK4 mRNA levels were increased in both genotype after diabetes (Fig. 6E), PDH serine 293 phosphorylation was augmented only in hearts from diabetic WT mice (Fig. 6F), suggesting increased glucose flux in hearts from MHC-ATGL mice.
FIG. 6.

Diabetes-induced metabolic switching is not altered in either genotype, and it is independent of GLUT levels and insulin signaling. A: Cardiac GLUT1 mRNA expression. B–D: Immunoblot analysis for protein expression of GLUT4 and AKT phosphorylation. Cardiac PDK4 mRNA expression (E) and PDH Ser293 phosphorylation (F) in WT and MHC ATGL mice in the absence or presence of diabetes. Mice were 13–15 weeks old (n = 6–8). +P < 0.001 vs. WT-SAL; *P < 0.001 vs. WT-SAL; **P < 0.001 vs. MHC ATGL-SAL; @P < 0.001 vs. WT STZ.
Because hearts from diabetic mice are reported to have augmented TAG turnover (7,15,16), we also measured the expression of diacylglycerol acyltransferase 2 (Dgat2), which is involved in TAG synthesis. As we reported previously (24), in the absence of diabetes, Dgat2 mRNA expression (Fig. 7A) was reduced in hearts from MHC-ATGL mice. Whereas Dgat2 mRNA expression increased in both genotypes after diabetes, Dgat2 mRNA transcript levels were still significantly lower in hearts from diabetic MHC-ATGL mice (Fig. 7A). In addition, cardiac PPARα protein expression was markedly decreased in hearts from SAL-treated MHC-ATGL mice compared with WT (Fig. 7B, C), which corresponded with reduced protein expression of CD36, UCP3, perilipin 5, and long-chain acyl CoA synthetase 1 (ACSL1) (Fig. 7B, D–G). Interestingly, diabetes led to increased protein expression of PPARα in hearts from WT but not from MHC-ATGL mice (Fig. 7B, C). Consistent with diminished activation of PPARα, protein expressions of CD36, UCP3, and perilipin 5 were significantly reduced in hearts from diabetic MHC-ATGL mice (Fig. 7B, D–G). Although diabetes did not influence ACSL1 protein expression (Fig. 7B, G), ACSL1 protein content was significantly lower in hearts from STZ-treated MHC-ATGL mice compared with WT (Fig. 7G). Together, these data show that hearts from MHC-ATGL mice displayed diminished PPARα activation and expression of proteins involved in FA utilization both at baseline and after diabetes. These findings provide a plausible explanation for the resistance to diabetes-induced myocardial lipotoxicity and metabolic remodeling in MHC-ATGL mice.
FIG. 7.

Diabetes-induced upregulation of PPARα is not observed in mice with ATGL overexpression. A: Cardiac Dgat2 mRNA expression. B–G: Immunoblot analysis for protein expression of PPARα (C), CD36 (D), UCP3 (E), perilipin 5 (F), and ACSL1 (G) in WT and MHC ATGL mice in the absence or presence of diabetes. Mice were 13–15 weeks old (n = 6–8). +P < 0.001 vs. WT-SAL; *P < 0.001 vs. WT-SAL; **P < 0.001 vs. MHC ATGL-SAL; @P < 0.001 vs. WT STZ.
MHC-ATGL hearts are resistant to diabetes-induced alterations in specific electron transport chain complex protein expression.
Because lipotoxicity-induced cardiac dysfunction could be secondary to changes in the mitochondrial proteome (43), we next examined the expression of relevant mitochondrial electron transport chain (ETC) proteins. In the absence of diabetes, complex II (Fig. 8A, B), complex IV (mitochondrial cytochrome c oxidase 1; Fig. 8A, B), and complex V content (Fig. 8A, B) were unchanged between genotypes. However, protein expression of complex I (NADH dehydrogenase [ubiquinone] 1α subcomplex subunit 9) and complex III were decreased (Fig. 8A, B) and increased (Fig. 8A, B), respectively, in hearts from MHC-ATGL mice. Furthermore, diabetes significantly reduced myocardial protein expression of NADH dehydrogenase (ubiquinone) 1α subcomplex subunit 9 (Fig. 8A, B), complex V (Fig. 8A, B), and mitochondrial cytochrome oxidase 1 (Fig. 8A, B) in hearts from WT mice subjected to diabetes but not in hearts from diabetic MHC-ATGL mice. Interestingly, protein expression of complex III (Fig. 8A, B) was increased in hearts from WT diabetic mice, an effect not observed in hearts from STZ-treated MHC-ATGL mice. Given similar citrate synthase activity (Fig. 8C) and nuclear respiratory factor-1 mRNA expression (Fig. 8D) between genotypes, these changes in mitochondrial ETC protein complexes also were independent of changes in mitochondrial mass or DNA content. In light of the fact that endoplasmic reticulum (ER) stress contributes to mitochondrial dysfunction during diabetes (44), we also examined ER stress markers in hearts from WT and MHC-ATGL mice. Protein expression of GRP94, CHOP, calreticulin, ERO1α, calnexin, and phosphorylated PKR-like eukaryotic initiation factor-2α kinase as well as p38MAPK phosphorylation (Fig. 8E, F) were significantly reduced in hearts from diabetic MHC-ATGL mice compared with WT mice. Together, these findings suggest that improved cardiac function in hearts from diabetic MHC-ATGL mice may be attributable to reduced ER stress and the prevention of mitochondrial ETC complex protein remodeling after diabetes.
FIG. 8.

ATGL overexpression prevents ETC complex remodeling and reduces ER stress after diabetes. Cardiac NADH dehydrogenase (ubiquinone) 1α subcomplex subunit (NDUFA9), complex II, complex III, complex V, and mitochondrial cytochrome oxidase-1 protein expression (A, B), citrate synthase enzyme activity (C), cardiac NRF1 mRNA expression (D), cardiac GRP94, CHOP, calreticulin, ERO1α, calnexin, phosphorylated PKR-like eukaryotic initiation factor-2α kinase (PERK) Thr980, p38MAPK phosphorylation, and p38MAPK protein expression (E, F) in WT and MHC ATGL mice shown as percent of WT-SAL. Mice were 13–15 weeks old (n = 7–8). +P < 0.001 vs. WT-SAL; *P < 0.001 vs. WT-SAL; @P < 0.05 vs. WT STZ.
DISCUSSION
Consistent with previous studies (32,39,40,45), we show that cardiac dysfunction in both the Akita and STZ models of diabetes (32,40) was associated with robust increases in cardiac TAG content and enhanced mRNA and protein expression of ATGL. Therefore, to ascertain whether increased myocardial ATGL expression during diabetes is protective or detrimental to the heart, we studied mice with either whole-body heterozygous deficiency of ATGL or cardiac-specific ATGL overexpression. In ATGL KO Het mice, diabetes-induced diastolic dysfunction was correlated with increased TAG levels, as well as palmitoyl CoA and ceramides accumulation. In contrast, the pathological increases in cardiac TAG, palmitoyl CoA, and ceramides observed in hearts from WT mice were blunted in hearts from MHC-ATGL mice, suggesting a profound resistance to diabetes-induced lipotoxic cardiac remodeling in MHC-ATGL mice. In the diabetic MHC-ATGL mice, reduced myocardial lipid content corresponded with improved cardiac function and blunted cardiac dilatation, suggesting a cardiomyocyte-autonomous beneficial effect of ATGL overexpression in the diabetic heart.
Although our data suggest otherwise, previous studies have indicated that TAG per se is not lipotoxic (46) and that TAG accumulation may be protective by channeling FA away from toxic metabolic pathways (46). However, our findings are consistent with studies demonstrating that decreases in myocardial TAG content are associated with improved cardiac function in patients with diabetes and obesity (46,47). Our findings are also in agreement with other mouse models, such as cardiac apolipoprotein B overexpressors (48) and hormone-sensitive lipase transgenic mice (45), which show that enhanced intramyocardial TAG clearance reduces lipotoxicity and prevents histological and biochemical remodeling of the heart after diabetes. As such, we propose that enhanced TAG catabolism in hearts from diabetic MHC-ATGL mice protects from lipotoxicity and cardiac dysfunction. That said, we recognize that TAG accumulation could be secondary to altered TAG synthesis or increased FA uptake independent of changes in TAG catabolism. However, hearts from diabetic WT or MHC-ATGL mice both displayed induction of Dgat2, a TAG-synthesizing enzyme (49), indicating that alterations in TAG synthesis are unlikely to be responsible for diminished lipid accumulation within the cardiomyocyte.
Consistent with impaired myocardial glucose utilization in diabetes (4,5,10), hearts from diabetic WT mice exhibited diminished glucose oxidation and hyperphosphorylation of PDH. This impaired glucose utilization also was reflected by an increased reliance on FA oxidation and upregulated β-hydroxyacyl-CoA dehydrogenase activity as well as PPAR-γ coactivator-1α mRNA levels. In contrast, hearts from control and diabetic MHC-ATGL mice exhibited decreased FA oxidation that resulted in increased glucose utilization at baseline (24) and following uncontrolled diabetes. Interestingly, despite enhanced FA availability induced by diabetes, hearts from MHC-ATGL mice displayed decreased expression of PPARα and its downstream targets perilipin 5, CD36, UCP3, and ACSL1 at baseline and after diabetes compared with WT mice (a finding that was not observed in hearts from WT and ATGL KO Het mice). These opposing diabetic phenotypes observed in mice with loss and gain of ATGL activity are reminiscent of cardiomyocyte-specific PPARα-overexpressing mice that develop cardiac steatosis, dilatation, and systolic dysfunction, whereas diabetic PPARα KO mice do not (10,12,13). Similar to ATGL-overexpressing mice, cardiomyocyte-specific hormone-sensitive lipase transgenic mice also display diminished induction of PPARα mRNA expression in response to diabetes (45), suggestive of a mechanism that likely inhibits diabetes-induced TAG accumulation and cardiac dysfunction. As such, we propose that the inability to expand the myocardial TAG pool in hearts from MHC-ATGL mice leads to decreased PPARα signaling, reduced FA oxidation, and increased reliance on glucose for ATP production not only at baseline (24) but also after uncontrolled diabetes. MHC-ATGL mice also exhibited decreased CD36 levels, suggesting that in addition to reduced TAG accumulation, decreased FA uptake also could be involved in the protection against diabetes-induced cardiac dysfunction. Although we cannot provide evidence why our findings appear to be contrary to the observation that ATGL provides ligands to activate PPARα (21), we proposed that this disparity may be attributable to the fact that ATGL overexpression in our study is constitutive with unchanged ATGL levels in all other cell types and without alterations in plasma lipids.
Mitochondrial dysfunction during diabetes is likely attributable to altered expression of ETC respiratory complexes (5,43,50,51) secondary to ER stress (44,52,53). Loss of ATGL during diabetes precipitated mitochondrial cristae dysmorphosis, an effect not observed in ATGL overexpressors. Furthermore, hearts from diabetic MHC-ATGL mice exhibited reduced expression of ER stress markers, which likely prevented decreases in NADH dehydrogenase (ubiquinone) 1α subcomplex subunit 9, mitochondrial cytochrome oxidase-1, and complex V subunit expression independent of changes in markers of mitochondrial mass or biogenesis such as nuclear respiratory factor-1 mRNA levels and citrate synthase activity (54). Taken together, we propose that during early stages of diabetes (4 weeks), robust TAG clearance after ATGL overexpression reduces expression of ER stress markers, preserves ETC protein content, and protects hearts from cardiac dysfunction.
In conclusion, pathological intramyocardial TAG accumulation is a hallmark of diabetic heart disease (1,7–9,12). However, the role that ATGL plays in the pathogenesis of diabetic cardiomyopathy has not been previously studied. Herein, we show that partial loss of whole-body ATGL did not alter TAG accumulation or ameliorate lipotoxicity, PPARα activation, and diastolic dysfunction after diabetes. In contrast, mice with cardiomyocyte-specific ATGL overexpression were resistant to diabetes-induced increases in myocardial TAG and lipotoxic intermediate accumulation as well as PPARα activation. Together, the prevention of these effects corresponded with decreased reliance on FA oxidation and preserved cardiac function in diabetic mice. As such, our data suggest that increased cardiac ATGL expression during diabetes is an adaptive, albeit insufficient, response to compensate for the increased accumulation of intramyocardial TAG. We also conclude that further augmentation of ATGL expression in the heart is sufficient to ameliorate diabetes-induced steatosis and cardiomyopathy.
ACKNOWLEDGMENTS
This work was supported by grants from the Canadian Institutes of Health Research, the Heart and Stroke Foundation of Canada, and the Alberta Diabetes Institute to J.R.B.D., by postdoctoral fellowships from the Heart and Stroke Foundation of Canada and the Canadian Diabetes Association to P.C.K., and by an Alberta Innovates–Health Solutions postdoctoral award to T.P. and P.C.K.
No potential conflicts of interest relevant to this article were reported.
T.P., P.C.K., and J.R.B.D. designed the research. T.P., P.C.K., J.N., T.J.W., and M.E.Y. performed the research. G.K. provided the reagents. G.H. and R.Z. contributed the mouse models. T.P., P.C.K., E.E.K., and J.R.B.D. analyzed data and wrote the manuscript. J.R.B.D. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
REFERENCES
- 1.Lteif AA, Mather KJ, Clark CM. Diabetes and heart disease an evidence-driven guide to risk factors management in diabetes. Cardiol Rev 2003;11:262–274 [DOI] [PubMed] [Google Scholar]
- 2.Sowers JR, Epstein M, Frohlich ED. Diabetes, hypertension, and cardiovascular disease: an update. Hypertension 2001;37:1053–1059 [DOI] [PubMed] [Google Scholar]
- 3.Wilson PW. Diabetes mellitus and coronary heart disease. Endocrinol Metab Clin North Am 2001;30:857–881 [DOI] [PubMed] [Google Scholar]
- 4.Battiprolu PK, Gillette TG, Wang ZV, Lavandero S, Hill JA. Diabetic Cardiomyopathy: Mechanisms and Therapeutic Targets. Drug Discov Today Dis Mech 2010;7:e135–e143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord 2010;11:31–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miki T, Yuda S, Kouzu H, Miura T. Diabetic cardiomyopathy: pathophysiology and clinical features. Heart Fail Rev 2013;18:149–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stanley WC, Lopaschuk GD, McCormack JG. Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc Res 1997;34:25–33 [DOI] [PubMed] [Google Scholar]
- 8.Rodrigues B, Cam MC, McNeill JH. Metabolic disturbances in diabetic cardiomyopathy. Mol Cell Biochem 1998;180:53–57 [PubMed] [Google Scholar]
- 9.Pulinilkunnil T, Rodrigues B. Cardiac lipoprotein lipase: metabolic basis for diabetic heart disease. Cardiovasc Res 2006;69:329–340 [DOI] [PubMed] [Google Scholar]
- 10.Fang ZY, Prins JB, Marwick TH. Diabetic cardiomyopathy: evidence, mechanisms, and therapeutic implications. Endocr Rev 2004;25:543–567 [DOI] [PubMed] [Google Scholar]
- 11.Klöting I, Berg S, Kovács P, Voigt B, Vogt L, Schmidt S. Diabetes and hypertension in rodent models. Ann N Y Acad Sci 1997;827:64–84 [DOI] [PubMed] [Google Scholar]
- 12.Lopaschuk GD, Belke DD, Gamble J, Itoi T, Schönekess BO. Regulation of fatty acid oxidation in the mammalian heart in health and disease. Biochim Biophys Acta 1994;1213:263–276 [DOI] [PubMed] [Google Scholar]
- 13.Pulinilkunnil T, Abrahani A, Varghese J, et al. Evidence for rapid “metabolic switching” through lipoprotein lipase occupation of endothelial-binding sites. J Mol Cell Cardiol 2003;35:1093–1103 [DOI] [PubMed] [Google Scholar]
- 14.Pulinilkunnil T, Qi D, Ghosh S, et al. Circulating triglyceride lipolysis facilitates lipoprotein lipase translocation from cardiomyocyte to myocardial endothelial lining. Cardiovasc Res 2003;59:788–797 [DOI] [PubMed] [Google Scholar]
- 15.Saddik M, Lopaschuk GD. Triacylglycerol turnover in isolated working hearts of acutely diabetic rats. Can J Physiol Pharmacol 1994;72:1110–1119 [DOI] [PubMed] [Google Scholar]
- 16.Paulson DJ, Crass MF., 3rd Endogenous triacylglycerol metabolism in diabetic heart. Am J Physiol 1982;242:H1084–H1094 [DOI] [PubMed] [Google Scholar]
- 17.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev 2010;90:207–258 [DOI] [PubMed] [Google Scholar]
- 18.Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev 2002;23:201–229 [DOI] [PubMed] [Google Scholar]
- 19.An D, Rodrigues B. Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol 2006;291:H1489–H1506 [DOI] [PubMed] [Google Scholar]
- 20.Haemmerle G, Lass A, Zimmermann R, et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006;312:734–737 [DOI] [PubMed] [Google Scholar]
- 21.Haemmerle G, Moustafa T, Woelkart G, et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat Med 2011;17:1076–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zechner R, Kienesberger PC, Haemmerle G, Zimmermann R, Lass A. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J Lipid Res 2009;50:3–21 [DOI] [PubMed] [Google Scholar]
- 23.Zimmermann R, Strauss JG, Haemmerle G, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004;306:1383–1386 [DOI] [PubMed] [Google Scholar]
- 24.Kienesberger PC, Pulinilkunnil T, Sung MM, et al. Myocardial ATGL overexpression decreases the reliance on fatty acid oxidation and protects against pressure overload-induced cardiac dysfunction. Mol Cell Biol 2012;32:740–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Severson DL. Diabetic cardiomyopathy: recent evidence from mouse models of type 1 and type 2 diabetes. Can J Physiol Pharmacol 2004;82:813–823 [DOI] [PubMed] [Google Scholar]
- 26.Deutsch J, Grange E, Rapoport SI, Purdon AD. Isolation and quantitation of long-chain acyl-coenzyme A esters in brain tissue by solid-phase extraction. Anal Biochem 1994;220:321–323 [DOI] [PubMed] [Google Scholar]
- 27.Larson TR, Graham IA. Technical Advance: a novel technique for the sensitive quantification of acyl CoA esters from plant tissues. Plant J 2001;25:115–125 [DOI] [PubMed] [Google Scholar]
- 28.Bose R, Kolesnick R. Measurement of ceramide levels by the diacylglycerol kinase reaction and by high-performance liquid chromatography-fluorescence spectrometry. Methods Enzymol 2000;322:373–378 [DOI] [PubMed] [Google Scholar]
- 29.Tsai JY, Kienesberger PC, Pulinilkunnil T, et al. Direct regulation of myocardial triglyceride metabolism by the cardiomyocyte circadian clock. J Biol Chem 2010;285:2918–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 2005;112:2686–2695 [DOI] [PubMed] [Google Scholar]
- 31.Ito M, Jaswal JS, Lam VH, et al. High levels of fatty acids increase contractile function of neonatal rabbit hearts during reperfusion following ischemia. Am J Physiol Heart Circ Physiol 2010;298:H1426–H1437 [DOI] [PubMed] [Google Scholar]
- 32.Basu R, Oudit GY, Wang X, et al. Type 1 diabetic cardiomyopathy in the Akita (Ins2WT/C96Y) mouse model is characterized by lipotoxicity and diastolic dysfunction with preserved systolic function. Am J Physiol Heart Circ Physiol 2009;297:H2096–H2108 [DOI] [PubMed] [Google Scholar]
- 33.Holness MJ, Samsuddin S, Sugden MC. The role of PPARs in modulating cardiac metabolism in diabetes. Pharmacol Res 2009;60:185–194 [DOI] [PubMed] [Google Scholar]
- 34.Burkart EM, Sambandam N, Han X, et al. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest 2007;117:3930–3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finck BN. The role of the peroxisome proliferator-activated receptor alpha pathway in pathological remodeling of the diabetic heart. Curr Opin Clin Nutr Metab Care 2004;7:391–396 [DOI] [PubMed] [Google Scholar]
- 36.Pedraza N, Rosell M, Villarroya J, et al. Developmental and tissue-specific involvement of peroxisome proliferator-activated receptor-alpha in the control of mouse uncoupling protein-3 gene expression. Endocrinology 2006;147:4695–4704 [DOI] [PubMed] [Google Scholar]
- 37.Young ME, Patil S, Ying J, et al. Uncoupling protein 3 transcription is regulated by peroxisome proliferator-activated receptor (alpha) in the adult rodent heart. FASEB J 2001;15:833–845 [DOI] [PubMed] [Google Scholar]
- 38.Wolins NE, Quaynor BK, Skinner JR, et al. OXPAT/PAT-1 is a PPAR-induced lipid droplet protein that promotes fatty acid utilization. Diabetes 2006;55:3418–3428 [DOI] [PubMed] [Google Scholar]
- 39.Finck BN, Han X, Courtois M, et al. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci USA 2003;100:1226–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finck BN, Lehman JJ, Leone TC, et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest 2002;109:121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morio B, Hocquette JF, Montaurier C, et al. Muscle fatty acid oxidative capacity is a determinant of whole body fat oxidation in elderly people. Am J Physiol Endocrinol Metab 2001;280:E143–E149 [DOI] [PubMed] [Google Scholar]
- 42.Nikolić N, Rhedin M, Rustan AC, Storlien L, Thoresen GH, Strömstedt M. Overexpression of PGC-1α increases fatty acid oxidative capacity of human skeletal muscle cells. Biochem Res Int 2012;2012:714074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bugger H, Chen D, Riehle C, et al. Tissue-specific remodeling of the mitochondrial proteome in type 1 diabetic akita mice. Diabetes 2009;58:1986–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lim JH, Lee HJ, Ho Jung M, Song J. Coupling mitochondrial dysfunction to endoplasmic reticulum stress response: a molecular mechanism leading to hepatic insulin resistance. Cell Signal 2009;21:169–177 [DOI] [PubMed] [Google Scholar]
- 45.Ueno M, Suzuki J, Zenimaru Y, et al. Cardiac overexpression of hormone-sensitive lipase inhibits myocardial steatosis and fibrosis in streptozotocin diabetic mice. Am J Physiol Endocrinol Metab 2008;294:E1109–E1118 [DOI] [PubMed] [Google Scholar]
- 46.Listenberger LL, Han X, Lewis SE, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA 2003;100:3077–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hammer S, Snel M, Lamb HJ, et al. Prolonged caloric restriction in obese patients with type 2 diabetes mellitus decreases myocardial triglyceride content and improves myocardial function. J Am Coll Cardiol 2008;52:1006–1012 [DOI] [PubMed] [Google Scholar]
- 48.Nielsen LB, Bartels ED, Bollano E. Overexpression of apolipoprotein B in the heart impedes cardiac triglyceride accumulation and development of cardiac dysfunction in diabetic mice. J Biol Chem 2002;277:27014–27020 [DOI] [PubMed] [Google Scholar]
- 49.Yen CL, Stone SJ, Koliwad S, Harris C, Farese RV., Jr Thematic review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J Lipid Res 2008;49:2283–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol 2009;54:1891–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bugger H, Boudina S, Hu XX, et al. Type 1 diabetic akita mouse hearts are insulin sensitive but manifest structurally abnormal mitochondria that remain coupled despite increased uncoupling protein 3. Diabetes 2008;57:2924–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee JW, Kim WH, Yeo J, Jung MH. ER stress is implicated in mitochondrial dysfunction-induced apoptosis of pancreatic beta cells. Mol Cells 2010;30:545–549 [DOI] [PubMed] [Google Scholar]
- 53.Tang C, Koulajian K, Schuiki I, et al. Glucose-induced beta cell dysfunction in vivo in rats: link between oxidative stress and endoplasmic reticulum stress. Diabetologia 2012;55:1366–1379 [DOI] [PubMed] [Google Scholar]
- 54.van Bilsen M, Smeets PJ, Gilde AJ, van der Vusse GJ. Metabolic remodelling of the failing heart: the cardiac burn-out syndrome? Cardiovasc Res 2004;61:218–226 [DOI] [PubMed] [Google Scholar]