

Abstract
Lipid metabolism is important for health and insulin action, yet the fundamental process of regulating lipid metabolism during muscle contraction is incompletely understood. Here, we show that liver kinase B1 (LKB1) muscle-specific knockout (LKB1 MKO) mice display decreased fatty acid (FA) oxidation during treadmill exercise. LKB1 MKO mice also show decreased muscle SIK3 activity, increased histone deacetylase 4 expression, decreased NAD+ concentration and SIRT1 activity, and decreased expression of genes involved in FA oxidation. In AMP-activated protein kinase (AMPK)α2 KO mice, substrate use was similar to that in WT mice, which excluded that decreased FA oxidation in LKB1 MKO mice was due to decreased AMPKα2 activity. Additionally, LKB1 MKO muscle demonstrated decreased FA oxidation in vitro. A markedly decreased phosphorylation of TBC1D1, a proposed regulator of FA transport, and a low CoA content could contribute to the low FA oxidation in LKB1 MKO. LKB1 deficiency did not reduce muscle glucose uptake or oxidation during exercise in vivo, excluding a general impairment of substrate use during exercise in LKB1 MKO mice. Our findings demonstrate that LKB1 is a novel molecular regulator of major importance for FA oxidation but not glucose uptake in muscle during exercise.
The interplay between lipids and insulin action has been the focus in diabetes research for several years. Importantly, exercise leads to increased insulin sensitivity (1,2) and oxidation of fatty acids (FAs) (3) in skeletal muscle. However, by which mechanisms FA oxidation is regulated by muscle contractions remains unsolved. Based on the original observations that the AMP-activated protein kinase (AMPK) activating substance 5-aminoimidazole-4-carboxamide riboside (AICAR) increased FA oxidation in perfused rat skeletal muscle (4) and the finding that exercise increases AMPK activity in muscle (5,6), it was originally thought that AMPK plays an important role in enhancing FA oxidation during exercise. In contrast, it was recently reported that FA oxidation during treadmill running was not decreased in muscle-specific AMPK β1β2 knockout mice (7) or during contractions ex vivo in muscle overexpressing a dominant negative AMPK construct (8). Also, women have higher FA oxidation but lower AMPK activity during submaximal exercise than men (9).
Liver kinase B1 (LKB1) is upstream of AMPK and a family of 12 other Ser/Thr kinases closely related to AMPK, which would potentially be regulated by LKB1 in vivo in skeletal muscle. These include sucrose nonfermenting AMPK-related kinase (SNARK) (10,11) and salt-inducible kinase (SIK)1 and -3 (12,13). Recently, the LKB1-regulated kinase SIK3 was in Drosophila shown to regulate FOXO activity by modulating its deacetylation by histone deacetylase (HDAC)4 (14). In skeletal muscle, FOXO1 activity was increased with fasting (15), which increased pyruvate dehydrogenase kinase 4–mediated inactivation of pyruvate dehydrogenase, resulting in increased FA oxidation (16,17). Furthermore, when FOXO1 activity was genetically increased in C2C12 myotubes, this markedly upregulated FA oxidation (18). Thus, it could be hypothesized that LKB1 might be involved in regulation of FA oxidation in muscle as well. Interestingly, deletion of LKB1 resulted in blunted AICAR-induced increase in FA oxidation in isolated extensor digitorum longus (EDL) mouse muscle (19). This is in contrast to EDL muscle overexpressing a dominant negative AMPKα2 isoform in which the AICAR effect on FA oxidation was normal (8). This could suggest that LKB1 is involved in regulation of FA oxidation through downstream targets other than AMPK.
LKB1 has been shown to play a key role in mediating both AICAR and contraction-induced muscle glucose uptake (12,20). However, the dependence of LKB1 in glucose uptake during muscle contraction has only been investigated using intense electrical stimulation. These stimulation regimes differ from physiological exercise in many ways (21), which was illustrated by severe fatigue leading to impaired contractile force development of both isolated soleus and EDL muscles within 2 min after the onset of electrical stimulation (12,20).
As the role of LKB1 in regulation of muscle metabolism under physiological conditions is unclear, we examined whether LKB1 is important for the cellular control of substrate turnover during physiological exercise in mice and electrical stimulation of isolated mouse muscle.
RESEARCH DESIGN AND METHODS
All reagents were from Sigma-Aldrich unless stated otherwise. Sixteen- to 20-week-old mice with muscle-specific knockout of LKB1 (LKB1 MKO) or whole-body AMPKα2 knockout (KO) and their respective wild-type (WT) littermates were used. The mice were generated as earlier described (19,22). Mice were maintained on a 10:14-h light-dark cycle and received standard chow (Altromin, cat. no. 1324; Chr. Pedersen, Ringsted, Denmark) and water ad libitum. All experiments were approved by the Danish Animal Experimental Inspectorate. Breeding protocols were approved by the Institutional Animal Care and Use Committee at Brigham Young University (LKB1 MKO mice) and the Danish Animal Experimental Inspectorate (AMPKα2 KO mice). Genotyping was performed by PCR analysis as previously described (19,23).
Treadmill exercise test, respiratory exchange ratio, and oxygen uptake.
Acclimatization to the treadmill and subsequently the maximal running test was performed as previously described (24). O2 uptake and CO2 production were measured using a CaloSys apparatus (TSE Systems, Bad Homburg, Germany) at rest (24 h) or during exercise. The respiratory exchange ratio (RER) was calculated as Vco2 production/Vo2 uptake. LKB1 MKO and WT mice were run for 24 min on the treadmill. The LKB1 MKO mice were assigned to run at 60% (12.5 m/min) of their maximal running speed, whereas WT mice were randomized to run at either 30% (12.5 m/min) or 60% (25 m/min) of their maximal running speed. AMPKα2 KO and respective WT mice were exercised at 50% of their maximal running speed, corresponding to 13 and 18 m/min, respectively.
FA oxidation in isolated muscle.
FA metabolism experiments were conducted in incubation reservoirs (Radnoti, CA) using procedures previously described (8,25). Isolated EDL muscles were incubated at 30°C in Krebs-Henseleit Ringer buffer, pH 7.4, containing 2 mmol/L pyruvate, 2% FA free BSA, and 0.5 mmol/L palmitic acid. After initial incubation at resting tension (4–5 mN), incubation buffer was refreshed and supplemented with 0.5 μCi/mL [1-14C]palmitate (Amersham BioSciences, Buckinghamshire, U.K.). Oxidation of [1-14C]palmitate was measured in resting or contracting EDL muscle (50 Hz, 350 ms pulse duration, 6 tetani ⋅ min−1) over 25 min.
Glucose transport in isolated muscle.
Isolated EDL muscle from LKB1 MKO and WT mice was quickly excised and suspended by ligatures at resting tension (4–5 mN) in incubation chambers (Multi Myograph system; Danish Myo Technology, Aarhus, Denmark) as previously described (23). After preincubation, muscles were stimulated electrically to contract for 10 min (moderate protocol: 2 s/15 s, 0.2 ms pulses, 100 Hz, 40 V; intense protocol: 10 s/30 s, 0.1 ms pulses, 100 Hz, 100 V). 2-deoxyglucose uptake was measured for 10 min either during stimulation (moderate protocol) or immediately after stimulation (intense protocol).
Glucose clearance during in vivo exercise.
Fasted mice (2 h) were given an intraperitoneal injection with a bolus of saline (800 µL/100 g body wt) containing 0.1 mmol/L 2-deoxyglucose and 60 µCi/mL 2-[3H]deoxyglucose corresponding to ∼12 µCi/mouse as previously described (24). The muscle glucose clearance calculation was based on the muscle accumulation of 2-[3H]deoxyglucose-6-phosphate activity related to the area under the curve of the plasma 3H activity at time points −2, 10, and 20 min (Supplementary Table 1) using the trapezoid method as previously described (24,26,27).
Kinase activity assays.
Isoform-specific AMPK activity was measured on muscle lysate immunoprecipitates as previously described (23). LKB1 or SIK3 activities were measured on immunoprecipitates using LKBtide and Sakamototide as previously described (28,29).
Microscopy.
Confocal microscopy was performed on single muscle fibers as previously described (30). Mitochondrial networks were visualized by using antibody against COXIV (Abcam, Cambridge, U.K.). Confocal images were collected with a TCS SP2 microscope (Leica) and were analyzed using Metamorph software (Universal Imaging). Transmission electron microscopy (TEM) was performed on ultrathin muscle sections stained with uranyl acetate and lead citrate and subsequently examined with a Philips CM 100 TEM (Philips, Eindhoven, the Netherlands), operated at an accelerating voltage of 80 kV and equipped with an OSIS Veleta digital slow-scan 2k × 2k CCD camera. Digital images were recorded with the ITEM software package.
Microarray labeling and analysis.
RNA was extracted from WT and LKB1 MKO muscle using TRIzol. Labeling of 100 ng total RNA was performed using the Gene-Chip Whole Transcript Sense Target Labeling Assay (Affymetrix) followed by hybridization to the GeneChip Mouse Gene 1.0 ST Arrays. RMA16 quantile normalization was performed using GeneSpring 12 software (Agilent). Only probe sets with a false discovery rate–corrected P value <0.05 and an absolute fold change >1.4 were selected for further analysis. Hierarchical clustering was performed using Euclidean distance and centroid linkage. Metabolomics pathway analysis was performed using Ingenuity Pathway Analysis software (Ingenuity Systems).
Muscle metabolites, enzyme activities, and Western blot.
Muscle glycogen, adenosine nucleotides, and lactate were determined as previously described (31). NAD+ and NADH content was determined using a photometrically NAD/NADH quantification kit (Biovision, Mountain View, CA). Malonyl-CoA, acetyl-CoA, succinyl-CoA, and free CoA were assayed by high-pressure liquid chromatography with ultraviolet detection as previously described (32). Maximal activity of citrate synthase and β-hydroxyacyl-CoA dehydrogenase (HAD) were measured as previously described (33). Protein extraction and Western blot was performed as previously described (23). A detailed list of antibodies is provided in the Supplementary Data.
Statistics.
Data are expressed as means ± SEM. Statistical evaluations were performed by using Student t test, two-way ANOVA, or one-way (intensity effects within WT in vivo groups) and two-way ANOVA with repeated measurements. The Studentized Newman-Keuls post hoc test was used when appropriate. Differences between groups were considered statistically significant if P < 0.05. Statistical analyses of microarray data were performed with an unpaired t test followed by Benjamini-Hochberg multiple-testing correction.
RESULTS
LKB1 MKO mice have reduced exercise tolerance and AICAR sensitivity.
The maximum running speed achieved by the WT mice was ∼41 m/min, whereas it was reduced by ∼50% in the LKB1 MKO mice (Fig. 1A). In accordance with a previous report (34), the relative decrease in blood glucose levels was less pronounced in the LKB1 MKO compared with WT mice during an AICAR tolerance test (Supplementary Fig. 1). Furthermore, LKB1 MKO mice had reduced fasting blood glucose concentration (WT 6.1 ± 0.5 mmol/L vs. LKB1 MKO 4.2 ± 0.4 mmol/L; n = 10–11; P < 0.05). Collectively, these in vivo tests demonstrate that our mouse cohort displays the same in vivo phenotypic characteristics as previously shown in mouse cohorts generated from the same founders (19,34).
FIG. 1.
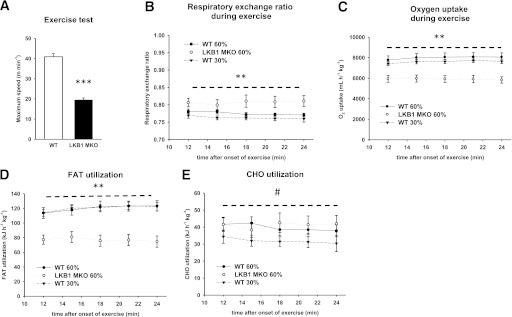
LKB1 MKO mice are exercise intolerant and display reduced oxygen uptake and limited FA oxidation during exercise in vivo. A: Maximal-speed running test of LKB1 MKO and WT mice was performed. B and C: RER (B) and oxygen uptake (C) (mL ⋅ h−1 ⋅ kg−1) during treadmill exercise at 60% of maximal running speed and at the same absolute intensity. Whole-body FA oxidation (kJ ⋅ h−1 ⋅ kg−1) (D) and carbohydrate (CHO) oxidation (kJ ⋅ h−1 ⋅ kg−1) (E) were calculated from RER values as described in research design and methods. Data are presented as means ± SEM (n = 11–12). **P < 0.01, ***P < 0.001, significantly different from WT; #P < 0.05, significant difference between WT groups.
Indirect calorimetry reveals a defect in regulation of FA metabolism in LKB1 MKO mice during in vivo exercise.
During submaximal treadmill exercise, RER was higher in the LKB1 MKO compared with WT mice at the same relative and absolute intensity (Fig. 1B). O2 uptake was generally lower in LKB1 MKO (Fig. 1C). Carbohydrate oxidation increased with increasing intensity in WT mice (Fig. 1E), whereas FA oxidation was similar between intensities in WT mice (Fig. 1D). FA oxidation during exercise was lower in LKB1 MKO compared with WT mice at the same relative and absolute exercise intensity (Fig. 1D). Differences in substrate use were not apparent at rest under fed conditions (Supplementary Fig. 2). In contrast to the LKB1 MKO mice, we did not observe differences in FA oxidation when AMPKα2 KO and WT littermates were compared at the same relative exercise intensity (Supplementary Fig. 3).
LKB1 deficiency causes a robust decrease in muscle activities of AMPKα2 and SIK3 but maintains normal malonyl-CoA concentration.
In LKB1 MKO mice, LKB1 protein was almost ablated (Fig. 2A) resulting in a 95% decrease in LKB1 activity in resting muscle (Fig. 2B). In contrast, the LKB1 substrates SIK3 and AMPK were similar to levels observed in WT mice (Fig. 2A). We observed a robust reduction in SIK3 (−46%) and AMPKα2 (−80%) activity, but not AMPKα1 activity, in resting LKB1 MKO muscle compared with WT (Fig. 2C–E). LKB1 and SIK3 activity was not significantly altered with treadmill exercise in vivo (Fig. 2B and C). In contrast, AMPKα1 activity increased modestly (50–60%) in both LKB1 MKO and WT mice (Fig. 2E) and AMPKα2 activity increased markedly (260%) in WT mice but was almost undetectable in LKB1 MKO mice (Fig. 2D). AMPK Thr172 phosphorylation increased by 120% in WT muscle, whereas it was reduced by 50% in the resting LKB1 MKO muscle and failed to increase in response to exercise (Fig. 2F). Even though mean ACC2 Ser212 phosphorylation was increased by ∼35% in WT muscle by exercise, this increase did not reach statistical significance (Fig. 2G). This was similar for the apparent decrease of ∼19 and 27% in ACC2 Ser212 phosphorylation in resting and exercising muscle, respectively, from LKB1 MKO compared with WT mice (Fig. 2G). Malonyl-CoA content was similar in WT and LKB1 MKO mice and did not change with exercise in either genotype (Fig. 2H). In contrast, the cellular free CoA content was significantly lower in LKB1 MKO compared with WT mice both at rest and during exercise and did not change with exercise in either genotype (Fig. 2I).
FIG. 2.
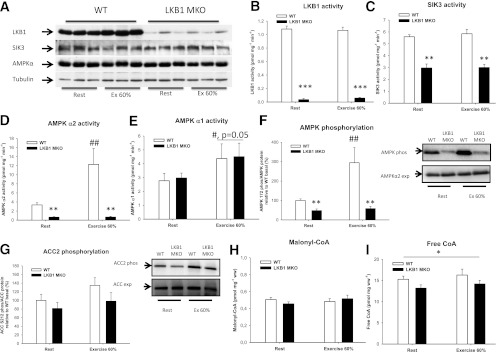
LKB1 signaling in LKB1 MKO and WT mice during in vivo exercise. A: LKB1 protein was almost completely ablated in LKB1 MKO mice. B–E: LKB1 activity and activity of downstream targets were determined in gastrocnemius muscle from LKB1 MKO and WT mice. F and G: AMPK Thr172 and ACC2 Ser212 phosphorylation (phos) in gastrocnemius muscle from LKB1 MKO and WT mice. Malonyl-CoA (H) and free CoA (I) content in gastrocnemius muscle in WT and LKB1 MKO mice. Data are presented as means ± SEM (n = 6–13). *P < 0.05, **P < 0.01, ***P < 0.001, significantly different from WT; #P = 0.05, significant effect of exercise, ##P < 0.01, significant difference between resting and exercising WT mice. Ex; exercise, exp; protein expression, ww; wet weight.
LKB1-deficient muscle has decreased FA oxidation capacity at rest and during contraction in isolated skeletal muscle ex vivo.
FA oxidation was suppressed in EDL muscle from LKB1 MKO compared with WT mice at rest and during contractions (Fig. 3A). During muscle contractions, FA oxidation increased in EDL muscle from both LKB1 MKO and WT mice, but total FA oxidation was still markedly reduced in LKB1 MKO muscle compared with WT (Fig. 3A). AMPK Thr172 phosphorylation was abolished in LKB1 MKO EDL muscle, and increased as expected, only in WT EDL muscle with contraction (Fig. 3B). ACC2 Ser212 phosphorylation was reduced in EDL muscle from LBK1 MKO mice (Fig. 3C) but increased in muscle from both LKB1 MKO and WT mice in response to muscle contraction (Fig. 3C).
FIG. 3.
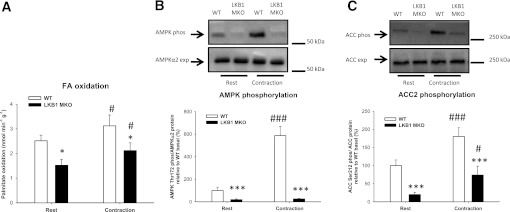
Lack of LKB1 reduces the capacity for FA oxidation in isolated EDL muscle. A: FA oxidation was determined at rest and after contractions in EDL muscle from LKB1 MKO and WT mice. B: AMPK Thr172 and ACC2 phosphorylation (phos) in resting and contracted EDL muscle from LKB1 MKO and WT mice. Data are presented as means ± SEM (n = 11–12). *P < 0.05, ***P < 0.001, significantly different from WT. #P < 0.05, ###P < 0.001, significant difference between WT groups.
LKB1 MKO mice have reduced NAD+ concentration and SIRT1 activity but display increased HDAC4 expression and reduced FA oxidative gene expression.
There were no differences in SIRT1 protein expression (Fig. 4A), but cellular NAD+ content was reduced by 20% in LKB1 MKO muscle compared with WT (Fig. 4B). As acetylation of p53 protein was increased, this indicates a lower SIRT1 activity in LKB1-deficient muscle (Fig. 4C). Additionally, HDAC4 protein content was 70% higher in LKB1 MKO compared with WT muscle (Fig. 4D), accompanied by a reduction (P = 0.08) in HDAC4 phosphorylation on Ser629 in LKB1 MKO (Fig. 4E). To evaluate downstream effects of these findings, we extracted RNA from LKB1 MKO and WT muscle and subjected this to mouse expression array analysis. Based on their fold change (>1.4 fold) and false discovery rate P value (P < 0.05), we identified 212 differentially expressed probe sets (Supplementary Table 2). Using these probe sets in Ingenuity software for pathway analysis, we observed that the most pronounced differences between genotypes were molecular and cellular functions related to energy production and lipid metabolism with the most functional annotation for both being on FA oxidation (P = 8.01 × 10−6). Heat map and supervised hierarchical cluster analysis using the 27 genes involved in lipid metabolism were able to perfectly separate LKB1 MKO and WT muscles (Fig. 4F). A detailed list of the 27 genes is shown in Supplementary Table 3.
FIG. 4.
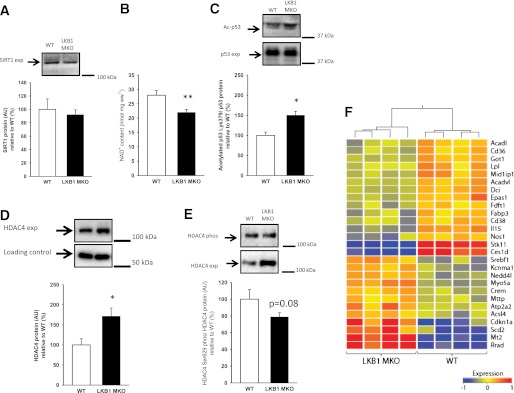
LKB1-deficient muscle has lower SIRT1 activity, increased HDAC4 expression (exp), and reduced FA oxidative gene expression. SIRT1 protein (A), NAD+ concentration (B), and the SIRT1 substrate acetylated p53 Lys379 (C) were determined in gastrocnemius muscle. HDAC4 protein expression (D) and phosphorylated (phos) HDAC4 Ser632 (E) were determined in gastrocnemius muscle from LKB1 MKO and WT mice. β-Tubulin, which was similar between genotypes, was used as loading control for HDAC4 expression. F: Heat map of 27 genes found to be differentially expressed between WT and LKB1 MKO (n = 4) muscle biopsies and involved in lipid metabolism. The fold change in gene expression is color coded: red, upregulation; blue, downregulation. Based on their expression, biopsies are separated in two distinct clusters: one containing WT muscles and the other LKB1 MKO muscles. The dendrogram above the heat map displays hierarchical clustering of the columns based on a distances matrix where the height of lines indicates the degree of separation between clusters. Data are presented as means ± SEM (n = 7–12). *P < 0.05, **P < 0.01, significantly different from WT. AU, arbitrary units.
LKB1 MKO mice display reduced mitochondrial enzyme activity but unchanged mitochondrial morphology.
Muscle from LKB1 MKO mice displayed a reduced citrate synthase activity (Fig. 5A), and there was a tendency (P = 0.09) toward a reduction in HAD activity (Fig. 5B). To evaluate whether LKB1 deletion would influence mitochondrial morphology, we investigated the mitochondrial networks by fluorescent staining of mitochondrial complex IV (COXIV) in single muscle fibers. The lack of LKB1 in muscle did not influence the mitochondrial network either at the subsarcolemmal region or in the intermyofibrillar regions (Fig. 5C). Cross-sections of muscle were investigated by TEM to evaluate mitochondrial ultrastructure. As with our observations in single muscle fibers, we did not detect a phenotype on mitochondrial morphology in muscle deficient of LKB1 (Fig. 5D).
FIG. 5.
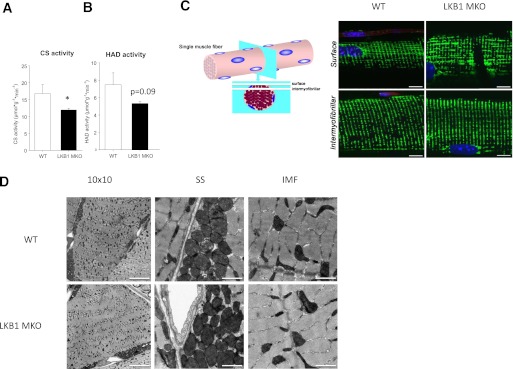
Skeletal muscle deficient in LKB1 has reduced mitochondrial enzyme activity but displays normal mitochondrial network and ultrastructural morphology. Citrate synthase (CS) activity (A) and HAD activity (B) (n = 9–10) in gastrocnemius muscle from LKB1 MKO and WT mice. Data are presented as means ± SEM. *P < 0.05, significantly different from WT. C: Mitochondrial network in single muscle fibers from WT and LKB1 MKO mice visualized using immunocytochemical staining of COXIV and imaged by confocal immunofluorescence microscopy (n = 3); bar = 10 μm. D: TEM on cross-sections from quadriceps muscle from WT and LKB1 MKO mice was performed. Left panel: A stitched image combined of 100 individual images (10 × 10); bar = 10 μm. Middle panel: Representative images of subsarcolemmal (SS) mitochondria; bar = 1 μm. Right panel: Representative images of intermyofibrillar (IMF) mitochondria; bar = 1 μm.
Muscle glucose clearance is not reduced in LKB1 MKO mice during exercise.
We measured muscle glucose uptake during running exercise at the same absolute and relative exercise intensity. In WT muscle, glucose clearance was intensity dependent during treadmill exercise (Fig. 6A and B). During 20 min of treadmill exercise at 60% of maximum speed, muscle glucose clearance increased similarly in WT and LKB1 MKO mice (Fig. 6A and B). When we compared muscle glucose clearance at the same absolute speed, glucose clearance was higher in LKB1 MKO quadriceps (77%) and EDL (44%) muscles compared with WT muscle (Fig. 6A and B). There was no difference in resting muscle glycogen and muscle lactate between genotypes (Fig. 6C and D). In response to exercise, muscle glycogen decreased and lactate increased to a similar extent in the two genotypes. The muscle glycogen data combined with a similar increase in muscle lactate in response to exercise and equal AMP-to-ATP and ADP-to-ATP ratios (Supplementary Fig. 4A and B) support that the mice were subjected to similar metabolic stress/work loads in relative terms.
FIG. 6.

LKB1 MKO mice display normal increases in muscle glucose clearance in response to treadmill exercise. 2-deoxyglucose (2DG) clearance (mL ⋅ g−1 ⋅ min−1) was determined in quadriceps (A) and EDL muscle (B) from LKB1 MKO and WT mice at rest and in response to treadmill exercise at the same relative intensity. Muscle glycogen (C) (µmol ⋅ g wet wt−1) and muscle lactate content (D) in quadriceps muscle from WT and LKB1 MKO mice at rest and in response to 20 min treadmill running exercise at the same relative intensity. Data are presented as means ± SEM (n = 6–13). #P < 0.05, ##P < 0.01, significantly different from rest within genotype. ND, not determined.
Glucose uptake in response to electrical stimulation of isolated EDL muscle.
We investigated 2-deoxyglucose uptake during both moderate and intense stimulation protocol ex vivo. 2-deoxyglucose uptake was not reduced in EDL muscle from LKB1 MKO compared with WT in response to this moderate stimulation protocol (Fig. 7A). However, when applying the intense protocol, used previously by others (12,20), we also observed that 2-deoxyglucose uptake was significantly reduced in LKB1 MKO EDL in response to contraction (Fig. 7B).
FIG. 7.

LKB1 MKO mice display reduced glucose uptake during intense, but not moderate, muscle contractions ex vivo. A: 2-deoxyglucose (2DG) uptake (µmol ⋅ g−1 ⋅ h−1) in EDL muscle from WT and LKB1 MKO mice at basal and in response to 10 min moderate electrical stimulation (n = 9–11). B: 2-deoxyglucose uptake (µmol ⋅ g−1 ⋅ h−1) in EDL muscle from LKB1 MKO and WT mice subjected to intense electrically stimulated muscle contraction for 10 min (n = 6–9). C: TBC1D1 Ser237 phosphorylation (phos) in EDL muscles in response to moderate and intense electrically induced muscle contraction (n = 7–11). Data are presented as means ± SEM. *P < 0.05, **P < 0.01, significantly different from WT. #P < 0.05, ##P < 0.01, significantly different from rest.
TBC1D1 Ser237 phosphorylation in response to ex vivo contraction.
In response to the moderate and the intense stimulation protocol, TBC1D1 Ser237 phosphorylation increased in muscle from WT mice by 136 and 120%, respectively (Fig. 7C). In the LKB1 MKO muscle, TBC1D1 Ser237 phosphorylation was reduced in the basal muscles. Even though TBC1D1 Ser237 phosphorylation was increased by contractions in the LKB1 MKO muscle, the response was significantly lower compared with WT muscle. Of note, TBC1D1 protein expression was similar in WT and LKB1 MKO muscle (Fig. 7C), indicating that compared with WT muscle the reduced phosphorylation of TBC1D1 in the LKB1 MKO means a markedly lower amount of TBC1D1 being phosphorylated and therefore inactivated during contractions.
DISCUSSION
During exercise, lipid and carbohydrate oxidation increases several fold to accommodate the increased energy demand by the working muscle. The mechanisms behind this regulation have for the last several decades received a lot of attention, since they could be critical in understanding the beneficial effects of exercise on insulin resistance conditions and thus be key to understanding and overcoming the metabolic derangement in the pathogenesis of type 2 diabetes (35,36). Here, we demonstrate that the lack of LKB1 protein in skeletal muscle severely reduces FA oxidation both during in vivo exercise and during contraction in isolated muscle ex vivo. The latter finding clearly indicates that the reduced FA oxidation observed during in vivo exercise was caused not by systemic factors but, rather, by muscle limitations in pathways involved in FA oxidation. Thus, we for the first time identify LKB1 as a molecular regulator of FA oxidation in muscle during exercise. Surprisingly, in contrast to what has been suggested previously based on intense electrical stimulation of muscle (12,20), we also show that LKB1 does not seem to regulate muscle glucose uptake during exercise in vivo or during moderate electrical stimulation.
It has been demonstrated that SIRT1 is a major regulator of FA oxidative gene expression in skeletal muscle (37,38). Also, in MEF- and C2C12 cell lines lacking SIRT1, low-glucose conditions failed to increase FA oxidation and catabolic gene expression (39). In the current study, NAD+ concentration was reduced in LKB1 MKO muscle, which was associated with a hyperacetylation of p53 protein, a well-known SIRT1 target, indicating decreased SIRT1 activity in LKB1 MKO muscle. Using mouse expression array analysis, we showed that this reduced SIRT1 activation was associated with a markedly reduced FA oxidative gene expression in LKB1-deficient muscle. Furthermore, LKB1 MKO mice displayed a markedly decreased SIK3 activity, which was accompanied by a decreased HDAC4 phosphorylation (P = 0.08) and higher HDAC4 protein expression. Therefore, LKB1-SIK3 signaling could play a crucial role for maintaining FA metabolic capacity in skeletal muscle, as demonstrated in Drosophila (14). In the current study, the Ingenuity pathway analysis indicated that the FA oxidation pathway was the most downregulated pathway in the LKB1 MKO mice, and this could be a potential explanation for the reduced FA oxidation during exercise. When we evaluated the mitochondrial networks in single muscle fibers, structural abnormalities of the mitochondria were not apparent. Furthermore, using TEM we showed no major differences in mitochondrial ultrastructure between LKB1 MKO and WT mice. The question is to what extent the decreased FA oxidative gene expression in LKB1 MKO mice actually is responsible for the decreased FA oxidation during exercise and muscle contractions. In AMPKα2 KO mice (Supplementary Fig. 3) and in AMPK β1β2 MKO mice (7), FA oxidation during exercise was not decreased despite decreased mitochondrial enzymes and altered mitochondrial morphology (7). Furthermore, in mice overexpressing a dominant negative AMPK construct, FA oxidation was also normal during exercise and during contractions in vitro (8,40). This suggests that the decreased FA oxidation during exercise in LKB1 MKO mice is independent of AMPK. Malonyl-CoA content was previously shown to decrease more in WT muscle after intense in situ muscle contractions than in LKB1 MKO muscle (41), suggesting that LKB1-mediated control of malonyl-CoA levels may play a role in regulation of FA oxidation under some experimental conditions. However, we show here that malonyl-CoA content was similar in LKB1 MKO and WT muscle both at rest and after in vivo exercise, indicating that the reduced FA oxidation in LKB1 MKO mice was not due to an impaired ability to relieve the malonyl-CoA inhibition of CPT1. Interestingly, the amount of free cellular CoA was reduced in LKB1 MKO muscle, which might result in reduced availability of FA-CoA substrate to carnitine and CPT1, in turn lowering FA availability for mitochondrial oxidation and could therefore be of importance for the decrease in FA oxidation.
Interestingly, mice carrying a mutation in the TBC1D1 gene inhibiting TBC1D1 function in muscle exhibited increased FA uptake and oxidation compared with mice expressing normal levels of TBC1D1 protein when fed a high-fat diet (42). In addition, both palmitate uptake and oxidation in C2C12 cells were increased when TBC1D1 protein was deleted by siRNA (42). This suggests that TBC1D1 functions as a brake on FA metabolism. Furthermore, when phosphorylation and thereby inactivation of AS160 (TBC1D4), another member of the TBC1 family, was impaired owing to the 4P mutation in TBC1D4, fatty acid translocase (FAT)/CD36 translocation to the plasma membrane was impaired in HL-1 cardiac myocytes (43). Since FAT/CD36 translocation has been shown to be critical for sarcolemmal FA transport and FA oxidation in skeletal muscle (44) and occurs independently of AMPK during in vivo exercise (45), it could be speculated that TBC1D1 and TBC1D4 function is important in regulation of FA transport and, in turn, FA oxidation. Because TBC1D1 is the TBC1 domain family member that is predominantly expressed in fast-twitch muscle like EDL (46), the decreased FA oxidation during exercise in LKB1 MKO mice (dominated by fast-twitch white muscle) (47) and during in vitro incubation of isolated EDL muscle could be related to the markedly decreased TBC1D1 phosphorylation, which in essence results in inability to reduce the brake on FA transport during muscle contractions. Importantly, the reduction in TBC1D1 phosphorylation was not driven by a reduction in TBC1D1 protein expression, which has previously been demonstrated in various mouse models with reduced AMPKα2 activity (7,46,48). Thus, a reduced FA transport together with a low free CoA content and thus FA availability to the mitochondria in the LKB1 MKO could add to the low FA oxidation in these mice.
We here demonstrate that LKB1 is not crucial for glucose uptake during physiological exercise and during moderate electrical stimulation. These findings contrast previous reports demonstrating reduced glucose uptake measured during intense contraction in isolated muscles (12,20). In agreement with those findings, we did observe modestly reduced glucose uptake in response to contraction in EDL muscle when using the same intense stimulation. Magnitude of reduction in muscle glucose uptake during intense contractions in our LKB1 MKO mice was moderate compared with the previous work (12), which could be due to more substantial deficiency of LKB1 using hypomorphic LKB1 loxp mice as background when they generated their LKB1 MKO mice. Still, it is likely that the residual LKB1 activity measured in the present LKB1 MKO mice is due to nonmuscle tissue in the muscle homogenate, which contributes much less in the hypomorphic background.
The current study indicates that AMPKα2 is dispensable for mouse muscle glucose uptake during exercise, as we observed that basal activity was negligible and activation in response to exercise was abolished in LKB1 MKO mice. This confirms our previous findings in mice overexpressing a dominant negative α2 AMPK (24). However, when both α1 and α2 AMPK activations are ablated by genetic deletion of both β1 and β2 AMPK subunits, exercise- and contraction-induced glucose uptake are both substantially impaired (7). Since the protein expression of AMPKα1 is elevated in LKB1-deficient mouse muscle (12,34), similar to what was observed in AMPKα2 KO mice (23), and since AMPKα1 activity was not decreased in the LKB1 MKO mice in the current study, AMPKα1 could possibly compensate for the lack of AMPKα2. Although Koh et al. (34) reported reduced activation of AMPKα1 in response to contraction in LKB1 MKO muscle compared with WT, α1 activity was still significantly elevated. The idea that low levels of AMPK activity might be sufficient to promote glucose uptake in response to different stimuli is supported by findings in the hypomorphic LKB1 mice (12) in which glucose uptake increased normally in response to both AICAR and electrical stimulation despite 50–60% reduction in AMPKα2 and >50% reduction in AMPKα1 activity. Furthermore, in AMPKα1 KO soleus muscle, glucose transport was reduced by 20% in response to tetanic contraction (23), as well as in response to twitch contractions (49). AMPKα1 could therefore play an important role in response to more physiological exercise regimes, whereas AMPKα2 may become important in situations of severe metabolic stress in mouse muscle.
In summary, we have shown that LKB1 is critical for regulation of FA oxidation in muscle during exercise and that this occurs independently of AMPK. Furthermore, we have demonstrated that LKB1 deficiency does not affect the normal increase in glucose transport or oxidation during in vivo exercise. The lack of LKB1 in muscle did not cause detectable alterations in mitochondrial morphology, but expression of genes involved in FA metabolism was decreased expectedly as a result of increased HDAC4 expression and decreased SIRT1 activity. In addition to the transcriptional response controlled by LKB1, the markedly impaired TBC1D1 phosphorylation in LKB1-deficient muscle during contractions likely contributes to the impaired FA oxidation during exercise and contractions.
Supplementary Material
ACKNOWLEDGMENTS
B.K., J.F.P.W., and E.A.R. are supported by grants from the Danish Medical Research Council, the Novo Nordisk Foundation, and the Lundbeck Foundation and are members of the UNIK - Food, Fitness, & Pharma, supported by the Danish Ministry of Science, Technology, and Innovation. J.J. is supported by a Research Fellowship from the Alfred Benzon Foundation. K.S. is funded by the British Medical Research Council. R.W.H. is supported by the British Heart Foundation. S.J.M. is an employee of Novo Nordisk A/S. No other potential conflicts of interest relevant to this article were reported.
J.J., S.J.M., E.A.R., and B.K. designed the study. J.J., S.J.M., A.B.J., and A.M.F. conducted the experiments. J.J., S.J.M., A.B.J., A.M.F., C.Pe., L.S., A.K.S., N.J., K.T., C.Pr., K.Q., J.R.B.D., R.W.H., K.S., D.M.T., P.S., and J.F.P.W. performed the laboratory analyses and commented on the manuscript. J.J., S.J.M., K.S., E.A.R., and B.K. wrote the manuscript. E.A.R. and B.K. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
The authors are grateful for the skilled technical assistance of I.B. Nielsen and B. Bolmgren, Molecular Physiology Group, Department of Exercise and Sport Sciences, University of Copenhagen. The authors also thank Prof. B. Voillet, Department of Endocrinology, Metabolism and Cancer, Institut Cochin, Université Paris Decartes, Paris, France, for providing the AMPKα2 KO founder mice.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1160/-/DC1.
REFERENCES
- 1.Richter EA, Garetto LP, Goodman MN, Ruderman NB. Muscle glucose metabolism following exercise in the rat: increased sensitivity to insulin. J Clin Invest 1982;69:785–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richter EA, Mikines KJ, Galbo H, Kiens B. Effect of exercise on insulin action in human skeletal muscle. J Appl Physiol 1989;66:876–885 [DOI] [PubMed] [Google Scholar]
- 3.Kiens B, Essen-Gustavsson B, Christensen NJ, Saltin B. Skeletal muscle substrate utilization during submaximal exercise in man: effect of endurance training. J Physiol 1993;469:459–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol 1997;273:E1107–E1112 [DOI] [PubMed] [Google Scholar]
- 5.Wojtaszewski JF, Nielsen P, Hansen BF, Richter EA, Kiens B. Isoform-specific and exercise intensity-dependent activation of 5′-AMP-activated protein kinase in human skeletal muscle. J Physiol 2000;528:221–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol 1996;270:E299–E304 [DOI] [PubMed] [Google Scholar]
- 7.O’Neill HM, Maarbjerg SJ, Crane JD, et al. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci USA 2011;108:16092–16097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dzamko N, Schertzer JD, Ryall JG, et al. AMPK-independent pathways regulate skeletal muscle fatty acid oxidation. J Physiol 2008;586:5819–5831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roepstorff C, Thiele M, Hillig T, et al. Higher skeletal muscle alpha2AMPK activation and lower energy charge and fat oxidation in men than in women during submaximal exercise. J Physiol 2006;574:125–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lizcano JM, Göransson O, Toth R, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J 2004;23:833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaleel M, McBride A, Lizcano JM, et al. Identification of the sucrose non-fermenting related kinase SNRK, as a novel LKB1 substrate. FEBS Lett 2005;579:1417–1423 [DOI] [PubMed] [Google Scholar]
- 12.Sakamoto K, McCarthy A, Smith D, et al. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J 2005;24:1810–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berdeaux R, Goebel N, Banaszynski L, et al. SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat Med 2007;13:597–603 [DOI] [PubMed] [Google Scholar]
- 14.Wang B, Moya N, Niessen S, et al. A hormone-dependent module regulating energy balance. Cell 2011;145:596–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furuyama T, Kitayama K, Yamashita H, Mori N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem J 2003;375:365–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furuyama T, Yamashita H, Kitayama K, Higami Y, Shimokawa I, Mori N. Effects of aging and caloric restriction on the gene expression of Foxo1, 3, and 4 (FKHR, FKHRL1, and AFX) in the rat skeletal muscles. Microsc Res Tech 2002;59:331–334 [DOI] [PubMed] [Google Scholar]
- 17.Holness MJ, Sugden MC. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans 2003;31:1143–1151 [DOI] [PubMed] [Google Scholar]
- 18.Bastie CC, Nahlé Z, McLoughlin T, et al. FoxO1 stimulates fatty acid uptake and oxidation in muscle cells through CD36-dependent and -independent mechanisms. J Biol Chem 2005;280:14222–14229 [DOI] [PubMed] [Google Scholar]
- 19.Thomson DM, Porter BB, Tall JH, Kim HJ, Barrow JR, Winder WW. Skeletal muscle and heart LKB1 deficiency causes decreased voluntary running and reduced muscle mitochondrial marker enzyme expression in mice. Am J Physiol Endocrinol Metab 2007;292:E196–E202 [DOI] [PubMed] [Google Scholar]
- 20.Koh HJ, Toyoda T, Fujii N, et al. Sucrose nonfermenting AMPK-related kinase (SNARK) mediates contraction-stimulated glucose transport in mouse skeletal muscle. Proc Natl Acad Sci USA 2010;107:15541–15546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jorgensen SB, Rose AJ. How is AMPK activity regulated in skeletal muscles during exercise? Front Biosci 2008;13:5589–5604 [DOI] [PubMed] [Google Scholar]
- 22.Viollet B, Andreelli F, Jørgensen SB, et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest 2003;111:91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jørgensen SB, Viollet B, Andreelli F, et al. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem 2004;279:1070–1079 [DOI] [PubMed] [Google Scholar]
- 24.Maarbjerg SJ, Jørgensen SB, Rose AJ, et al. Genetic impairment of AMPKalpha2 signaling does not reduce muscle glucose uptake during treadmill exercise in mice. Am J Physiol Endocrinol Metab 2009;297:E924–E934 [DOI] [PubMed] [Google Scholar]
- 25.Steinberg GR, Bonen A, Dyck DJ. Fatty acid oxidation and triacylglycerol hydrolysis are enhanced after chronic leptin treatment in rats. Am J Physiol Endocrinol Metab 2002;282:E593–E600 [DOI] [PubMed] [Google Scholar]
- 26.Fueger PT, Bracy DP, Malabanan CM, Pencek RR, Wasserman DH. Distributed control of glucose uptake by working muscles of conscious mice: roles of transport and phosphorylation. Am J Physiol Endocrinol Metab 2004;286:E77–E84 [DOI] [PubMed] [Google Scholar]
- 27.Kraegen EW, James DE, Jenkins AB, Chisholm DJ. Dose-response curves for in vivo insulin sensitivity in individual tissues in rats. Am J Physiol 1985;248:E353–E362 [DOI] [PubMed] [Google Scholar]
- 28.Henriksson E, Jones HA, Patel K, et al. The AMPK-related kinase SIK2 is regulated by cAMP via phosphorylation at Ser358 in adipocytes. Biochem J 2012;444:503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakamoto K, Göransson O, Hardie DG, Alessi DR. Activity of LKB1 and AMPK-related kinases in skeletal muscle: effects of contraction, phenformin, and AICAR. Am J Physiol Endocrinol Metab 2004;287:E310–E317 [DOI] [PubMed] [Google Scholar]
- 30.Ploug T, van Deurs B, Ai H, Cushman SW, Ralston E. Analysis of GLUT4 distribution in whole skeletal muscle fibers: identification of distinct storage compartments that are recruited by insulin and muscle contractions. J Cell Biol 1998;142:1429–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jørgensen SB, Wojtaszewski JF, Viollet B, et al. Effects of alpha-AMPK knockout on exercise-induced gene activation in mouse skeletal muscle. FASEB J 2005;19:1146–1148 [DOI] [PubMed] [Google Scholar]
- 32.Stanley WC, Hernandez LA, Spires D, Bringas J, Wallace S, McCormack JG. Pyruvate dehydrogenase activity and malonyl CoA levels in normal and ischemic swine myocardium: effects of dichloroacetate. J Mol Cell Cardiol 1996;28:905–914 [DOI] [PubMed] [Google Scholar]
- 33.Jørgensen SB, Treebak JT, Viollet B, et al. Role of AMPKalpha2 in basal, training-, and AICAR-induced GLUT4, hexokinase II, and mitochondrial protein expression in mouse muscle. Am J Physiol Endocrinol Metab 2007;292:E331–E339 [DOI] [PubMed] [Google Scholar]
- 34.Koh HJ, Arnolds DE, Fujii N, et al. Skeletal muscle-selective knockout of LKB1 increases insulin sensitivity, improves glucose homeostasis, and decreases TRB3. Mol Cell Biol 2006;26:8217–8227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richter EA, Ruderman NB. AMPK and the biochemistry of exercise: implications for human health and disease. Biochem J 2009;418:261–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koh HJ, Brandauer J, Goodyear LJ. LKB1 and AMPK and the regulation of skeletal muscle metabolism. Curr Opin Clin Nutr Metab Care 2008;11:227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cantó C, Jiang LQ, Deshmukh AS, et al. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab 2010;11:213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cantó C, Gerhart-Hines Z, Feige JN, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009;458:1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gerhart-Hines Z, Rodgers JT, Bare O, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J 2007;26:1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee-Young RS, Griffee SR, Lynes SE, et al. Skeletal muscle AMP-activated protein kinase is essential for the metabolic response to exercise in vivo. J Biol Chem 2009;284:23925–23934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomson DM, Brown JD, Fillmore N, et al. LKB1 and the regulation of malonyl-CoA and fatty acid oxidation in muscle. Am J Physiol Endocrinol Metab 2007;293:E1572–E1579 [DOI] [PubMed] [Google Scholar]
- 42.Chadt A, Leicht K, Deshmukh A, et al. Tbc1d1 mutation in lean mouse strain confers leanness and protects from diet-induced obesity. Nat Genet 2008;40:1354–1359 [DOI] [PubMed] [Google Scholar]
- 43.Samovski D, Su X, Xu Y, Abumrad NA, Stahl PD. Insulin and AMPK regulate FA translocase/CD36 plasma membrane recruitment in cardiomyocytes via Rab GAP AS160 and Rab8a Rab GTPase. J Lipid Res 2012;53:709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McFarlan JT, Yoshida Y, Jain SS, et al. In vivo, fatty acid translocase (CD36) critically regulates skeletal muscle fuel selection, exercise performance, and training-induced adaptation of fatty acid oxidation. J Biol Chem 2012;287:23502–23516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeppesen J, Albers PH, Rose AJ, et al. Contraction-induced skeletal muscle FAT/CD36 trafficking and FA uptake is AMPK independent. J Lipid Res 2011;52:699–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pehmøller C, Treebak JT, Birk JB, et al. Genetic disruption of AMPK signaling abolishes both contraction- and insulin-stimulated TBC1D1 phosphorylation and 14-3-3 binding in mouse skeletal muscle. Am J Physiol Endocrinol Metab 2009;297:E665–E675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Allen DL, Harrison BC, Sartorius C, Byrnes WC, Leinwand LA. Mutation of the IIB myosin heavy chain gene results in muscle fiber loss and compensatory hypertrophy. Am J Physiol Cell Physiol 2001;280:C637–C645 [DOI] [PubMed] [Google Scholar]
- 48.Frøsig C, Pehmøller C, Birk JB, Richter EA, Wojtaszewski JF. Exercise-induced TBC1D1 Ser237 phosphorylation and 14-3-3 protein binding capacity in human skeletal muscle. J Physiol 2010;588:4539–4548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jensen TE, Schjerling P, Viollet B, Wojtaszewski JF, Richter EA. AMPK alpha1 activation is required for stimulation of glucose uptake by twitch contraction, but not by H2O2, in mouse skeletal muscle. PLoS ONE 2008;3:e2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.