

Abstract
Impaired cardiac microvascular function contributes to cardiovascular complications in diabetes. Glucagon-like peptide-1 (GLP-1) exhibits potential cardioprotective properties in addition to its glucose-lowering effect. This study was designed to evaluate the impact of GLP-1 on cardiac microvascular injury in diabetes and the underlying mechanism involved. Experimental diabetes was induced using streptozotocin in rats. Cohorts of diabetic rats received a 12-week treatment of vildagliptin (dipeptidyl peptidase-4 inhibitor) or exenatide (GLP-1 analog). Experimental diabetes attenuated cardiac function, glucose uptake, and microvascular barrier function, which were significantly improved by vildagliptin or exenatide treatment. Cardiac microvascular endothelial cells (CMECs) were isolated and cultured in normal or high glucose medium with or without GLP-1. GLP-1 decreased high-glucose–induced reactive oxygen species production and apoptotic index, as well as the levels of NADPH oxidase such as p47phox and gp91phox. Furthermore, cAMP/PKA (cAMP-dependent protein kinase activity) was increased and Rho-expression was decreased in high-glucose–induced CMECs after GLP-1 treatment. In conclusion, GLP-1 could protect the cardiac microvessels against oxidative stress, apoptosis, and the resultant microvascular barrier dysfunction in diabetes, which may contribute to the improvement of cardiac function and cardiac glucose metabolism in diabetes. The protective effects of GLP-1 are dependent on downstream inhibition of Rho through a cAMP/PKA-mediated pathway.
Diabetes is recognized as a major risk factor for cardiovascular disease, the leading cause for morbidity and mortality in the diabetic population (1). Diabetic cardiovascular disease results from many causes such as microangiopathy, myocardial metabolic abnormalities, and fibrosis (2,3). Under microangiopathy, the vessel wall of microvessels becomes thicker and vulnerable to bleeding, protein leakage, and slow blood flow. Accumulating evidence has demonstrated that microvascular injury plays a very important role in the diabetic cardiovascular dysfunction (4,5). However, there are still few effective strategies to prevent the progress of microvascular dysfunction in diabetes.
Glucagon-like peptide-1 (GLP-1), synthesized and secreted by intestinal l-cells, is a peptide with many protective biological functions. GLP-1 receptor (GLP-1R) is widely expressed in islet cells, kidney, lung, brain, and, interestingly, heart (6,7). It has been shown that administration of GLP-1 at the time of reperfusion was effective in decreasing myocardial infarct size in patients with acute myocardial infarction (8). Furthermore, a study of diabetic patients found that infusion of GLP-1 was associated with improved endothelial function (9). More importantly, recent data have suggested that GLP-1 was capable of exerting a direct cytoprotective effect against oxidative stress in diabetic mice aorta (10). Therefore, GLP-1 is of potential interest as a possible therapeutic regimen for treatment of cardiac microvascular injury in diabetes. However, native GLP-1 has a short half-life of minutes before getting degraded rapidly by dipeptidyl peptidase-4 (DPP-4) (11). To assess the potential role of intact GLP-1, DPP-4 inhibitors such as vildagliptin and GLP-1 analog exenatide are used as a means of preventing its degradation.
It is well-known that diabetes and hyperglycemia could increase intracellular reactive oxygen species (ROS), subsequently inducing apoptotic cell death, inflammation, and injury in endothelial cells (12,13). NADPH oxidase is the most important enzyme responsible for superoxide production in vasculature (14). Recent evidence demonstrates that Rho plays a regulatory role in oxidative stress in many diseases (15,16). Inhibition of Rho may result in the cardioprotective effect on cardiovascular remodeling associated with oxidative stress (17). Therefore, we hypothesize that GLP-1 could protect cardiac microvessels by inhibition of Rho and subsequently induce NADPH oxidase suppression.
To this end, this study was designed to determine the protective effects of GLP-1 on cardiac microvessels in diabetes and to characterize the underlying molecular mechanism. In vivo and in vitro studies were performed to assess cardiac function, glucose metabolism, and microvascular barrier function. The effects of GLP-1 on oxidative stress and apoptosis in microvessels were evaluated in isolated cardiac microvascular endothelial cells (CMECs). Furthermore, the underlying mechanism for GLP-1–induced protective effects was investigated.
RESEARCH DESIGN AND METHODS
Animal preparation for in vivo experiments.
Male Sprague-Dawley rats (weight, 220–250 g) were made diabetic using intraperitoneal injection of streptozotocin (35 mg/kg) for 3 days (18). Blood glucose levels were tested 1 week after streptozotocin injection. Animals with glucose levels ≥16.6 mmol/L were considered diabetic. To determine the dosage of insulin to control blood glucose in the diabetic group at the similar level as vildagliptin-treated and exenatide-treated groups, diabetic rats were randomized into the following groups: vildagliptin group that received daily treatment of vildagliptin at 1 mg/kg of body weight; exenatide group that received daily treatment of exenatide at 1 nmol/kg of body weight; and insulin group that received daily treatment of insulin at 0.5 units, 1 units, 1.5 units, and 2 units. Blood glucose was measured once per day for 2 weeks. After having determined the dosage of insulin, diabetic rats were randomly assigned to the following groups: vehicle group (n = 15) that received only saline daily for 12 weeks; vildagliptin group (n = 15) that received daily treatment of vildagliptin (Beijing HuiKang BoYuan Chemical Tech) at 1 mg/kg of body weight for 12 weeks; exenatide group (n = 15) that received daily treatment of exenatide (Tocris Bioscience, Minneapolis, MN) at 1 nmol/kg of body weight for 12 weeks; and insulin group (n = 15) that received daily treatment of protamine zinc insulin (Jiangsu Wanbang Biochemical Medicine) at 1.5 units for 12 weeks. Age-matched normal rats (n = 15) were used as nondiabetic controls. All experiments were conducted under the National Institutes of Health Guidelines on the Use of Laboratory Animal and were approved by the Fourth Military Medical University Committee on Animal Care.
Measurement of blood pressure.
Blood pressure was monitored with a tail-cuff system (Noninvasive Blood Pressure System; PanLab) as described previously (19). Briefly, rats were placed in a warm chamber (37°C) for 10 min to rest, and then occluding cuffs and pneumatic pulse transducers were placed on the tail. Five readings were obtained from each rat.
Measurement of insulin levels and insulin resistance.
We used commercially available ELISA kits to determine plasma insulin levels (Cusabio Biotech). The euglycemic-hyperinsulinemic clamp technique was performed to assess insulin resistance. Five days before clamp experiments, an intravenous silicone catheter was implanted into the right jugular vein of rats under surgical anesthesia as previously described (20). Rats were overnight-fasted. A priming dose of insulin infusion was administered during the initial 10 min to acutely increase plasma insulin concentration to the desired level, at which it was maintained by continuous insulin infusion at a rate of 2.5 × 10−3 units/kg body weight per minute. Blood glucose was clamped at 5 mmol/L for the next 180 min by adjusting the rate of 20% glucose infusion according to blood glucose measurements performed every 5 min using a hand-held glucometer (Johnson & Johnson Medical). The M-value (mg/kg/min) expressing the rate of whole-body glucose uptake was calculated as the mean value for each 20-min interval during the last 60 min of the clamp. The higher the M-value, the better the peripheral insulin sensitivity.
Echocardiography to assess cardiac diastolic function.
Transthoracic echocardiography was performed by Vevo 2100 ultrasound device (VisualSonics, Toronto, Canada) with a 21-MHz linear transducer. To evaluate the left ventricular diastolic function, mitral valve inflow velocities were recorded from an apical four-chamber view. Measurements of peak flow velocity of the early rapid diastolic filling wave and late diastolic filling wave were made and the early mitral diastolic wave/late mitral diastolic wave ratios were calculated. Also, two-dimensionally guided M-mode recordings were obtained from the short-axis view at the level of the papillary muscles more than three beats and averaged. Left ventricular end-diastolic diameter and fraction shortening were measured from the M-mode tracings. All measurements were determined by a single echocardiographer who was blinded to the experimental groups.
18F-fluorodeoxyglucose positron emission tomography/computed tomography to assess cardiac glucose metabolism.
Whole-body 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography (PET)/computed tomography (CT) scanning was performed using an animal PET/CT scanner (Mediso Nano PET/CT) described previously (21). The rats were maintained under fasting condition for 12–14 h, and fasting blood glucose levels were maintained between 6.0 and 7.5 mmol/L. Animals were injected with ∼37 MBq (1 mCi) of 18F-FDG via the tail vein and allowed to recover. To obtain the best signal-to-background ratio, the PET/CT imaging acquisition was initiated 30 min after injection of the tracer. CT scan (45 kV, 179 μA) was obtained during 20 min in two frames. Static whole-body PET scan was performed during 30 min in two frames. The images were reconstructed using ordered subsets expectation maximization reconstruction algorithm with decay correction, attenuation correction, and random correction from raw framed sinograms. The data were reconstructed over a 170- × 170- × 108-matrix with 0.77-mm slice thickness. Processed images were displayed in sagittal, transverse, and coronal planes. In each scan, 3-dimensional regions of interest were drawn over the heart region on whole-body axial images. Relative accumulation of the radioactivity in particular regions of interest was expressed as identification % per gram (22).
Examination of integrity of cardiac microvessels.
Scanning electron microscopy was used to assess microvascular endothelial integrity within the vessel wall of rat’s heart. Animal hearts received 10 mL low-viscosity resin previously mixed with benzoyl peroxide as a catalyzer at perfusion pressure no more than 100 mmHg through the aorta and were corrupted in a solution consisting of 5% sodium hydroxide solution at room temperature; the solution was changed every 12 h for 4–5 days. After removal of all connective tissue components surrounding the blood vessels, regular pretreatments in accordance with standard instruction were used, including dehydration, desiccation, and gilding. Prepared samples were subsequently observed by a scanning electron microscope (Hitachi S-3400N).
Examination of cardiac microvascular barrier function.
Transmission electron microscopy was used to assess CMEC-to-cell junctions as previously described (23). In brief, hearts of anesthetized rats were perfused via the aorta with 50 mL of prefixative solution (100 mmol/L Tris, pH 7.4, 150 mmol/L NaCl, 1 mmol/L MgCl2, 2.5 mmol/L CaCl2, 5.6 mmol/L KCl, 3.7 mmol/L glucose, and 3.6 mmol/L procaine), followed by 250 mL fixing solution (2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 mol/L sodium cacodylate buffer, pH 7.4, containing 2% lanthanum nitrate) using a Langendorff system. After perfusion, the heart tissue was kept in the same fixative solution without lanthanum for 1 h. Then, specimens were rinsed with washing solution (0.15 mol/L NaCl plus 0.2 mol/L sucrose). Subsequently, tissue samples were dehydrated by graded ethanol and embedded in Agar 100. Ultra-thin sections (60-nm-thick) were cut, mounted on copper grids, and observed using a transmission electron microscope (JEOL JEM-2000EX) to check for the location of lanthanum nitrate. Cardiac microvessels were regarded as having increased permeability when the electron-dense marker penetrated into the junctional complex and electron-dense material was identified on the luminal surface.
Isolation, cultivation, and identification of CMECs for in vitro experiments.
CMECs were isolated as previously described, with minor modifications (24). Briefly, left ventricles were excised from male Sprague-Dawley rats (100–150 g) and rinsed with PBS supplemented with heparin. Left ventricle was immersed in 75% ethanol for 15 s to devitalize endocardial endothelial cells and epicardial mesothelial cells. The remaining tissue was then minced in PBS and incubated in 0.2% collagenase II (Sigma, St. Louis, MO) for 6 min, followed by 0.25% trypsin (Sigma) for another 6 min at 37°C in a shaking bath. After centrifugation, cells were resuspended in Dulbecco modified Eagle medium (Invitrogen, Cambridge, MA) supplemented with 15% (v/v) fetal calf serum (FCS) and plated on laminin (10 μg/mL) coated dishes. Primary cultures of CMECs were positively identified by Dil-acetylated low-density lipoprotein (Molecular Probes, Eugene, OR) intake assay. Passage 3–5 cells were used for further studies. Media were replaced to different conditions after confluence: normal glucose medium (5.5 mmol/L), high glucose medium (25 mmol/L) plus vehicle (DMSO), and high glucose plus GLP-1 (10−10, 10−9, 10−8, 10−7 mol/L).
Detection of GLP-1R expression in CMECs.
To detect GLP-1R expression in CMECs, immunofluorescence staining was performed. Cells were fixed with 4% paraformaldehyde at room temperature for 30 min and then blocked by goat serum. GLP-1R antibody (1:200; Tocris Bioscience) was incubated overnight at 4°C, followed by detection with corresponding fluorescent secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) for 1 h at 37 °C. Nuclei were counterstained with 4′, 6-diamidino-2-phenylindole (Beyotime). Samples were examined by confocal microscope (Olympus FV 1000).
Quantification of ROS production and NADPH oxidase activity in CMECs.
ROS production, an index of oxidative stress, in viable CMECs was measured by lucigenin-enhanced chemiluminescence assay under adaptation of the previously described method (25). ROS production was expressed as relative light units per second per million cells. Dihydroethidine staining was used to detect the in situ formation of superoxide according to the oxidative fluorescent microtopography, as described recently (26). Dihydroethidine staining was visualized under a confocal microscope (Olympus). Then, images of cells were analyzed with Image-Pro Plus software version 6.0. The mean fluorescence intensity of each cell was calculated, and the total cell emission signals per field were averaged for data analysis.
NADPH oxidase activity was measured by lucigenin-enhanced chemiluminescence with a commercial kit according to the manufacturer’s instructions (Genmed Scientifics).
Assessment of apoptosis of CMECs.
Apoptosis of CMECs was detected by terminal deoxynucleotidyl TUNEL (in green) assay using a Cell Death Detection Kit (Roche, Penzberg, Germany) according to the manufacturer’s instructions. The index of apoptosis was expressed as the proportion of the TUNEL-positive CMECs to the total CMECs in percentage.
Western blot analysis.
Western blot was performed as previously described. Briefly, CMECs from each group were cultured and harvested at the indicated time. Cells were washed and scraped using lysis buffer. Total proteins were loaded onto an SDS-PAGE gel and transferred electrophoretically to nitrocellulose membranes (Millipore, Billerica, MA). After blocking with 5% skim milk, the membranes were incubated with the appropriate primary antibody at 4°C overnight. The membranes were washed and further incubated with horseradish peroxidase–linked secondary antibody at 37°C for 60 min. The blots were developed with an enhanced chemiluminescence reagent kit (Millipore) and visualized with UVP Bio-Imaging Systems. Blot densities were analyzed with Vision Works LS Acquisition and Analysis Software.
Primary antibodies used in this study were as follows: GLP-1R (1:2,000; Santa Cruz Biotechnology), cleaved caspase-3 (1:200; Santa Cruz Biotechnology), Rho (1:2,000; Abcam), Rho-kinase (ROCK) (1:500; Abcam), p47phox (1:1,000, Abcam), gp91phox (1:500; Abcam), p22phox (1:500; Santa Cruz Laboratories), p40phox (1:1,000; Abcam), and β-actin (1:2,000; Cell Signaling). Secondary antibodies were horseradish peroxidase–conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology), rabbit anti-goat (Santa Cruz Biotechnology), and rabbit antimice (Santa Cruz Biotechnology) at 1:5,000 dilution.
Plasmids and DNA manipulations.
Site-directed mutagenesis was performed with Pfu Turbo polymerase (Stratagene). For adenovirus construction, cDNAs and short hairpin sequences were subcloned in the pAdTRACK vector. Complete viral vectors were generated by homologous recombination with the AdEASY vector as described (27).
Statistical analysis.
All data were expressed as mean ± SD and were analyzed using ANOVA, followed by a Bonferroni correction for post hoc t test (with exception of Western blot). Western blotting result was analyzed with the Kruskal-Wallis test, followed by Dunn’s post hoc test. A value of P < 0.05 was considered to be statistically significant. All statistical tests were performed using GraphPad Prism software version 5.0 (GraphPad Software, San Diego, CA).
RESULTS
Basic parameters of rats in different experimental groups.
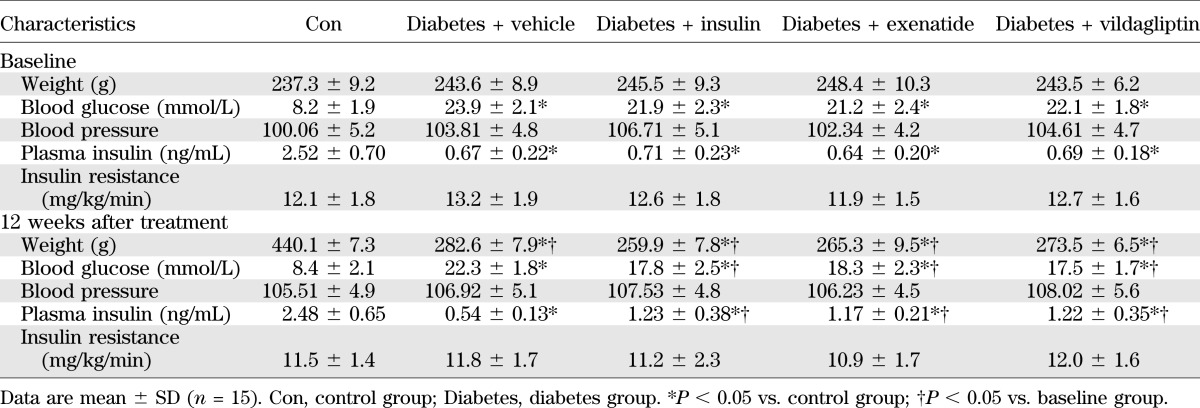
It has been reported that GLP-1 is an intestinally derived hormone acting in vivo to promote the level of circulating insulin, leading to reduced blood glucose levels. We performed experiments to determine the dosage of insulin administration to keep blood glucose in the diabetic group comparable with that in vildagliptin-treated and exenatide-treated groups. According to our results (Table 1), we found that insulin treatment at dosage of 1.5 units per day had the similar effect in blood glucose as those elicited by vildagliptin and exenatide in rats. As shown in Table 2, diabetic rats exhibited hyperglycemia (22.3 ± 1.8 mmol/L vs. 8.4 ± 2.1 mmol/L; P <0.05) and reduced body weight gain (282.6 ± 7.9 g vs. 440.1 ± 7.3 g; P <0.05) compared with control group. Administration of vildagliptin and exenatide for 12 weeks significantly decreased plasma glucose level (17.5 ± 1.7 mmol/L vs. 22.3 ± 1.8 mmol/L; P <0.05; 18.3 ± 2.3 mmol/L vs. 22.3 ± 1.8 mmol/L; P <0.05) and slightly decreased body weight compared with diabetic group (273.5 ± 6.5 g vs. 282.6 ± 7.9 g; P <0.05; 265.3 ± 9.5 g vs. 282.6 ± 7.9 g; P < 0.05). Vildagliptin or exenatide treatment increased the level of plasma insulin in diabetes (1.22 ± 0.35 ng/mL vs. 0.54 ± 0.13 ng/mL; P <0.05; 1.17 ± 0.21 ng/mL vs. 0.54 ± 0.13 ng/mL; P < 0.05). Administration of 1.5 units insulin per day had the similar effects in blood glucose and body weight as those elicited by vildagliptin and exenatide in rats. However, blood pressure and insulin resistance exhibited little difference between diabetic and the treatment groups.
TABLE 1.
Blood glucose levels in different experimental groups
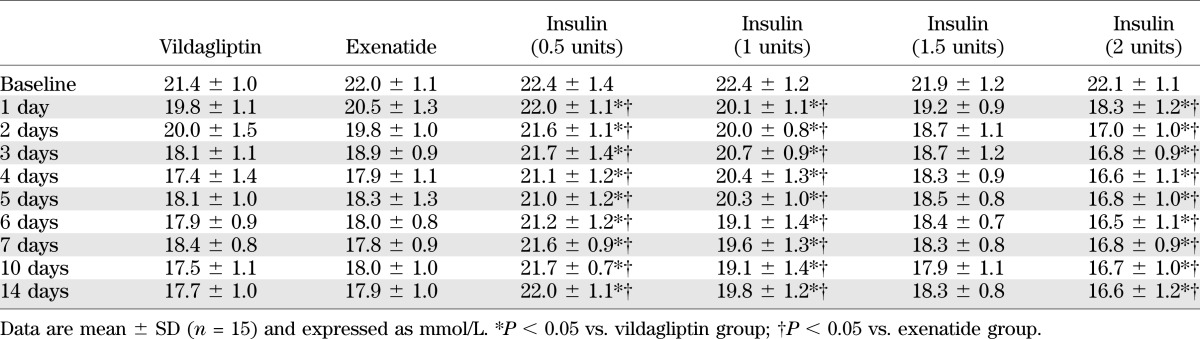
TABLE 2.
Basic parameters of rats in different experimental groups
Vildagliptin or exenatide treatment improved cardiac diastolic function in diabetes.
After 12 weeks of treatment with vildagliptin or exenatide, rats were anesthetized and cardiac function was determined using echocardiography. Compared with the control group, diabetic rats exhibited significantly dampened diastolic function, as manifested by increased E/A ratio (Fig. 1A, B) and left ventricular end-diastolic diameter (Fig. 1C), the effects of which were mitigated by vildagliptin or exenatide treatment. Compared with insulin-treated group, vildagliptin or exenatide further improved cardiac diastolic function in diabetic rats. However, there was little difference in fractional shortening among the four groups studied (Fig. 1D). In summary, vildagliptin or exenatide was capable of improving cardiac diastolic function in diabetes.
FIG. 1.

Effects of vildagliptin or exenatide on cardiac function and cardiac glucose metabolism in diabetes. A: Representative mitral flow patterns from pulsed Doppler. B: Quantification of E/A ratio. C: Measurement of left ventricular end-diastolic diameter after 12 weeks of drug treatment. D: Measurement of fractional shortening (FS) after 12 weeks of drug treatment. E: Representative PET/CT scan images from coronal, sagittal, and transverse view from each group. Higher glucose uptake level is shown as an increase in the intensity of red color as indicated on the color scale bar shown. F: Quantification of accumulated 18F-FDG in the heart. Data are expressed as mean ± SD (n = 6). *P <0.05 vs. control group; #P <0.05 vs. Diabetes + vehicle group; &P <0.05 vs. Diabetes + insulin group. DM, diabetes; E, early mitral diastolic wave; A, late mitral diastolic wave; LVEDD, left ventricular end-diastolic diameter.
Vildagliptin or exenatide treatment improved cardiac glucose metabolism in diabetes.
To examine the role of cardiac glucose metabolism after vildagliptin or exenatide treatment in diabetes, 18F-FDG was used to analyze the level of myocardial glucose uptake. 18F-FDG method has been widely used in both experimental and clinical settings to quantify cardiac glucose metabolism. Our data revealed that diabetes led to a defective 18F-FDG uptake in the heart, the effect of which was significantly improved by vildagliptin or exenatide treatment. Insulin treatment exhibited less 18F-FDG uptake in the heart compared with administration of vildagliptin or exenatide (Fig. 1E, F).
Vildagliptin or exenatide treatment maintained cardiac microvessel integrity in diabetes.
Scanning electron microscopy was used to evaluate cardiac microvascular integrity (28). In control animals, the surface of cardiac microvessels was smooth and well-integrated (Fig. 2A). However, numerous irregular exvaginations and invaginations were identified in cardiac microvessels from diabetic animals. Cell junction integrity was greatly compromised and cells were not well-integrated with the vessel (Fig. 2B). Vildagliptin or exenatide treatment effectively attenuated the changes, preserving cardiac microvascular integrity and vessel integration, in a manner more pronounced than the insulin treatment (Fig. 2C–E).
FIG. 2.

Effects of vildagliptin or exenatide on cardiac microvascular integrity and permeability. A: The surface of cardiac microvessels in control animals was smooth and well-integrated. B: Cardiac microvessels in diabetic animals were visualized to be highly irregular and rough. C: Cardiac microvessels treated with insulin. D and E: Cardiac microvessels with vildagliptin or exenatide treatment exhibited integrity and vessel integration. F: Lanthanum nitrate was regulated to the blood vessel lumen in control animals. G: Lanthanum nitrate was found to diffuse across vessel lumen to the basal lamina in diabetic animals. H: Cardiac microvessels were treated with insulin. I and J: Diffusion of lanthanum nitrate across CMECs was attenuated in vildagliptin or exenatide treatment (scale bar, 2 μm). The extracellular tracer lanthanum nitrate is highlighted with arrows. DM, diabetes.
Vildagliptin or exenatide treatment protected cardiac microvascular barrier function in diabetes.
Lanthanum nitrate has been widely used to assess the barrier function of endothelial cells (29). Under healthy conditions, lanthanum nitrate is unable to cross endothelial cell-to-cell junctions, but it may traverse cell boundaries via paracellular diffusion when such barriers are in pathologic conditions. Lanthanum nitrate was regulated to the blood vessel lumen in control animals (Fig. 2F); however, significant deficit in barrier integrity of cardiac microvessels was found in diabetic rats (Fig. 2G). Diffusion of lanthanum nitrate across endothelial cells was attenuated by administration of vildagliptin (Fig. 2I) or exenatide (Fig. 2J) in a manner more pronounced than that of the insulin (Fig. 2H). These findings indicated that vildagliptin or exenatide treatment was capable of protecting diabetes-induced cardiac endothelium injury independent of its glucose-lowering property.
Identification of CMECs and detection of GLP-1R.
The cells displayed “flagstone” morphology (Fig. 3A) and tested positive on Dil-acetylated low-density lipoprotein intake assay (Fig. 3B), indicating that the cultured cells were CMECs. Both immunofluorescence staining (Fig. 3C, D) and Western blot (Fig. 3E) showed that GLP-1R was expressed on CMECs.
FIG. 3.

Characterization of CMECs and detection of GLP-1R. A: CMEC monolayer presents cobblestone appearance by phase-contrast microscopy (scale bar, 40 μm). B: Uptake of Dil-acetylated low-density lipoprotein (Dil-Ac-LDL) by immunofluorescence (red, Dil-Ac-LDL; blue, DAPI). C: Expression of GLP-1R by immunofluorescence (green, GLP-1R; scale bar, 10 μm). D: Merge between B and C. E: Western blot analysis for GLP-1R. β-Cells were used as positive control.
GLP-1 attenuated high-glucose–induced oxidative stress in CMECs.
ROS are known to play a major role in diabetic cardiovascular disease. To determine whether GLP-1 attenuated high-glucose–induced oxidative stress in CMECs, ROS production and NADPH activity was measured. As shown in Fig. 4A and B, high glucose increased ROS production and NADPH activity after 24-h incubation in CMECs, whereas GLP-1 could dose-dependently suppress ROS production and NADPH activity in high-glucose–induced CMECs. We noticed that GLP-1 at 10−8 mol/L significantly suppressed ROS production and NADPH activity. Dihydroethidine staining also showed that ROS production was elevated in the high-glucose group, whereas it was decreased in the GLP-1–treated (10−8 mol/L) group (Fig. 4C, D). To further determine whether GLP-1 protected against high-glucose–induced oxidative stress, the protein expression of p47phox, gp91phox, p22phox, and p40phox subunits of NADPH oxidase were measured. The expression of p47phox, gp91phox, p22phox, and p40phox were significantly increased in high-glucose–induced CMECs compared with control group. After treatment with GLP-1 (10−8 mol/L), the expression of p47phox, gp91phox, p22phox, and p40phox was significantly decreased compared with high-glucose–induced CMECs (Fig. 4E–H).
FIG. 4.

Effects of GLP-1 on oxidative stress in high-glucose–induced CMECs. A: GLP-1 inhibits high-glucose–induced superoxide generation in CMECs dose dependently. B: GLP-1 inhibits high-glucose–induced NADPH oxidase activity in CMECs dose dependently. C: Representative images of dihydroethidine (DHE) staining of CMECs (red, DHE; blue, DAPI; scale bar, 25 μm). D: The average fluorescence intensity from five fields was summarized. E–H: Western blot assay for p47phox, gp91phox, p22phox, and p40phox protein expression. *P <0.01 vs. control group; #P <0.05 vs. high-glucose (HG) group; &P <0.01 vs. HG group. Con, control.
GLP-1 decreased high-glucose–induced apoptosis in CMECs.
To investigate the role of GLP-1 on high-glucose–induced apoptosis in CMECs, TUNEL assay was performed. High glucose induced a significant increase in TUNEL-positive cells. Compared with cells cultured in high glucose with vehicle, the GLP-1–treated group showed a significant decrease in apoptotic cells (46.1 ± 8.5% vs. 29.8 ± 5.3%; P < 0.05; Fig. 5A, B). We also detected the expression of caspase-3 and found that GLP-1 statistically decreased the caspase-3 expression in high-glucose–induced CMECs (P <0.05; Fig. 5C). Taken together, these results demonstrated that GLP-1 exerted an antiapoptotic effect on high-glucose–induced CMECs.
FIG. 5.

GLP-1 exerted antiapoptotic effect on high-glucose–induced CMECs. A: Representative images of immunostaining for apoptotic (TUNEL) cells. Nuclei were labeled with DAPI (scale bar, 25 μm). B: Quantification of apoptotic nuclei by Image-Pro Plus software. C: Western blot assay for cleaved caspase-3 protein expression. *P <0.05 vs. high-glucose (HG) plus vehicle group; #P <0.05 vs. control plus vehicle group. Con, control.
GLP-1 suppressed high-glucose–induced activation of Rho in CMECs through cAMP/PKA pathway.
Rho is a key regulator for oxidative stress (30). ROCK is a serine/threonine kinase, which is a known downstream effector of Rho. We examined the effect of GLP-1 on Rho expression in CMECs. Incubation of CMECs with high glucose significantly upregulated Rho expression and elicited a significant increase in ROCK expression. Treatment of CMECs with GLP-1 significantly alleviated high-glucose–induced increase in Rho and ROCK expression without affecting these parameters under normal glucose conditions. Incubation of cells with the PKA selective inhibitor H89 abrogated GLP-1–induced effect on Rho suppression (Fig. 6A, B).
FIG. 6.

GLP-1 suppressed high-glucose (HG)-induced activation of Rho. A: Western blot assay for Rho protein expression. B: Western blot assay for ROCK protein expression. *P <0.05 vs. control group; #P <0.05 vs. HG group; &P <0.05 vs. HG plus GLP-1 group. Con, control; H89, PKA inhibitor.
GLP-1 decreased high-glucose–induced ROS production in CMECs via a cAMP/PKA/Rho-dependent mechanism.
To further determine the mechanism of action behind the beneficial effect of GLP-1 on CMECs, fasudil (Selleck, Houston, TX) was used. Chronic exposure of CMECs to high glucose promoted intracellular ROS levels, the effect of which was obliterated by GLP-1. Incubation of CMECs with fasudil inhibited ROS production. However, combination of GLP-1 and fasudil failed to further attenuate ROS production. The inhibitory effect of GLP-1 on high-glucose–induced ROS accumulation was significantly attenuated by treatment with H89 (Fig. 7A–C). In line with the changes in intracellular ROS accumulation, the inhibitory effect of GLP-1 on high-glucose–induced activation of p47phox and gp91phox was reduced by H89. The combined treatment with GLP-1 and fasudil failed to produce any additive effects on high-glucose–induced p47phox and gp91phox expression and oxidative stress (Fig. 7D–F). Overexpression of Rho in CMECs potentiated p47phox and gp91phox activity and ROS production. Taken together, these results suggested that GLP-1 may decrease high-glucose–induced ROS accumulation in CMECs through a cAMP/PKA/Rho-dependent mechanism.
FIG. 7.

Effects of GLP-1, fasudil, and H89 on high-glucose (HG)-induced oxidative stress in CMECs. A: Representative diagram showing dihydroethidine (DHE) staining of CMECs (red, DHE; blue, DAPI; scale bar, 25 μm). B: The average fluorescence intensity from five fields was summarized. C: Superoxide generation of CMECs in different groups. D: Western blot assay for p47phox protein and gp91phox protein expression. E and F: Quantitative analysis of p47phox and gp91phox expression. *P <0.01 vs. control group; #P <0.01 vs. HG group; &P <0.05 vs. HG plus GLP-1 group. Con, control; H89, PKA inhibitor.
DISCUSSION
Pharmacological modulation of GLP-1 has emerged as a novel therapeutic option for diabetes (31–33). The therapeutic use of GLP-1 is severely compromised by its rapid degradation by DPP-4 (34). Exenatide is a peptide incretin mimetic that shares many biological functions with GLP-1. Clinical efficacy of DPP-4 inhibitor such as vildagliptin has been well-documented in many studies (35).
It has been reported that GLP-1 is an intestinally derived hormone acting in vivo to promote the level of circulating insulin, leading to reduced blood glucose levels (36). In addition to its glucose-lowering property, GLP-1 also exhibits potential cardioprotective properties (37). For example, GLP-1 is capable of protecting the heart against ischemia/reperfusion injury (38). In addition, GLP-1 was shown to retard atherosclerotic lesion formation through enhancing GLP-1 activity in macrophages and endothelium (39). In this study, GLP-1 was found to improve cardiac function and glucose metabolism in hearts from diabetic rats, consistent with a recent clinical report in which GLP-1–based therapy reduces overall cardiovascular events in diabetic patients (40).
Tight control of cardiac microvascular function is essential to the maintenance of circulatory homeostasis and physiological function of the heart. Impaired cardiac microvascular function is thought to contribute greatly to diabetic cardiovascular disease (41,42). There is convincing evidence suggesting that endothelial apoptosis occurs at an earlier time point compared with pathological changes in cardiomyocytes in diabetes (43). The increased rate of endothelial apoptosis in diabetes may contribute to microvascular barrier dysfunction and rarefaction, which lead to myocardial metabolism abnormalities and impaired intracellular calcium homeostasis (44). In addition, microvascular injury aggravates the myocardium stiffness because of reduced blood supply, which directly prompts diastolic dysfunction (45). Our results from the present study exhibited that vildagliptin or exentinade significantly attenuated microvascular barrier defect and diastolic dysfunction in the diabetic heart.
A number of scenarios may be considered that underscore the beneficial effects of these agents in diabetes. Physiological levels of intracellular ROS production are deemed necessary for normal cellular development and function, whereas excessive ROS generation may facilitate pathophysiological progression in many diseases, including diabetes. This is in line with the speculation for a pivotal role of oxidative stress in the onset and development of diabetes complications, both microvascular and macrovascular (46). High glucose facilitated ROS production in CMECs, mainly through NADPH oxidase activation, involving formation and translocation of cytosolic NADPH oxidase subunits (p47phox, p67phox, and p40phox) and membrane-bound subunits (p22phox and gp91phox). Intriguingly, GLP-1 significantly suppressed high-glucose–induced ROS accumulation in CMECs, as well as levels of p47phox and gp91phox. Similarly, GLP-1 was effective in rescuing against high-glucose–induced apoptosis, possibly through inhibition of oxidative stress in CMECs.
Our data further revealed that GLP-1 prevented high-glucose–induced CMECs oxidative stress, possibly through inhibition of activation of the Rho/ROCK pathway. Rho/ROCK has been considered a key mediator for oxidative stress–mediated cell injurious process. Burger et al. (47) reported an important role of Rho/ROCK in angiotensin II–induced upregulation of endothelial NADPH oxidase and subsequently increased endothelial superoxide production. Moreover, accumulating evidence has depicted that the adverse endothelial outcome in diabetes is corroborated with increased oxidative stress, whereas Rho/ROCK may govern the downstream effects of hyperglycemia-induced oxidative stress. We have shown that exposure of CMECs to high glucose promoted Rho/ROCK activity. Rho/ROCK activity was suppressed by GLP-1 in high-glucose–induced CMECs. These findings demonstrated that the protective effects of GLP-1 may be mediated through inhibition of Rho/ROCK activation, which in turn alleviated high-glucose–induced oxidative stress in CMECs.
It is noteworthy that the upstream molecules and mechanisms responsible for GLP-1–associated cardiac microvascular protection remain elusive in diabetes. It was recently revealed that activation of the cAMP/PKA pathway may be involved in GLP-1–offered protective effects. Xiao et al. (48) demonstrated that GLP-1 promoted voltage-gated l-type Ca2+ current in the heart possibly through a cAMP/PKA-dependent pathway and may contribute, in part, to prolonged action potential duration. DPP-4 inhibition was found to protect against vascular senescence, which was attenuated by GLP-1 in a receptor-dependent manner involving downstream PKA signaling (49). Interestingly, cAMP/PKA was an essential negative regulator of Rho (50). In this study, our results revealed that cAMP/PKA may contribute to GLP-1–mediated suppression of high-glucose–evoked Rho/ROCK activity in CMECs. This conclusion was supported by the fact that GLP-1–induced suppression of high-glucose–induced Rho/ROCK activation was abrogated when the cAMP/PKA pathway was inhibited. Also, data from our study showed that Rho inhibitor fasudil suppressed ROS production and NADPH oxidase upregulation. However, joint therapy with GLP-1 and fasudil failed to further suppress ROS production and NADPH oxidase upregulation. Taken together, these results suggested that GLP-1 may inhibit high-glucose–induced oxidative stress in CMECs through a cAMP/PKA/Rho-dependent mechanism.
This study provides both in vivo and in vitro evidence supporting the protective effects of GLP-1 against diabetes-induced microvascular defect. These findings indicate that pharmacological intervention targeting on GLP-1 may represent a promising therapeutic strategy to maintain microvascular function and cardiac function in patients with diabetes and cardiovascular disease. Nonetheless, it is noteworthy that our findings were mainly based on rodent models and in vitro experiments. Therefore, caution must be taken in evaluating the effects of GLP-1 on microvascular and cardiac function in patients with diabetes.
In conclusion, GLP-1 could protect the cardiac microvessels against oxidative stress, apoptosis, and the resultant microvascular barrier dysfunction in diabetes rats, en route to improved cardiac diastolic function and cardiac glucose metabolism. The protective effects of GLP-1 are dependent on downstream inhibition of Rho through a cAMP/PKA-dependent manner, resulting in a subsequent decrease in the expression of NADPH oxidase. These findings should provide important implications for diabetes with cardiovascular disease, for which GLP-1 may hold promise for prevention and treatment.
ACKNOWLEDGMENTS
This work was supported by the National Nature Science Foundation of China (No. 81270168, No. 81090274, No. 81227901; FCao BWS12J037), by an Innovation Team Development Grant from the China Department of Education (2010CXTD01, IRT1053), by the National Basic Research Program of China (2012CB518101), and by China’s Ministry of Science and Technology 863 Program (2012AA02A603).
No potential conflicts of interest relevant to this article were reported.
D.W., J.R., and F.C. designed the experiments. D.W., P.L., and Y.W. performed the experiments. D.W. wrote the manuscript. P.L. researched data. Y.W. and W.L. contributed to the discussion. C.W. and D.S. contributed to the CMEC isolation. R.Z. and T.S. assisted in the preparation of the experiments. X.M. and C.Z. contributed to PET/CT assessment. H.W. and W.L. reviewed the manuscript. J.R. and F.C. analyzed the results and edited the manuscript. F.C. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were presented in abstract form at the American Heart Association Scientific Sessions, Los Angeles, California, 4–6 November 2012.
REFERENCES
- 1.Mazzone T. Intensive glucose lowering and cardiovascular disease prevention in diabetes: reconciling the recent clinical trial data. Circulation 2010;122:2201–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Acar E, Ural D, Bildirici U, Sahin T, Yilmaz I. Diabetic cardiomyopathy. Anadolu Kardiyol Derg 2011;11:732–737 [DOI] [PubMed] [Google Scholar]
- 3.Voulgari C, Papadogiannis D, Tentolouris N. Diabetic cardiomyopathy: from the pathophysiology of the cardiac myocytes to current diagnosis and management strategies. Vasc Health Risk Manag 2010;6:883–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ismail-Beigi F, Craven T, Banerji MA, et al. ACCORD trial group Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: an analysis of the ACCORD randomised trial. Lancet 2010;376:419–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hao M, Li SY, Sun CK, et al. Amelioration effects of berberine on diabetic microendothelial injury model by the combination of high glucose and advanced glycation end products in vitro. Eur J Pharmacol 2011;654:320–325 [DOI] [PubMed] [Google Scholar]
- 6.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007;132:2131–2157 [DOI] [PubMed] [Google Scholar]
- 7.Lee CH, Yan B, Yoo KY, et al. Ischemia-induced changes in glucagon-like peptide-1 receptor and neuroprotective effect of its agonist, exendin-4, in experimental transient cerebral ischemia. J Neurosci Res 2011;89:1103–1113 [DOI] [PubMed] [Google Scholar]
- 8.Lønborg J, Vejlstrup N, Kelbæk H, et al. Exenatide reduces reperfusion injury in patients with ST-segment elevation myocardial infarction. Eur Heart J 2012;33:1491–1499 [DOI] [PubMed] [Google Scholar]
- 9.Nyström T, Gutniak MK, Zhang Q, et al. Effects of glucagon-like peptide-1 on endothelial function in type 2 diabetes patients with stable coronary artery disease. Am J Physiol Endocrinol Metab 2004;287:E1209–E1215 [DOI] [PubMed] [Google Scholar]
- 10.Shen M, Sun D, Li W, et al. The synergistic effect of valsartan and LAF237 [(S)-1-[(3-hydroxy-1-adamantyl)ammo]acetyl-2-cyanopyrrolidine] on vascular oxidative stress and inflammation in type 2 diabetic mice. Exp Diabetes Res 2012;2012:146194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen W, Zhou Y, Zhang H, et al. Stability and bioactivity studies on dipeptidyl peptidase IV resistant glucogan-like peptide-1 analogues. Protein Pept Lett 2012;19:203–211 [DOI] [PubMed] [Google Scholar]
- 12.Folli F, Corradi D, Fanti P, et al. The role of oxidative stress in the pathogenesis of type 2 diabetes mellitus micro- and macrovascular complications: avenues for a mechanistic-based therapeutic approach. Curr Diabetes Rev 2011;7:313–324 [DOI] [PubMed] [Google Scholar]
- 13.Naudi A, Jove M, Ayala V, et al. Cellular dysfunction in diabetes as maladaptive response to mitochondrial oxidative stress. Exp Diabetes Res 2012;2012:696215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang X, Zhang J, Liu J, et al. C-reactive protein promotes adhesion of monocytes to endothelial cells via NADPH oxidase-mediated oxidative stress. J Cell Biochem 2012;113:857–867 [DOI] [PubMed] [Google Scholar]
- 15.Ma Z, Zhang J, Ji E, Cao G, Li G, Chu L. Rho kinase inhibition by fasudil exerts antioxidant effects in hypercholesterolemic rats. Clin Exp Pharmacol Physiol 2011;38:688–694 [DOI] [PubMed] [Google Scholar]
- 16.Noma K, Goto C, Nishioka K, et al. Roles of rho-associated kinase and oxidative stress in the pathogenesis of aortic stiffness. J Am Coll Cardiol 2007;49:698–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takeshima H, Kobayashi N, Koguchi W, Ishikawa M, Sugiyama F, Ishimitsu T. Cardioprotective effect of a combination of Rho-kinase inhibitor and p38 MAPK inhibitor on cardiovascular remodeling and oxidative stress in Dahl rats. J Atheroscler Thromb 2012;19:326–336 [DOI] [PubMed] [Google Scholar]
- 18.Wei L, Yin Z, Yuan Y, et al. A PKC-beta inhibitor treatment reverses cardiac microvascular barrier dysfunction in diabetic rats. Microvasc Res 2010;80:158–165 [DOI] [PubMed] [Google Scholar]
- 19.Rodríguez A, Becerril S, Valentí V, et al. Short-term effects of sleeve gastrectomy and caloric restriction on blood pressure in diet-induced obese rats. Obes Surg 2012;22:1481–1490 [DOI] [PubMed] [Google Scholar]
- 20.Neschen S, Morino K, Dong J, et al. n-3 Fatty acids preserve insulin sensitivity in vivo in a peroxisome proliferator-activated receptor-alpha-dependent manner. Diabetes 2007;56:1034–1041 [DOI] [PubMed] [Google Scholar]
- 21.Eguchi M, Kim YH, Kang KW, et al. Ischemia-reperfusion injury leads to distinct temporal cardiac remodeling in normal versus diabetic mice. PLoS ONE 2012;7:e30450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stolc S, Jakubíková L, Kukurová I. Body distribution of C-methionine and FDG in rat measured by microPET. Interdiscip Toxicol 2011;4:52–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei L, Sun D, Yin Z, et al. A PKC-beta inhibitor protects against cardiac microvascular ischemia reperfusion injury in diabetic rats. Apoptosis 2010;15:488–498 [DOI] [PubMed] [Google Scholar]
- 24.Wang D, Wang H, Luo P, et al. Effects of ghrelin on homocysteine-induced dysfunction and inflammatory response in rat cardiac microvascular endothelial cells. Cell Biol Int 2012;36:511–517 [DOI] [PubMed] [Google Scholar]
- 25.Schuhmacher S, Wenzel P, Schulz E, et al. Pentaerythritol tetranitrate improves angiotensin II-induced vascular dysfunction via induction of heme oxygenase-1. Hypertension 2010;55:897–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zuo L, Youtz DJ, Wold LE. Particulate matter exposure exacerbates high glucose-induced cardiomyocyte dysfunction through ROS generation. PLoS ONE 2011;6:e23116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koo SH, Flechner L, Qi L, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 2005;437:1109–1111 [DOI] [PubMed] [Google Scholar]
- 28.Verli FD, Marinho SA, Rossi-Schneider TR, Yurgel LS, de Souza MA. Angioarchitecture of the ventral surface of the tongue from Wistar rats. Scanning 2008;30:414–418 [DOI] [PubMed] [Google Scholar]
- 29.Rapôso C, Zago GM, da Silva GH, da Cruz Höfling MA. Acute blood-brain barrier permeabilization in rats after systemic Phoneutria nigriventer venom. Brain Res 2007;1149:18–29 [DOI] [PubMed] [Google Scholar]
- 30.Rolfe BE, Worth NF, World CJ, Campbell JH, Campbell GR. Rho and vascular disease. Atherosclerosis 2005;183:1–16 [DOI] [PubMed] [Google Scholar]
- 31.Kazafeos K. Incretin effect: GLP-1, GIP, DPP4. Diabetes Res Clin Pract 2011;93(Suppl 1):S32–S36 [DOI] [PubMed] [Google Scholar]
- 32.Peterson G. Current treatments and strategies for type 2 diabetes: can we do better with GLP-1 receptor agonists? Ann Med 2012;44:338–349 [DOI] [PubMed] [Google Scholar]
- 33.Fonseca VA, Alvarado-Ruiz R, Raccah D, Boka G, Miossec P, Gerich JE, EFC6018 GetGoal-Mono Study Investigators Efficacy and safety of the once-daily GLP-1 receptor agonist lixisenatide in monotherapy: a randomized, double-blind, placebo-controlled trial in patients with type 2 diabetes (GetGoal-Mono). Diabetes Care 2012;35:1225–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacDonald PE, El-Kholy W, Riedel MJ, Salapatek AM, Light PE, Wheeler MB. The multiple actions of GLP-1 on the process of glucose-stimulated insulin secretion. Diabetes 2002;51(Suppl. 3):S434–S442 [DOI] [PubMed] [Google Scholar]
- 35.Dicker D. DPP-4 inhibitors: impact on glycemic control and cardiovascular risk factors. Diabetes Care 2011;34(Suppl. 2):S276–S278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parkes DG, Pittner R, Jodka C, Smith P, Young A. Insulinotropic actions of exendin-4 and glucagon-like peptide-1 in vivo and in vitro. Metabolism 2001;50:583–589 [DOI] [PubMed] [Google Scholar]
- 37.Vergès B, Bonnard C, Renard E. Beyond glucose lowering: glucagon-like peptide-1 receptor agonists, body weight and the cardiovascular system. Diabetes Metab 2011;37:477–488 [DOI] [PubMed] [Google Scholar]
- 38.Bose AK, Mocanu MM, Carr RD, Brand CL, Yellon DM. Glucagon-like peptide 1 can directly protect the heart against ischemia/reperfusion injury. Diabetes 2005;54:146–151 [DOI] [PubMed] [Google Scholar]
- 39.Arakawa M, Mita T, Azuma K, et al. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes 2010;59:1030–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sullivan SD, Alfonso-Cristancho R, Conner C, Hammer M, Blonde L. A simulation of the comparative long-term effectiveness of liraglutide and glimepiride monotherapies in patients with type 2 diabetes mellitus. Pharmacotherapy 2009;29:1280–1288 [DOI] [PubMed] [Google Scholar]
- 41.Avogaro A, Fadini GP, Gallo A, Pagnin E, de Kreutzenberg S. Endothelial dysfunction in type 2 diabetes mellitus. Nutr Metab Cardiovasc Dis 2006;16(Suppl. 1):S39–S45 [DOI] [PubMed] [Google Scholar]
- 42.La Fontaine J, Harkless LB, Davis CE, Allen MA, Shireman PK. Current concepts in diabetic microvascular dysfunction. J Am Podiatr Med Assoc 2006;96:245–252 [DOI] [PubMed] [Google Scholar]
- 43.Rosenson RS, Fioretto P, Dodson PM. Does microvascular disease predict macrovascular events in type 2 diabetes? Atherosclerosis 2011;218:13–18 [DOI] [PubMed] [Google Scholar]
- 44.An D, Rodrigues B. Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol 2006;291:H1489–H1506 [DOI] [PubMed] [Google Scholar]
- 45.Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med 2007;356:830–840 [DOI] [PubMed] [Google Scholar]
- 46.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res 2010;107:1058–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burger D, Montezano AC, Nishigaki N, He Y, Carter A, Touyz RM. Endothelial microparticle formation by angiotensin II is mediated via Ang II receptor type I/NADPH oxidase/ Rho kinase pathways targeted to lipid rafts. Arterioscler Thromb Vasc Biol 2011;31:1898–1907 [DOI] [PubMed] [Google Scholar]
- 48.Xiao YF, Nikolskaya A, Jaye DA, Sigg DC. Glucagon-like peptide-1 enhances cardiac L-type Ca2+ currents via activation of the cAMP-dependent protein kinase A pathway. Cardiovasc Diabetol 2011;10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oeseburg H, de Boer RA, Buikema H, van der Harst P, van Gilst WH, Silljé HH. Glucagon-like peptide 1 prevents reactive oxygen species-induced endothelial cell senescence through the activation of protein kinase A. Arterioscler Thromb Vasc Biol 2010;30:1407–1414 [DOI] [PubMed] [Google Scholar]
- 50.Brown JA, Diggs-Andrews KA, Gianino SM, Gutmann DH. Neurofibromatosis-1 heterozygosity impairs CNS neuronal morphology in a cAMP/PKA/ROCK-dependent manner. Mol Cell Neurosci 2012;49:13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]