

Abstract
Elevated fasting blood glucose (FBG) is associated with increased risk for the development of type 2 diabetes and cardiovascular-associated mortality. Genome-wide association studies (GWAS) have linked polymorphisms in G6PC2 with variations in FBG and body fat, although not insulin sensitivity or glucose tolerance. G6PC2 encodes an islet-specific, endoplasmic reticulum–resident glucose-6-phosphatase catalytic subunit. A combination of in situ perfused pancreas, in vitro isolated islet, and in vivo analyses were used to explore the function of G6pc2 in mice. G6pc2 deletion had little effect on insulin sensitivity and glucose tolerance, whereas body fat was reduced in female G6pc2 knockout (KO) mice on both a chow and high-fat diet, observations that are all consistent with human GWAS data. G6pc2 deletion resulted in a leftward shift in the dose-response curve for glucose-stimulated insulin secretion (GSIS). As a consequence, under fasting conditions in which plasma insulin levels were identical, blood glucose levels were reduced in G6pc2 KO mice, again consistent with human GWAS data. Glucose-6-phosphatase activity was reduced, whereas basal cytoplasmic calcium levels were elevated in islets isolated from G6pc2 KO mice. These data suggest that G6pc2 represents a novel, negative regulator of basal GSIS that acts by hydrolyzing glucose-6-phosphate, thereby reducing glycolytic flux.
Numerous studies have investigated the biological impact of variations in fasting blood glucose (FBG) levels in humans. Elevated FBG has been shown to be associated with increased risk for the development of type 2 diabetes (1). In addition, a number of studies have found that even mild variations in FBG can have significant consequences on the risk of cardiovascular-associated mortality (CAM). For example, a study performed on a European male population demonstrated that a small increase in FBG levels from just 90 mg/dL to between 99 and 108 mg/dL was associated with a 30% increase in the risk of CAM (2). Conversely, a study performed in an Asian population found that a reduction in FBG levels from 99 to 90 mg/dL was associated with a 25% reduction in CAM (3). The risk of CAM increases still further in individuals with the very high FBG levels characteristic of diabetes (2–5). Because of the apparent biological importance of tightly regulating FBG levels, there has been tremendous interest in understanding how this parameter is controlled.
Recent genome-wide association studies (GWAS) have shed light on this question by demonstrating that single nucleotide polymorphisms (SNPs) within the human G6PC2 gene are associated with variations in FBG (6,7). These GWAS data are consistent with the observation that global deletion of G6pc2 in mice on a mixed 129SvEv × C57BL/6J genetic background results in a ∼15% reduction in FBG levels compared with wild-type (WT) littermates (8). These observations raise the key question as to how G6pc2 modulates FBG. G6PC2, formerly known as IGRP, is primarily expressed in islet β-cells and encodes a glucose-6-phosphatase catalytic subunit isoform that catalyzes the hydrolysis of glucose-6-phosphate (G6P) to produce glucose and inorganic phosphate within the endoplasmic reticulum lumen (9,10).
Historically, the biological purpose and even existence of glucose-6-phosphatase activity in islets has been highly controversial (9,11,12). Although there is now general agreement that glucose-6-phosphatase activity is present in pancreatic islets, the issue as to whether the level of activity is enough to result in significant glucose cycling and hence affect glycolytic flux and glucose-stimulated insulin secretion (GSIS) and, therefore, be of physiological significance has remained unclear (13,14). To address this controversy over the importance of glucose-6-phosphatase activity in pancreatic islets, we have analyzed GSIS in G6pc2 knockout (KO) mice on a pure C57BL/6J genetic background. We provide evidence that G6pc2 acts as a novel, negative regulator of basal GSIS by hydrolyzing G6P and thereby opposing the action of the glucose sensor, glucokinase (15,16). This glucokinase/G6pc2 futile substrate cycle is predicted to reduce glycolytic flux and hence insulin secretion. Consistent with this model and human GWAS data, we show that a reduction in G6pc2 expression results in a leftward shift in the dose-response curve for GSIS such that under fasting conditions, blood glucose levels are reduced.
RESEARCH DESIGN AND METHODS
Animal care.
The animal housing and surgical facilities used for this study meet the standards set by the American Association for the Accreditation of Laboratory Animal Care standards. The Vanderbilt University Medical Center Animal Care and Use Committee approved all protocols used. Mice were maintained on either a standard rodent chow diet (calorie contributions: 28% protein, 12% fat, 60% carbohydrate [14% disaccharides]; LabDiet 5001; PMI Nutrition International) or a high-fat diet (calorie contributions: 15% protein, 59% fat, 26% carbohydrate [42% disaccharides]; Mouse Diet F3282; BioServ). High-fat–feeding studies were initiated at 8 weeks of age, and mice were maintained on the diet for 12 weeks. Food and water were provided ad libitum.
Generation of G6pc2 KO mice.
G6pc2 KO mice that had been generated on a mixed 129/SvEvBRD × C57BL/6J genetic background (8) were backcrossed onto a pure C57BL/6J genetic background using a speed congenic breeding strategy (17).
Radioisotopic glucose-6-phosphatase assay in permeabilized islets.
Isolated islets (∼100) were precipitated by centrifugation, resuspended in 20 μL 10% (weight for volume) sucrose in 10 mmol MES pH 6.5 buffer, and permeabilized by freeze/thawing. Glucose-6-phosphatase assays were performed for 2 h at 30°C in a final volume of 120 μL containing 4 mmol/L G6P, 150 mmol/L 2-glycerophosphate, 100 mmol/L MES pH 6.5, and 1 μCi [U-C14] G6P (262 mCi/mmol; Moravek Biochem, Brea, CA). A total of 200 μL 10% (weight for volume) sucrose in 10 mmol/L MES pH 6.5 was then added to each tube, the contents sonicated for 1 min (sonication water bath), and then mixed with 245 μL 1 mol/L ZnSO4 followed by 285 μL Ba(OH)2 and centrifugation at 20,000 g at 4°C for 5 min. A 300-μL sample of the supernatant was mixed with 3 mL Microscint scintillation fluid (PerkinElmer) and radioactivity determined by liquid scintillation counting. Enzyme activity was expressed as namomoles G6P hydrolyzed per minute per milligram cellular protein. Protein was determined by Bio-Rad dye binding assay (Bio-Rad) using BSA as standard.
Pancreas perfusions.
In situ pancreas perfusions were performed on ∼14-week-old mice following a 3-h fast according to the method of Bonnevie-Neilsen et al. (18), modified as previously described (19).
Intraperitoneal and oral glucose tolerance tests, insulin tolerance tests, islet isolations, and GSIS assays.
Intraperitoneal glucose tolerance tests (IPGTTs), oral glucose tolerance tests (OGTTs), insulin tolerance tests, and islet isolations for GSIS assays were performed on ∼13-week-old male mice as previously described (20).
Phenotypic analysis of fasted G6pc2 KO mice.
Mice were fasted for 5 h and then weighed. After an additional hour of fasting, mice were anesthetized using isofluorane, and blood samples were isolated from the retro-orbital venous plexus. Glucose concentrations were measured in whole blood using a glucose monitor (Accu-Chek Advantage; Roche, Indianapolis, IN). EDTA (5 μL; 0.5 mol/L) was then added to blood samples prior to isolation of plasma by centrifugation. Insulin samples were assayed using radioimmunoassay (21) by the Vanderbilt Diabetes Research and Training Center Hormone Assay & Analytical Services Core.
Measurement of cytoplasmic calcium.
Islet cytoplasmic calcium concentration was measured with the Ca2+-sensitive dye fura-2 acetoxymethyl ester (Molecular Probes, Eugene, OR) as previously described (22). Briefly, dispersed mouse islet cells were dye-loaded by incubation for 20 min at 37°C in a pH 7.3 solution containing 2 μmol/L fura-2, 119 mmol/L NaCl, 2.5 mmol/L CaCl2·[(H2O)6], 4.7 mmol/L KCl, 10 mmol/L HEPES, 1.2 mmol/L MgSO4, 1.2 mmol/L KH2PO4, and 5.6 mmol/L glucose. Fluorescence imaging was performed using a Nikon Eclipse TE2000-U microscope equipped with an epifluorescent illuminator (Sutter Instruments, Novato, CA), a CoolSNAP HQ2 camera (Photometrics, Tucson, AZ), and Nikon Elements software (Nikon, Tokyo, Japan). The cytoplasmic calcium concentration ratios of emitted fluorescence intensities at excitation wavelengths of 340 and 380 nm (F340/F380) were determined every 5 s with background subtraction. Cells were perifused at 37°C at a flow of 2 mL/min; the solutions used during the experiments were the same as the loading solution.
Statistical analyses.
Data were analyzed using a Student t test: two sample assuming equal variance. The level of significance was as indicated (two-sided Student t test).
RESULTS
Analysis of glucose-6-phosphatase activity in G6pc2 KO mouse islets in vitro.
Petrolonis et al. (10) have previously shown that G6PC2 can hydrolyze G6P. Consistent with this observation, Fig. 1A shows that glucose-6-phosphatase activity is reduced in islets isolated from G6pc2 KO mice. The level of glucose-6-phosphatase activity in islets (Fig. 1A) is much lower than in liver, again consistent with published data (9,11,12). Based on its ability to hydrolyze G6P, we hypothesized that G6pc2 opposes the action of the glucose sensor glucokinase, thereby creating a futile substrate cycle and reducing glycolytic flux, the ATP/ADP ratio, and hence insulin secretion (Fig. 1B). If correct, deletion of G6pc2 should result in a leftward shift in the dose-response curve for GSIS.
FIG. 1.

G6PC2: a negative regulator of basal GSIS. A: Glucose-6-phosphatase activity was compared in two independent islet preparations isolated from G6pc2 WT and KO mice as described in Research Design and Methods. The results show mean glucose-6-phosphatase activity ± SD. B: A simplified model for GSIS proposing the existence of a glucokinase/G6PC2 futile cycle. The best-characterized pathway for GSIS is shown, although other pathways clearly contribute (40). Overexpression of Gck increases glycolytic flux and results in a leftward shift in the S0.5 for GSIS (31,32). We hypothesize that G6PC2 is a negative regulator of basal GSIS such that a reduction in G6PC2 expression augments glycolytic flux and causes a leftward shift in the dose-response curve for GSIS. The G6P transporter (G6PT) is a G6P–Pi antiporter (41). ER, endoplasmic reticulum.
Analysis of GSIS from perfused G6pc2 KO mouse pancreata in situ.
To begin to assess our hypothesis, we measured insulin secretion in response to varying glucose concentrations from perfused pancreata in situ. When challenged with a 6.5 mmol/L submaximal concentration of glucose, the pancreata of G6pc2 KO mice secreted ∼2.5-fold more insulin than WT mouse pancreata (Fig. 2). A trend toward increased insulin secretion was also observed at 10 mmol/L glucose (Fig. 2). These data suggest that there is a leftward shift in the dose-response curve for GSIS in G6pc2 KO mice.
FIG. 2.

In situ perfused pancreas experiments demonstrate that G6pc2 deletion results in a leftward shift in the dose-response curve for GSIS. GSIS from perfused ∼14-week-old male WT and G6pc2 KO mouse pancreata was assayed in situ as described in Research Design and Methods. The results show the mean insulin concentrations ± SEM determined using three WT and five KO animals. *P < 0.05 vs. WT. Max., maximum.
Analysis of GSIS in isolated G6pc2 KO mouse islets in vitro.
To directly assess the role of G6pc2 in islet function, GSIS from isolated male WT and G6pc2 KO mouse islets was compared in static 30-min incubations. Islets were stimulated with submaximal glucose concentrations (5 and 11 mmol/L) to address the hypothesis that deletion of G6pc2 affects the dose-response curve for GSIS (Fig. 3A). GSIS from G6pc2 KO mouse islets was enhanced ∼2.2-fold at 5 mmol/L glucose (Fig. 3B) and ∼2.2-fold at 11 mmol/L glucose (Fig. 3C) relative to GSIS from WT mouse islets. No differences in insulin content were detected between WT and G6pc2 KO mouse islets (Fig. 3B and C). These observations are consistent with the in situ pancreas perfusion data (Fig. 2) and support the presence of a leftward shift in the dose-response curve for GSIS following G6pc2 deletion (Fig. 3A).
FIG. 3.

In vitro isolated islet experiments demonstrate that G6pc2 deletion results in a leftward shift in the dose-response curve for GSIS. A and D: Schematics to explain the effect of G6pc2 deletion on the dose-response curve for GSIS. The model in A proposes that at submaximal glucose concentrations, insulin secretion from G6pc2 KO mouse islets will be enhanced relative to WT islets due to a leftward shift in the dose-response curve for GSIS. The model in D proposes that at high glucose concentrations, flux through glucokinase is much greater than flux through G6pc2 such that deletion of G6pc2 does not affect the Vmax of GSIS. B, C, and E: GSIS from and insulin content in isolated ∼13-week-old male WT and G6pc2 KO mouse islets were assayed in vitro as described in Research Design and Methods following stimulation with 5 (B), 11 (C), or 16.7 mmol/L (E) glucose. The results in B, C, and E show the mean insulin concentrations ± SEM determined using six (at 5 mmol/L) or three (at 11 and 16.7 mmol/L) independent islet preparations. The experiments shown in B, C, and E were done at separate times and because the actual level of insulin secretion can vary significantly between different islet preparations the level of secretion between the graphs is not directly comparable. *P < 0.05 vs. WT. IEQ, islet equivalents.
Because the extent of organ perfusion is highly variable between individual pancreata, the data in Fig. 2 have to be expressed as a percentage of the insulin secretion observed in the presence of 16.7 mmol/L glucose and 20 mmol/L arginine. As such, the pancreas perfusion assay cannot provide information as to whether the absence of G6pc2 also affects the maximum rate (Vmax) of GSIS. We hypothesized that at high glucose concentrations, flux through glucokinase would be much greater than flux through G6pc2 such that deletion of G6pc2 would not affect the Vmax of GSIS (Fig. 3D). To address this hypothesis, islets were stimulated with a high glucose concentration (16.7 mmol/L). Fig. 3E shows that no difference in insulin secretion or insulin content was observed between WT and G6pc2 KO mouse islets at 16.7 mmol/L glucose. This observation suggests that the absence of G6pc2 does not affect the Vmax of GSIS.
Analysis of FBG in G6pc2 KO mice in vivo and basal cytoplasmic calcium in G6pc2 KO mouse islets in vitro.
If G6pc2 only affects the dose-response curve for GSIS, it would be predicted that, under fasting conditions, insulin levels should be identical regardless of the level of G6pc2 expression, and a leftward shift in the dose-response curve for GSIS should result in a reduction in FBG in G6pc2 KO mice in vivo (Fig. 4A). Fig. 4B shows that, following a 6- h fast, blood glucose levels were indeed reduced in both male and female G6pc2 KO mice relative to their WT littermates (16.4 and 14.4%, respectively), whereas fasting insulin levels were unchanged (Fig. 4C). This reduction in FBG is consistent with our previous data obtained with mixed genetic background mice (8). The absence of a difference in fasting insulin is consistent with the results of insulin tolerance tests showing little difference in insulin sensitivity between male WT and G6pc2 KO mice (Fig. 4D) and no differences in body weights between male mice (Fig. 4E). Similarly, human GWAS data show no association between G6PC2 and insulin sensitivity (23–26).
FIG. 4.

FBG is reduced in chow-fed G6pc2 KO mice. A: A schematic to explain the effect of G6pc2 deletion on FBG levels. The model proposes that in 6-h–fasted mice, in which insulin levels are the same in WT and G6pc2 KO mice (C), FBG is reduced in G6pc2 KO mice due to a leftward shift in the dose-response curve for GSIS relative to WT mice. B, C, and E: At 17 weeks of age, mice were fasted for 5 h and then weighed (E). Weight results are the mean ± SEM of data from the following number of animals: male WT, 31; male heterozygous (HET), 52: male KO, 17; female WT, 12; female HET, 37; and female KO, 11. One hour later, mice were anesthetized and blood isolated. Blood glucose (B) and plasma insulin (C) were determined as described in Research Design and Methods. Glucose results are the mean ± SEM of data from the following number of animals: male WT, 31; male HET, 52; male KO, 17; female WT, 12; female HET, 37; and female KO, 11. Insulin results are the mean ± SEM of data from the following number of animals: male WT, 29; male HET, 46; male KO, 14; female WT, 10; female HET, 35; and female KO, 11. D: Insulin tolerance tests using 0.75 units/kg insulin were performed on 5-h–fasted conscious ∼14-week-old WT and G6pc2 KO male mice as described in Research Design and Methods. The results show the mean glucose concentrations ± SEM determined using eight WT and seven KO animals. F and G: Cytoplasmic calcium levels at 5.6 mmol/L glucose were compared in dispersed cells generated from three independent WT and G6pc2 KO mouse islet preparations as described in Research Design and Methods. Results are the mean ± SEM of 288 measurements from WT cells and 236 measurements from KO cells. *P < 0.05 vs.WT. AUC, area under the curve; F, female; M, male.
Our model (Fig. 1B) predicts that this reduction in FBG resulting from the absence of G6pc2 should be associated with enhanced glycolytic flux and hence cytoplasmic calcium levels. Fig. 4F and G demonstrate that basal cytoplasmic calcium levels are enhanced in islets isolated from G6pc2 KO mice.
Overall, these observations are consistent with the in situ pancreas perfusion data (Fig. 2) and in vitro–isolated islet data (Fig. 3B and C) and further support the presence of a leftward shift in the dose-response curve for GSIS following G6pc2 deletion.
Analysis of the effect of G6pc2 deletion on body weight and composition.
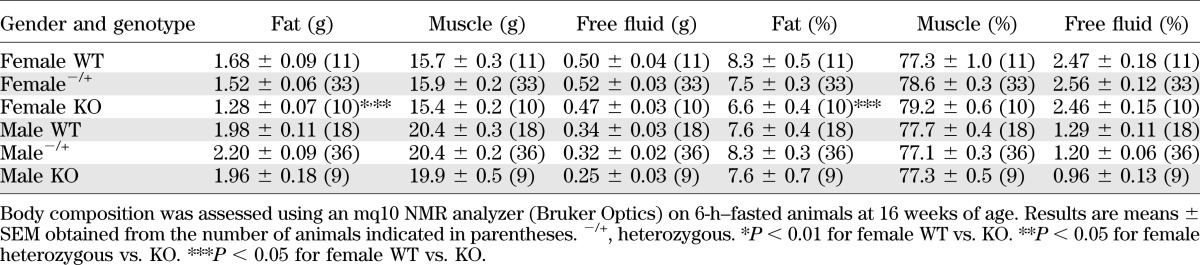
Human GWAS data show that the SNP within the G6PC2 gene (specifically the A allele of rs560887) that is associated with reduced fasting glycemia is also associated with reduced BMI and body fat (23). No differences in weight (Fig. 4E) or body fat (Table 1) were observed between chow-fed male G6pc2 KO mice and WT littermates (Fig. 4E) following a 6-h fast. In contrast, chow-fed female G6pc2 KO mice were slightly lighter than WT littermates (Fig. 4E) and had reduced body fat (Table 1).
TABLE 1.
Nuclear magnetic resonance analysis of chow-fed G6pc2 KO mouse body composition
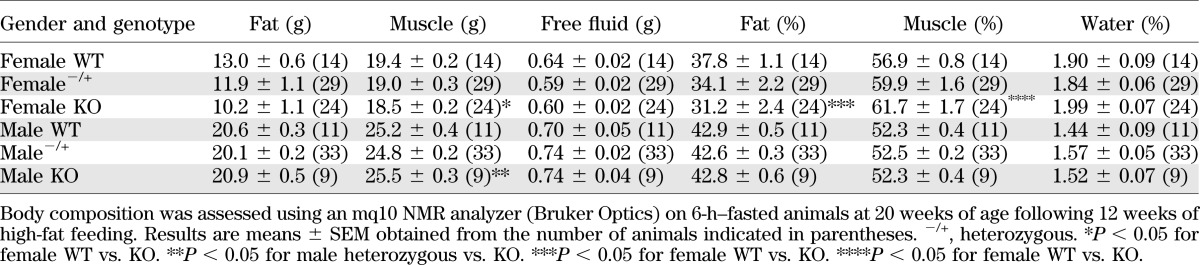
To further explore this difference in body fat, we investigated the effect of high-fat feeding, a standard nutritional challenge in the field of obesity and diabetes research that induces insulin resistance and is considered to model human disease (27). High-fat feeding was started at 8 weeks of age and continued for 12 weeks. Weekly measurements of body weight in nonfasted mice showed that female (Fig. 5A) but not male (Fig. 5B) G6pc2 KO mice were protected from diet-induced obesity, relative to WT and heterozygous mice. An analysis of body weights and fat composition following a 6-h fast confirmed that high-fat–fed female G6pc2 KO mice were lighter than WT littermates (Fig. 5C), and a difference in body fat was again apparent (Table 2). These results are consistent with the human GWAS data (23), at least with respect to female mice.
FIG. 5.

FBG is reduced in high-fat–fed G6pc2 KO mice. Starting at 8 weeks of age, female (A) and male (B) mice were fed a high-fat diet with nonfasting body weights measured weekly. Results are the mean ± SEM of data from the following number of animals: male WT, 14; male heterozygous (HET), 37; male KO, 9; female WT, 16; female HET, 29; and female KO, 23. C–E: At 21 weeks of age, mice were fasted for 5 h and then weighed (C). One hour later, mice were anesthetized and blood isolated. Weight results are the mean ± SEM of data from the following number of animals: male WT, 25; male HET, 36; male KO, 24; female WT, 17; female HET, 30; and female KO, 24. Blood glucose (D) and plasma insulin (E) were determined as described in Research Design and Methods. Glucose results are the mean ± SEM of data from the following number of animals: male WT, 25; male HET, 36; male KO, 23; female WT, 17; female HET, 30; and female KO, 24. Insulin results are the mean ± SEM of data from the following number of animals: male WT, 25; male HET, 36; male KO, 23; female WT, 16; female HET, 30; and female KO, 24. *P < 0.05 vs. WT. F, female; M, male.
TABLE 2.
Nuclear magnetic resonance analysis of high-fat–fed G6pc2 KO mouse body composition
FBG (Fig. 5D) and plasma insulin (Fig. 5E) were both markedly elevated in high-fat–fed mice compared with chow-fed animals (Fig. 4B and C), consistent with the presence of insulin resistance. In high-fat–fed mice there was no difference in fasting plasma insulin between WT and G6pc2 KO mice (Fig. 5E), whereas FBG was higher in WT mice (Fig. 5D), again consistent with the chow-fed mouse data (Fig. 4B and C) and a leftward shift in the dose-response curve for GSIS.
Analysis of GSIS in G6pc2 KO mice in vivo during glucose tolerance tests.
Glucose tolerance tests were performed to determine the effect of G6pc2 deletion on insulin secretion in mice in vivo following a glucose challenge. Because G6pc2 deletion resulted in no change in GSIS in isolated islets at high glucose concentrations (Fig. 3E), we hypothesized that no difference in glucose tolerance or insulin secretion would be detected between G6pc2 KO mice and WT mice following a challenge with a high glucose concentration. IPGTTs were performed to explore this hypothesis. Following a 6-h fast, male WT and KO mice were injected with a 2.0 g/kg body weight dose of glucose, and blood glucose was then measured over a 120-min period. Fig. 6A shows that surprisingly a slight impairment in glucose tolerance was observed in G6pc2 KO mice relative to WT mice, but only at one time point. We next directly measured insulin secretion 15 min following intraperitoneal injection of 2.0 g/kg glucose. Fig. 6B–D demonstrate that no difference in insulin secretion was detected between G6pc2 KO mice relative to WT mice. This observation is consistent with isolated islet experiments that showed no difference in insulin secretion between WT and G6pc2 KO mouse islets at high glucose concentrations (Fig. 3E).
FIG. 6.

G6pc2 deletion has complex effects on glucose tolerance and insulin secretion in vivo. IPGTTs using 2.0 g/kg glucose (A–D), 0.4 g/kg glucose (E–H), or 0.75 g/kg glucose (I–L) and OGTTs using 2.0 g/kg glucose (M–P) were performed on 6-h–fasted conscious WT and G6pc2 KO male mice as described in Research Design and Methods. The results show the mean glucose or insulin concentrations ± SEM. The number of animals used in experiments in which only glucose was measured were as follows: A: 15 WT, 12 KO; E: 10 WT, 9 KO; I: 12 WT, 13 KO; and M: 11 WT, 10 KO. The number of animals used in experiments in which both glucose and insulin were measured were as follows: B–D: 12 WT, 15 KO; F–H: 9 WT, 6 KO; J–L: 12 WT, 16 KO; and N–P: 10 WT, 10 KO. *P < 0.05 vs. WT. AUC, area under the curve.
Because G6pc2 deletion resulted in improved GSIS in perfused pancreata (Fig. 2) and in isolated islets (Fig. 3) at submaximal glucose concentrations, we hypothesized that glucose tolerance would be improved in the G6pc2 KO mice relative to WT mice following a challenge with a moderate glucose concentration due to enhanced insulin secretion. IPGTTs were performed to explore this hypothesis. Following a 6-h fast, male WT and KO mice were injected with a 0.4 g/kg body weight dose of glucose, and blood glucose was then measured over a 90-min period. Fig. 6E shows that a slight improvement in glucose tolerance was observed in the G6pc2 KO mice relative to WT mice. We next directly measured insulin secretion 15 min following intraperitoneal injection of 0.4 g/kg glucose. Fig. 6F–H show that no difference in insulin secretion was detected between G6pc2 KO mice and WT mice. When IPGTTs were repeated using a higher 0.75 g/kg body weight dose of glucose, no differences in glucose tolerance or insulin secretion were detected between G6pc2 KO and WT mice (Figs. 6I–L).
OGTTs were performed to further explore the effect of G6pc2 deletion on insulin secretion in vivo. Following a 6-h fast, male WT and G6pc2 KO mice were challenged with a 2.0 g/kg body weight dose of glucose administered by oral gavage, and blood glucose was then measured over a 120-min period. Fig. 6M shows that a small improvement in glucose tolerance was observed in the G6pc2 KO mice relative to WT mice. We next directly measured insulin secretion 15 min following oral gavage with 2.0 g/kg glucose. Fig. 6N–P show that no difference in insulin secretion was detected between G6pc2 KO mice and WT mice. Overall, these data show that deletion of G6pc2 has surprisingly little effect on glucose tolerance or insulin secretion in vivo. Potential explanations for the absence of a change in insulin secretion are discussed below; however, the absence of marked changes in glucose tolerance is consistent with human GWAS data showing no association between G6PC2 and glucose tolerance (23–26), and it suggests that G6pc2 primarily regulates FBG in vivo.
DISCUSSION
In the current study, we have investigated the effect of a global deletion of G6pc2 in vivo, a mouse model that is directly relevant to the control of FBG in humans (6,7). Data from in situ pancreas perfusion (Fig. 2), in vitro isolated islet (Fig. 3), and in vivo fasting studies (Fig. 4) demonstrate that deletion of G6pc2 in C57BL/6J mice results in a leftward shift in the dose-response curve for GSIS. This results in increased insulin secretion at submaximal glucose concentrations (Figs. 2 and 3) and reduced blood glucose levels during fasting (Fig. 4). Importantly, these observations are consistent with human GWAS data in which the rs560887-A and rs13431652-G alleles, which are predicted to reduce G6PC2 expression, were found to associate with reduced FBG levels (6,7,28). These data are also consistent with a model in which G6PC2 opposes the action of glucokinase, the β-cell glucose sensor (15,16), by hydrolyzing G6P (Fig. 1). This would create a futile cycle, thereby reducing glycolytic flux, the ATP/ADP ratio, and hence GSIS. To support this conclusion, future studies will directly compare glucose cycling and glycolytic flux in WT and G6pc2 KO mouse islets. The latter will be important because the consumption of ATP by glucokinase in β-cells is negligible when compared with ATP consumption by Na+/K+ ATPase and other ATPase pumps (29). As such, we hypothesize that the absence of G6pc2 affects ATP levels in β-cells by enhancing glycolytic flux (Fig. 1) rather than ATP consumption by glucokinase. This model is consistent with the concept that glucose phosphorylation rather than transport is the major control point in GSIS (15). This conclusion is supported by studies demonstrating that a reduction in glucose transporter 2 by >90% is required to affect GSIS (30). In contrast, overexpression of glucokinase increases GSIS (31,32), while reducing glucokinase expression impairs GSIS (33). Although overexpression of G6pc2 leads to diabetes, this is likely due to the induction of endoplasmic reticulum stress rather than a direct effect on the kinetics of GSIS (34).
Human GWAS data show that G6PC2 is also associated with altered body fat and BMI (23). Both chow-fed (Table 1 and Fig. 4E) and high-fat–fed (Table 2 and Fig. 5C) female G6pc2 KO mice show reduced body fat and body weight, consistent with the human GWAS data (23). The reason why similar differences are not observed in male mice is unclear. Of greater interest will be future studies exploring how deletion of G6pc2, which is selectively expressed in islets (9), could impact body fat and body weight.
Neither IPGTTs nor OGTTs show major differences in glucose tolerance or insulin secretion between WT and G6pc2 KO mice over a range of glucose concentrations (Fig. 6). These observations were surprising given the expectation from the perfused pancreas (Fig. 2) and isolated islet (Fig. 3) data that increased insulin secretion at submaximal glucose concentrations would result in improved glucose tolerance in G6pc2 KO mice. However, these observations are consistent with human GWAS data that show no association between G6PC2 and glucose tolerance (23–26). These data are not explained by a difference in insulin sensitivity in G6pc2 KO mice (Fig. 4D). Similarly, G6PC2 is not associated with variations in insulin sensitivity in humans (23–26). Although these data suggest that G6pc2 primarily regulates FBG in vivo, the key issue is why insulin secretion is not enhanced in G6pc2 KO mice during submaximal glucose tolerance tests. Insight into this question is provided by human GWAS data showing that the SNP within the G6PC2 gene (specifically the A allele of rs560887) that is associated with reduced fasting glycemia is also associated with a paradoxical reduction in insulin secretion during glucose tolerance tests (23–26). This observation is paradoxical for two reasons. First, based on its association with reduced fasting glycemia it was expected that the A allele of rs560887 would be associated with increased rather than reduced insulin secretion in vivo. Second, as mentioned above, despite the association with reduced insulin secretion in vivo, this SNP is not associated with a counterbalancing change in glucose tolerance or insulin sensitivity (23–26). These data suggest that G6pc2 not only opposes the action of glucokinase but also influences other aspects of β-cell function and GSIS. The observed reduction in insulin secretion during glucose tolerance tests in humans has been hypothesized to indicate that either G6PC2 affects the pulsatility of insulin secretion (23) or that it affects hepatic glucose production rather than β-cell function (26). The latter explanation appears less likely since human G6PC2 (6,35) and mouse G6pc2 (9,36) are only expressed in islets and not in liver. A similar reduction in insulin secretion was not observed in G6pc2 KO mice during glucose tolerance tests (Fig. 6), but this may simply be due to the relatively low number of animals analyzed (n = 6–16) relative to the vast number of humans analyzed in GWAS studies (n > 5,000) (23) and the fact that significant variations in insulin sensitivity, and hence insulin secretion, are observed even within inbred C57BL/6J mice (37). Based on these observations in humans and mice, future studies will be designed to assess whether the absence of G6pc2 influences other aspects of β-cell function such as pulsatile insulin secretion.
The severe diabetic phenotype observed in mice with a β-cell–specific deletion of the Gck gene (38) contrasts with the mild phenotype of G6pc2 KO mice (Table 1), suggesting that glucokinase is the major determinant of glycolytic flux with G6pc2 playing a modulatory role. Whether the same applies in humans is unclear since mutations resulting in the activation or inactivation of G6PC2 have not been reported. In contrast, multiple glucokinase mutations have been identified in humans, and the data clearly indicate a major role for glucokinase in glucose sensing (39). Thus, heterozygous inactivating mutations in glucokinase are one cause of maturity-onset diabetes of the young, which is characterized by mild fasting hyperglycemia, whereas homozygous inactivating glucokinase mutations result in permanent neonatal diabetes mellitus, which is characterized by severe hyperglycemia (39). In contrast, glucokinase-activating mutations result in hyperinsulinemia, leading to hypoglycemia (39).
In conclusion, the data presented in this study show that G6pc2 acts as a negative regulator of basal GSIS. However, the utility of regulating the set point for basal GSIS remains unresolved. The function of G6pc2 is clearly not to block insulin secretion during fasting (Fig. 4), and although the regulation of FBG levels is important for disease risk, the diseases involved are those of old age and therefore not subject to evolutionary pressure. This suggests that perhaps controlling FBG is more important for other aspects of metabolism than appreciated. We have also considered the possibility that, although G6pc2 is not required for blocking insulin secretion under basal conditions, activation of G6pc2 might be important for blocking insulin secretion under specific physiological conditions (for example, during exercise or in the fetal state). Future studies will examine these ideas.
ACKNOWLEDGMENTS
L.D.P. was supported by the Vanderbilt Molecular Endocrinology Training Program Grant 5T32 DK-07563. Research in the laboratory of D.A.J. was supported by National Institutes of Health (NIH) Grants DK-081666 and DK-20593. Research in the laboratory of J.C.H. was supported by NIH Grant R56-DK-052068, a Juvenile Diabetes Research Foundation Autoimmunity Prevention Center grant, and the Barbara Davis Center Diabetes and Endocrinology Research Center (P30 DK-57516). Research in the laboratory of O.P.M. was supported by NIH Grants DK-043748 and DK-078188. Research in the laboratory of M.S. was supported by NIH Grant DK-060667. The Vanderbilt Hormone Assay & Analytical Services Core and the Vanderbilt Islet Procurement & Analysis Core are both supported by NIH Grant P60 DK-20593 to the Vanderbilt Diabetes Research and Training Center and NIH Grant DK-59637 to the Vanderbilt Mouse Metabolic Phenotyping Center. Research in the laboratory of R.M.O. was supported by NIH Grant DK-92589.
No potential conflicts of interest relevant to this article were reported.
L.D.P. took the primary lead on all in vivo mouse studies and wrote the paper. J.K.O. and Y.W. assisted with in vivo mouse studies. T.P.O. assisted with pancreas perfusion studies. C.J.F. and P.K.D. assisted with isolated islet calcium studies. D.A.J. assisted with isolated islet calcium studies and wrote the paper. J.C.H. and O.P.M. provided input on multiple aspects of these studies and wrote the paper. M.S. performed pancreas perfusion studies and wrote the paper. R.M.O. took the primary role directing studies and wrote the paper. R.M.O. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank P. Jacobson (Abbott Pharmaceuticals) for assistance with the initiation of this project. The authors also thank Erin Stewart and Randall Wong (Barbara Davis Center for Childhood Diabetes, Anschutz Medical Center, University of Colorado Denver, Aurora, CO) for help with the speed congenic backcrossing procedure; W. Snead, G. Poffenberger, and B. Trivedi (Vanderbilt University) for performing insulin and glucagon assays; and M. Brissova and A. Coldren (Vanderbilt University) for performing islet isolations and GSIS analyses. The authors also thank Pierre Pirot (Barbara Davis Center for Childhood Diabetes) for help with the glucose-6-phosphatase assays.
REFERENCES
- 1.Abdul-Ghani MA, DeFronzo RA. Plasma glucose concentration and prediction of future risk of type 2 diabetes. Diabetes Care 2009;32(Suppl. 2):S194–S198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khaw KT, Wareham N, Luben R, et al. Glycated haemoglobin, diabetes, and mortality in men in Norfolk cohort of european prospective investigation of cancer and nutrition (EPIC-Norfolk). BMJ 2001;322:15–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawes CM, Parag V, Bennett DA, et al. Asia Pacific Cohort Studies Collaboration Blood glucose and risk of cardiovascular disease in the Asia Pacific region. Diabetes Care 2004;27:2836–2842 [DOI] [PubMed] [Google Scholar]
- 4.Coutinho M, Gerstein HC, Wang Y, Yusuf S. The relationship between glucose and incident cardiovascular events. A metaregression analysis of published data from 20 studies of 95,783 individuals followed for 12.4 years. Diabetes Care 1999;22:233–240 [DOI] [PubMed] [Google Scholar]
- 5.DECODE Study Group, European Diabetes Epidemiology Group Is the current definition for diabetes relevant to mortality risk from all causes and cardiovascular and noncardiovascular diseases? Diabetes Care 2003;26:688–696 [DOI] [PubMed] [Google Scholar]
- 6.Bouatia-Naji N, Rocheleau G, Van Lommel L, et al. A polymorphism within the G6PC2 gene is associated with fasting plasma glucose levels. Science 2008;320:1085–1088 [DOI] [PubMed] [Google Scholar]
- 7.Chen WM, Erdos MR, Jackson AU, et al. Variations in the G6PC2/ABCB11 genomic region are associated with fasting glucose levels. J Clin Invest 2008;118:2620–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Martin CC, Oeser JK, et al. Deletion of the gene encoding the islet-specific glucose-6-phosphatase catalytic subunit-related protein autoantigen results in a mild metabolic phenotype. Diabetologia 2007;50:774–778 [DOI] [PubMed] [Google Scholar]
- 9.Hutton JC, O’Brien RM. Glucose-6-phosphatase catalytic subunit gene family. J Biol Chem 2009;284:29241–29245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petrolonis AJ, Yang Q, Tummino PJ, et al. Enzymatic characterization of the pancreatic islet-specific glucose-6-phosphatase-related protein (IGRP). J Biol Chem 2004;279:13976–13983 [DOI] [PubMed] [Google Scholar]
- 11.Ashcroft SJ, Randle PJ. Glucose-6-phosphatase activity of mouse pancreatic islets. Nature 1968;219:857–858 [DOI] [PubMed] [Google Scholar]
- 12.van Schaftingen E, Gerin I. The glucose-6-phosphatase system. Biochem J 2002;362:513–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan A, Chandramouli V, Ostenson CG, et al. Evidence for the presence of glucose cycling in pancreatic islets of the ob/ob mouse. J Biol Chem 1989;264:9732–9733 [PubMed] [Google Scholar]
- 14.Sweet IR, Najafi H, Li G, Grodberg J, Matschinsky FM. Measurement and modeling of glucose-6-phosphatase in pancreatic islets. Am J Physiol 1997;272:E696–E711 [DOI] [PubMed] [Google Scholar]
- 15.Matschinsky FM. Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes 1996;45:223–241 [DOI] [PubMed] [Google Scholar]
- 16.Iynedjian PB. Molecular physiology of mammalian glucokinase. Cell Mol Life Sci 2009;66:27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serreze DV, Chapman HD, Varnum DS, et al. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new “speed congenic” stock of NOD.Ig mu null mice. J Exp Med 1996;184:2049–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonnevie-Nielsen V, Steffes MW, Lernmark A. A major loss in islet mass and B-cell function precedes hyperglycemia in mice given multiple low doses of streptozotocin. Diabetes 1981;30:424–429 [DOI] [PubMed] [Google Scholar]
- 19.Shiota C, Rocheleau JV, Shiota M, Piston DW, Magnuson MA. Impaired glucagon secretory responses in mice lacking the type 1 sulfonylurea receptor. Am J Physiol Endocrinol Metab 2005;289:E570–E577 [DOI] [PubMed] [Google Scholar]
- 20.Pound LD, Sarkar SA, Ustione A, et al. The physiological effects of deleting the mouse SLC30A8 gene encoding zinc transporter-8 are influenced by gender and genetic background. PLoS One 2012;7:e40972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgan CR, Lazarow AL. Immunoassay of insulin: two antibody system: plasma insulin of normal, subdiabetic, and diabetic rats. Diabetes 1963;12:115–126 [Google Scholar]
- 22.Jacobson DA, Weber CR, Bao S, Turk J, Philipson LH. Modulation of the pancreatic islet beta-cell-delayed rectifier potassium channel Kv2.1 by the polyunsaturated fatty acid arachidonate. J Biol Chem 2007;282:7442–7449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X, Shu YH, Xiang AH, et al. Additive effects of genetic variation in GCK and G6PC2 on insulin secretion and fasting glucose. Diabetes 2009;58:2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rose CS, Grarup N, Krarup NT, et al. A variant in the G6PC2/ABCB11 locus is associated with increased fasting plasma glucose, increased basal hepatic glucose production and increased insulin release after oral and intravenous glucose loads. Diabetologia 2009;52:2122–2129 [DOI] [PubMed] [Google Scholar]
- 25.Ingelsson E, Langenberg C, Hivert MF, et al. MAGIC investigators Detailed physiologic characterization reveals diverse mechanisms for novel genetic Loci regulating glucose and insulin metabolism in humans. Diabetes 2010;59:1266–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heni M, Ketterer C, t’Hart LM, et al. The impact of genetic variation in the G6PC2 gene on insulin secretion depends on glycemia. J Clin Endocrinol Metab 2010;95:E479–E484 [DOI] [PubMed] [Google Scholar]
- 27.Young GS, Kirkland JB. Rat models of caloric intake and activity: relationships to animal physiology and human health. Appl Physiol Nutr Metab 2007;32:161–176 [DOI] [PubMed] [Google Scholar]
- 28.Bouatia-Naji N, Bonnefond A, Baerenwald DA, et al. Genetic and functional assessment of the role of the rs13431652-A and rs573225-A alleles in the G6PC2 promoter that are strongly associated with elevated fasting glucose levels. Diabetes 2010;59:2662–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fridlyand LE, Ma L, Philipson LH. Adenine nucleotide regulation in pancreatic beta-cells: modeling of ATP/ADP-Ca2+ interactions. Am J Physiol Endocrinol Metab 2005;289:E839–E848 [DOI] [PubMed] [Google Scholar]
- 30.Tal M, Wu YJ, Leiser M, et al. [Val12] HRAS downregulates GLUT2 in beta cells of transgenic mice without affecting glucose homeostasis. Proc Natl Acad Sci USA 1992;89:5744–5748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, Iynedjian PB. Modulation of glucose responsiveness of insulinoma beta-cells by graded overexpression of glucokinase. Proc Natl Acad Sci USA 1997;94:4372–4377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H, Iynedjian PB. Acute glucose intolerance in insulinoma cells with unbalanced overexpression of glucokinase. J Biol Chem 1997;272:25731–25736 [DOI] [PubMed] [Google Scholar]
- 33.Efrat S, Leiser M, Wu YJ, et al. Ribozyme-mediated attenuation of pancreatic beta-cell glucokinase expression in transgenic mice results in impaired glucose-induced insulin secretion. Proc Natl Acad Sci USA 1994;91:2051–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shameli A, Yamanouchi J, Thiessen S, Santamaria P. Endoplasmic reticulum stress caused by overexpression of islet-specific glucose-6-phosphatase catalytic subunit-related protein in pancreatic Beta-cells. Rev Diabet Stud 2007;4:25–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin CC, Bischof LJ, Bergman B, et al. Cloning and characterization of the human and rat islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) genes. J Biol Chem 2001;276:25197–25207 [DOI] [PubMed] [Google Scholar]
- 36.Arden SD, Zahn T, Steegers S, et al. Molecular cloning of a pancreatic islet-specific glucose-6-phosphatase catalytic subunit-related protein. Diabetes 1999;48:531–542 [DOI] [PubMed] [Google Scholar]
- 37.Koza RA, Nikonova L, Hogan J, et al. Changes in gene expression foreshadow diet-induced obesity in genetically identical mice. PLoS Genet 2006;2:e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Postic C, Shiota M, Niswender KD, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem 1999;274:305–315 [DOI] [PubMed] [Google Scholar]
- 39.Osbak KK, Colclough K, Saint-Martin C, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat 2009;30:1512–1526 [DOI] [PubMed] [Google Scholar]
- 40.Jensen MV, Joseph JW, Ronnebaum SM, Burgess SC, Sherry AD, Newgard CB. Metabolic cycling in control of glucose-stimulated insulin secretion. Am J Physiol Endocrinol Metab 2008;295:E1287–E1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen SY, Pan CJ, Nandigama K, Mansfield BC, Ambudkar SV, Chou JY. The glucose-6-phosphate transporter is a phosphate-linked antiporter deficient in glycogen storage disease type Ib and Ic. FASEB J 2008;22:2206–2213 [DOI] [PubMed] [Google Scholar]