

Abstract
The normal β-cell response to obesity-associated insulin resistance is hypersecretion of insulin. Type 2 diabetes develops in subjects with β-cells that are susceptible to failure. Here, we investigated the time-dependent gene expression changes in islets of diabetes-prone db/db and diabetes-resistant ob/ob mice. The expressions of adaptive unfolded protein response (UPR) genes were progressively induced in islets of ob/ob mice, whereas they declined in diabetic db/db mice. Genes important for β-cell function and maintenance of the islet phenotype were reduced with time in db/db mice, whereas they were preserved in ob/ob mice. Inflammation and antioxidant genes displayed time-dependent upregulation in db/db islets but were unchanged in ob/ob islets. Treatment of db/db mouse islets with the chemical chaperone 4-phenylbutyric acid partially restored the changes in several β-cell function genes and transcription factors but did not affect inflammation or antioxidant gene expression. These data suggest that the maintenance (or suppression) of the adaptive UPR is associated with β-cell compensation (or failure) in obese mice. Inflammation, oxidative stress, and a progressive loss of β-cell differentiation accompany diabetes progression. The ability to maintain the adaptive UPR in islets may protect against the gene expression changes that underlie diabetes development in obese mice.
The critical contribution of deficient insulin secretion to the pathogenesis of type 2 diabetes is beyond doubt (1–3). The normal β-cell response to excess nutrient and obesity-associated insulin resistance is hypersecretion of insulin that maintains blood glucose levels within the normal range. This is associated with both expansion of β-cell mass and enhanced β-cell function such that the amount of insulin secreted per given unit of β-cell mass is increased (2,3). Normoglycemia can be maintained for decades in obese subjects with robust β-cells that cope with this increased demand and sustain the compensatory response. Mechanisms for β-cell compensation have been proposed (2,3), but there has been little exploratory investigation. Type 2 diabetes only develops in subjects unable to sustain the β-cell compensatory response. This is associated with a progressive deterioration of β-cell function, particularly impairment of glucose-stimulated insulin secretion (GSIS), and a loss of β-cell mass through an increased rate of apoptosis (1–5). Thus, type 2 diabetes arises in subjects with islets that are susceptible to dysfunction and apoptosis under stressful conditions. The current knowledge of the molecular differences between robust and susceptible β-cells is poor.
The db/db and ob/ob mouse models of obesity have been cornerstones in research of the mechanisms of insulin resistance and β-cell failure (6,7). In these models, the absence of leptin signaling results in similar hyperphagia, obesity, hyperlipidemia, and insulin resistance. However, opposing predisposition to diabetes development is displayed by these mice when bred on the C57BL/6J or C57BL/KsJ background. The ob/ob mouse on the C57BL/6J background strain exhibits resistance to diabetes because of successful β-cell compensation, whereas the db/db mouse on the C57BL/KsJ background strain displays time-dependent progression to overt diabetes because of the failure of β-cell compensation. The differences in β-cell phenotype (propensity to compensation or failure) are revealed only in the setting of obesity and insulin resistance. We have used these mouse models of obesity with opposing disposition to development of diabetes to study the mechanisms of β-cell compensation (diabetes-resistant ob/ob mice) and failure (diabetes-prone db/db mice).
Endoplasmic reticulum (ER) stress has been proposed as a mechanism for β-cell dysfunction and death in type 2 diabetes (8–11). ER stress activates a signaling cascade known as the unfolded protein response (UPR), which has roles alleviating the ER stress through the upregulation of ER chaperones and folding enzymes and, paradoxically, activating apoptosis via deleterious UPR signaling if the stress is too severe or prolonged. We found, unexpectedly, that the presence of ER stress in islets was not unique to the model of β-cell failure. Rather, ER stress was indicated in islets of both diabetes-resistant ob/ob and diabetes-prone db/db mice. However, the models differed in the pattern of the ER stress response, whereas the adaptive UPR was progressively upregulated with β-cell compensation, it declined with β-cell failure. We also tested the influence of improving the chaperone activity of the ER on the gene expression changes that were found exclusively in islets of diabetes-prone db/db mice, namely the upregulation of inflammation and oxidative stress gene expression and the progressive loss of β-cell differentiation.
RESEARCH DESIGN AND METHODS
Mice.
C57BL/KsJ db/db and C57BL/6J ob/ob mice and their age-matched lean control mice (C57BL/KsJ or C57BL/6J, respectively) were taken from the Garvan Institute breeding colonies. Animals were kept under conventional conditions with free access to food and water. All procedures were approved by the Garvan Institute/St. Vincent’s Hospital Animal Experimentation Ethics Committee and followed guidelines issued by the National Health and Medical Research Council of Australia. To assess the time course changes in islet gene expression, mice were studied at 6 and 16 weeks of age. Blood samples were taken via tail prick for measurement of glucose levels. Blood collected in EDTA via a terminal heart bleed was used for measurement of plasma insulin, triglyceride, and nonesterified fatty acid levels. Mice were anesthetized and their islets were isolated with liberase RI (Roche) digestion of the pancreas. Islets were further separated with a Ficoll-Paque PLUS gradient (Amersham Biosciences) and handpicked under a stereomicroscope. Immediately after collection, islets were used for extraction of RNA.
Metabolic studies and assays.
Intraperitoneal glucose tolerance tests (2 g/kg glucose) were performed in conscious mice after 6 h of fasting. Blood glucose was measured using an Accu-Chek Performa glucose monitor (Roche Diagnostics, Castle Hill, Australia). Plasma insulin was measured using ELISA (Crystal Chem, Downers Grove, IL). Plasma triglyceride was measured using an enzymatic colorimetric method (GPO-PAP reagent; Roche Diagnostics). Plasma nonesterified fatty acid was measured by an acyl-CoA oxidase–based colorimetric method (Wako Pure Chemical Industries, Osaka, Japan).
Insulin secretion assay.
Isolated islets were washed in Krebs-Ringer HEPES buffer (containing 5 mmol/L NaHCO3, 1 mmol/L CaCl2, 2.8 mmol/L glucose, 10 mmol/L HEPES, and 0.1% BSA). Groups of five islets, with three to four replicates per animal, were incubated for 1 h at 37°C in Krebs-Ringer HEPES buffer containing 2.8 or 16.7 mmol/L glucose. Insulin was measured in an aliquot of the buffer by radioimmunoassay (Millipore, Billerica, MA).
Treatment of islets.
Isolated islets from lean nondiabetic C57BL/KsJ control and obese diabetic db/db mice were cultured in RPMI supplemented with 0.2 mmol/L glutamine, 10% heat-inactivated FBS 100 units/mL penicillin, and 100 μg/mL streptomycin at 37°C. Islets were treated with 4-phenylbutyric acid (PBA; 2.5 mmol/L; Sigma, St. Louis, MO) or trimethylamine N-oxide (TMAO; 100 mmol/L) for 24 h. Isolated islets from C57BL/6J control and ob/ob mice were treated with salubrinal (75 μmol/L; Merck, Kilsyth, Victoria, Australia) for 24 h. Insulin secretion was assessed and RNA was extracted for gene expression analysis.
RNA analysis.
Total RNA was extracted using RNeasy Mini Kit (Qiagen, Doncaster, Australia) and cDNA was synthesized using the QuantiTect Reverse Transcription Kit (Qiagen, Victoria, Australia). Real-time PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) on a 7900HT Real-Time PCR System (Applied Biosystems). Primer sequences are provided in Supplementary Table 1. The value obtained for each specific product was normalized to a control gene (cyclophilin A) and expressed as a fold change of the value in control extracts. Xbp1 splicing was assessed as previously described (12).
Statistical analysis.
All results are presented as means ± SEM. Statistical analyses were performed using unpaired two-tailed Student's t test or ANOVA.
RESULTS
Metabolic characteristics at 6 and 16 weeks of age.
The db/db and ob/ob mice displayed increased body weight at both 6 and 16 weeks of age as compared with their respective age-matched control mice (Supplementary Figure 1A and B). At 6 weeks of age, blood glucose levels were not different among the groups (Supplementary Fig. 1C and D). Thus, db/db and ob/ob mice were nondiabetic at 6 weeks of age, although both groups displayed impaired glucose tolerance compared with their respective controls (Supplementary Fig. 2A and B). At 16 weeks of age, blood glucose levels were elevated in db/db mice but remained unaltered in ob/ob mice (Supplementary Fig. 1C and D). Plasma nonesterified fatty acid levels were unchanged at 6 weeks but tended to be increased at 16 weeks of age in both db/db and ob/ob mice (Supplementary Fig. 1E and F). There was a tendency for increased plasma triglyceride levels in db/db and ob/ob mice at both 6 and 16 weeks of age (Supplementary Fig. 1G and H). Plasma insulin levels were increased in db/db and ob/ob mice at both 6 and 16 weeks of age (Supplementary Fig. 1I and J). In ob/ob mice, plasma insulin levels were significantly higher at 16 weeks compared with 6 weeks of age (Supplementary Fig. 1J). In islets isolated from db/db mice, GSIS was enhanced at 6 weeks but was reduced at 16 weeks compared with islets from age-matched control mice (Fig. 1A). In islets isolated from ob/ob mice, GSIS was enhanced at 6 weeks but was unaltered at 16 weeks compared with their age-matched controls (Fig. 1B). Having established these metabolic indices, we next sought to measure the expression of islet-associated transcription factors, genes involved in β-cell function, as well as stress-response genes, in islets from 6- and 16-week-old obese db/db and ob/ob mice compared with their respective lean controls.
FIG. 1.
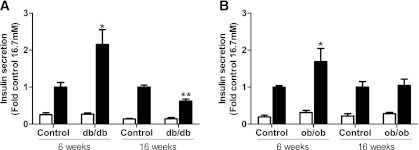
Glucose-stimulated insulin secretion ex vivo in islets isolated from C57BL/KsJ control and db/db mice (A), and C57BL/6J control and ob/ob mice (B) at 6 and 16 weeks of age. Batches of islets were cultured in Krebs-Ringer HEPES buffer containing 0.1% BSA and 2.8 mmol/L (white bars) or 16.7 mmol/L glucose (black bars) for 1 h. Insulin was measured in an aliquot of the buffer by radioimmunoassay. Insulin secretion was expressed as fold change of the level in age-matched control islets cultured in 16.7 mmol/L glucose. All results are mean ± SEM. n ≥ 4 in each group. *P < 0.05, **P < 0.01 genotype effect in each age group.
Changes in levels of islet-associated transcription factor mRNA.
After normalization of the gene of interest to a control gene (cyclophilin A), mRNA levels in db/db islets were quantitated as fold change of their respective age-matched lean C57BL/KsJ control mice. The expression levels of several transcription factors important for islet development and the maintenance of β-cell differentiation were altered in db/db islets. Beta2 and MafA mRNA levels were significantly reduced at 6 weeks of age (Fig. 2A). Thus, the downregulation of Beta2 and MafA preceded the onset of diabetes in db/db mice. Beta2 and MafA mRNA levels were further reduced at 16 weeks of age, showing a significant time-dependent effect (P < 0.05 for each gene). Pdx1 and Nkx6.1 mRNA levels were unchanged at 6 weeks but were significantly reduced at 16 weeks in db/db islets (Fig. 2A). Thus, the downregulation of Pdx1 and Nkx6.1 was associated with diabetes in db/db mice. We also tested Id1 (inhibitor of differentiation/DNA binding), which was identified recently as a negative regulator of insulin secretion and β-cell gene expression (13). Id1 mRNA levels were increased at both 6 and 16 weeks of age in db/db islets. We also assessed changes in the expression of islet-associated transcription factors in islets of obese ob/ob mice compared with their age-matched lean C57BL/6J control mice (Fig. 2B). No significant difference was observed in the mRNA levels of Beta2, MafA, Pdx1, Nkx6.1, and Id1 at both 6 and 16 weeks of age in ob/ob islets, although there was a tendency for reduced expression of Beta2 and MafA (Fig. 2B). For all genes tested in this study, expression levels in the C57BL/6J and C57BL/KsJ control islets were similar.
FIG. 2.

Changes in mRNA expression of islet-associated transcription factors and genes that optimize β-cell function in islets of db/db and ob/ob mice at 6 and 16 weeks of age. Islets were isolated from 9 to 10 C57BL/KsJ control and 8 db/db mice at 6 weeks of age, 13 to 16 C57BL/KsJ control and 9 to 12 db/db mice at 16 weeks of age, 6 C57BL/6J control and 6 ob/ob mice at 6 weeks of age, and 4 to 5 C57BL/6J control and 7 ob/ob mice at 16 weeks of age. Total RNA was extracted, reverse-transcribed, and analyzed by real-time RT-PCR. mRNA levels were expressed as fold change of the levels in respective age-matched controls (represented by the dashed line). Shown are changes for the indicated genes in islets of db/db (A, C, E) and ob/ob (B, D, F) mice at 6 (white bars) and 16 (black bars) weeks of age. All results are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 genotype effect in each age group.
mRNA levels of β-cell function genes.
Several genes involved in β-cell glucose metabolism were evaluated (Fig. 2C and D). The glucose transporter, Glut2, the anaplerotic enzyme, pyruvate carboxylase (PC), the rate-limiting enzyme of the glycerol-phosphate shuttle, mitochondrial glycerol phosphate dehydrogenase (mGPDH), and the enzyme responsible for the majority of β-cell glucose phosphorylation, glucokinase (Gk), were downregulated in islets of db/db mice. Glut2 and PC were significantly reduced in db/db islets at 6 weeks and were reduced further at 16 weeks (Fig. 2C). The downregulation of mGPDH and Gk was observed only at 16 weeks (Fig. 2C). Thus, the expression of β-cell glucose metabolism genes showed a tendency for a time-dependent deterioration in db/db islets. In ob/ob islets, Glut2, PC, and mGPDH mRNA levels were significantly reduced at 6 weeks (Fig. 2D). However, in contrast to db/db islets, the mRNA levels of glucose metabolism genes did not reduce further with time in ob/ob islets. Moreover, we failed to detect a difference in Gk mRNA levels at both ages in ob/ob islets (Fig. 2D).
The expression of Kir6.2, the pore-forming subunit of the ATP-sensitive K+ channel also was evaluated (Fig. 2E and F). The mRNA levels of Kir6.2 were significantly reduced in db/db mice at 6 weeks, and they were reduced further at 16 weeks (Fig. 2E). In contrast, Kir6.2 mRNA levels were not significantly altered in ob/ob islets, although there was a tendency for reduced expression at 6 weeks (Fig. 2F). The G-protein-coupled receptor GPR40 may play a role in both fatty acid and glucose stimulation of insulin secretion (14–16). In db/db islets, Gpr40 mRNA levels were unchanged at 6 weeks but were significantly reduced at 16 weeks (Fig. 2E). In ob/ob islets, no significant difference was detected in Gpr40 mRNA levels, although there was a tendency for reduced expression at 6 weeks (Fig. 2F). A similar pattern of expression was observed for the incretin GLP-1 receptor, Glp1r; mRNA levels were unchanged at 6 weeks but reduced at 16 weeks in db/db islets (Fig. 2E), whereas we failed to measure a difference in ob/ob islets (Fig. 2F). The GIP receptor, Gipr, was reduced at both 6 and 16 weeks in db/db islets (Fig. 2E). There was a nonsignificant tendency for reduced mRNA expression of Gipr in ob/ob islets (Fig. 2F). These results suggest that in contrast to the progressive downregulation of genes important for β-cell function in db/db islets, transcript levels are maintained better with time in ob/ob islets.
The proportion of β-cells within islets was similar in 16-week-old control and diabetic db/db mice (control: 86.4 ± 2.0%; db/db: 88.6 ± 0.9%; n = 3 in each group). Thus, the changes in gene expression observed in islets of diabetic db/db mice are indicative of β-cell dedifferentiation rather than β-cell loss. This is consistent with studies suggesting the importance of β-cell dedifferentiation as a mechanism of diabetic β-cell failure (3,7,17–19).
mRNA levels of UPR genes.
The role of cellular stress and stress-response mediators in the failure of β-cells in diabetes has been the subject of much recent attention (9,20). We previously reported the upregulation of several ER stress genes in islets of diabetic db/db mice (10). Here, prediabetic db/db mice showed significant upregulation of adaptive (BiP, p58, Erp72, Fkbp11, and Grp94; Fig. 3A) and deleterious (Atf3, Chop, and Trib3; Fig. 3C) UPR genes. Interestingly, expression levels of the adaptive UPR genes were higher at 6 weeks than at 16 weeks, showing a reduction with time in db/db islets (Fig. 3A). In ob/ob islets, expression of adaptive UPR genes were increased at 6 weeks and, in contrast to db/db islets, they were maintained or increased further at 16 weeks (Fig. 3B). Furthermore, the deleterious ER stress genes were not induced in ob/ob islets (Fig. 3D). The transcription factor XBP1 regulates the expression of many adaptive UPR genes (21,22). In concert with the changes in the adaptive UPR, the level of Xbp1 splicing (activation) was increased in ob/ob and prediabetic db/db mouse islets, and it was lowered in diabetic db/db mice (Fig. 3E and F). These differential responses provide the first evidence that the maintenance (or suppression) of the adaptive UPR is associated with β-cell compensation (or failure) in obese mice.
FIG. 3.

Changes in mRNA expression of UPR genes in islets of db/db and ob/ob mice at 6 and 16 weeks of age. Islets were isolated from 9 to 10 C57BL/KsJ control and 8 db/db mice at 6 weeks of age, 13 to 16 C57BL/KsJ control and 9 to 12 db/db mice at 16 weeks of age, 6 C57BL/6J control and 6 ob/ob mice at 6 weeks of age, and 4 to 5 C57BL/6J control and 7 ob/ob mice at 16 weeks of age. Shown are changes for the indicated genes in islets of db/db (A, C, E) and ob/ob (B, D, F) mice at 6 (white bars) and 16 (black bars) weeks of age. A–D: Total RNA was extracted, reverse-transcribed, and analyzed by real-time RT-PCR. mRNA levels were expressed as fold change of the levels in respective age-matched controls (represented by the dashed line). E and F: Xbp1 cDNA was amplified by PCR and digested with PstI, which cuts unprocessed Xbp1 into fragments. Processed (activated) Xbp1 lacks the restriction site and remains intact. Processed (intact) and unprocessed (cut) Xbp1 were quantified by densitometry. The value obtained for processed Xbp1 was expressed as a ratio of the total (processed + unprocessed) Xbp1 mRNA level for each sample. These ratios are expressed as fold change of the ratio in respective age-matched controls. All results are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 genotype effect in each age group.
mRNA levels of antioxidant genes.
Oxidative stress has been proposed as a mechanism for gluco-lipotoxicity of β-cells (20,23). We assessed the expression of oxidative stress–inducible genes. mRNA levels of the antioxidant enzymes heme oxygenase-1 (HO-1), glutathione peroxidase (GPx), catalase, and superoxide dismutase 1 (Sod1) were upregulated in islets of db/db mice; time-dependent increases were displayed for HO-1, GPx, and catalase (Fig. 4A). In contrast, we failed to detect differences in the antioxidant genes tested in ob/ob islets (Fig. 4B).
FIG. 4.

Changes in mRNA expression of antioxidant and inflammation genes in islets of db/db and ob/ob mice at 6 and 16 weeks of age. Islets were isolated from 9 to 10 C57BL/KsJ control and 8 db/db mice at 6 weeks of age, 13 to 16 C57BL/KsJ control and 9 to 12 db/db mice at 16 weeks of age, 6 C57BL/6J control and 6 ob/ob mice at 6 weeks of age, and 4 to 5 C57BL/6J control and 7 ob/ob mice at 16 weeks of age. Total RNA was extracted, reverse-transcribed, and analyzed by real-time RT-PCR. mRNA levels were expressed as fold change of the levels in respective age-matched controls (represented by the dashed line). Shown are changes for the indicated genes in islets of db/db (A, C, E) and ob/ob (B, D, F) mice at 6 (white bars) and 16 (black bars) weeks of age. All results are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 genotype effect in each age group.
mRNA levels of inflammation genes.
Emerging evidence implicates systemic and tissue inflammation with obesity and type 2 diabetes (24,25). Here, time-dependent increases in the expression of chemokines and cytokines were observed in db/db islets (Fig. 4C and E). At 6 weeks, the expressions of IL-6, Cxcl1, and Ccl2 were significantly increased. Minor nonsignificant increases were observed for IL-1β (P = 0.09) and TNF-α (P = 0.17), but these are worth highlighting because of the interest of low-grade inflammation in type 2 diabetes islets (24). At 16 weeks, the expression of all the tested chemokines and cytokines were markedly increased in db/db islets (Fig. 4C and E), as were the mRNA levels for CD68, a commonly used marker for macrophages (Fig. 4E). In complete contrast, the expressions of these inflammatory genes were unaltered in ob/ob islets at 6 and 16 weeks of age (Fig. 4D and F). These data provide the first demonstration of an escalating inflammatory response in association with β-cell failure in diabetes-prone db/db mice, juxtaposed with an absence of islet inflammation in association with successful β-cell compensation in obese diabetes-resistant ob/ob mice. Furthermore, mRNA levels of proapoptotic genes, Bax and Bak1, were selectively increased in islets of db/db mice (Fig. 4E and F).
Changes in gene expression in islets of C57BL/6J db/db mice.
We also evaluated gene expression in islets of 6-week-old C57BL/6J db/db mice compared with control C57BL/6J (Supplementary Figs. 3–5). In islets of C57BL/6J db/db mice, the adaptive UPR genes were upregulated (Supplementary Fig. 4), whereas differences in oxidative stress and inflammatory gene expression were not detected (Supplementary Fig. 5). These results are broadly consistent with the findings in ob/ob mice, demonstrating that the different background strains, rather than the ob (Lep) and db (Lepr) mutations, are critically important for the β-cell response to obesity in these models.
Enhanced signaling downstream of PERK/eIF2α does not affect the changes in gene expression in islets of ob/ob mice.
We next examined the effects of enhancing signaling downstream of PERK/eIF2α on the mRNA changes in islets of obese ob/ob mice. Isolated islets from ob/ob mice were treated with salubrinal, an inhibitor of eIF2α dephosphorylation (26). As shown in Fig. 5A, salubrinal treatment led to the expected increase in Chop expression in ob/ob islets. However, salubrinal treatment had no effect on the changes in the expression of islet-associated transcription factors and genes that optimize β-cell function in ob/ob islets (Fig. 5A). Moreover, salubrinal treatment did not affect GSIS, which was increased in ob/ob islets (Fig. 5B). These findings suggest that the induction of signaling downstream of PERK/eIF2α is insufficient for the loss of β-cell phenotype that accompanies β-cell decompensation.
FIG. 5.

Effect of enhancing signaling downstream of PERK/eIF2α on the changes in gene expression and insulin secretion in islets of ob/ob mice. A: Islets isolated from ob/ob and age-matched control mice were cultured in the absence (control [cont], white bars; ob/ob, black bars) or presence (ob/ob+S, striped bars) of salubrinal (75 μmol/L) for 24 h. Total RNA was extracted, reverse-transcribed, and analyzed by real-time RT-PCR. mRNA levels were expressed as fold change of the level in control islets. n = 3–6 in each group. B: Batches of islets were cultured in Krebs-Ringer HEPES buffer containing 0.1% BSA and 2.8 mmol/L (white bars) or 16.7 mmol/L glucose (black bars) for 1 h. Insulin was measured in an aliquot of the buffer by radioimmunoassay. Insulin secretion was expressed as fold change of the level in control islets cultured in 16.7 mmol/L glucose. n = 3–6 in each group. All results are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 genotype effect; †P < 0.05 salubrinal treatment effect in ob/ob mouse islets.
Effect of chemical chaperone treatment on the changes in gene expression in islets of db/db mice.
We next examined the effects of chemical chaperone treatment on the mRNA changes in islets of diabetic db/db mice. PBA acts as a chemical chaperone in the ER by improving folding capacity and trafficking (12,27). As shown in Fig. 6A, PBA treatment of db/db islets partially restored the abundance of islet-associated transcription factors toward the levels apparent in islets from nondiabetic control mice. The lowered mRNA levels for Pdx1, MafA, and Nkx6.1 in untreated db/db islets were significantly reversed toward normal in PBA-treated db/db islets (Fig. 6A). Beta2 mRNA levels displayed a tendency for restoration toward normal in PBA-treated db/db islets. PBA treatment of db/db islets also led to the partial restoration toward normal of several genes involved in β-cell glucose metabolism and function. The reduced mRNA levels of PC and mGPDH in untreated db/db islets were significantly restored toward normal in PBA-treated db/db islets (Fig. 6A). In contrast, PBA treatment had no effect on the reduced expression of Kir6.2 or Gpr40 in db/db islets (Fig. 6A). However, Glp1r mRNA levels were normalized in PBA-treated db/db islets to levels equivalent to nondiabetic control mouse islets (Fig. 6A). These changes in gene expression were associated with increased insulin secretion in PBA-treated db/db islets (Fig. 6B). Furthermore, these changes were associated with upregulation of the adaptive UPR and XBP1 splicing in PBA-treated db/db islets (Fig. 7). The data demonstrate that improving the adaptive UPR gene expression in db/db islets leads to the partial recovery of β-cell gene expression and insulin secretion.
FIG. 6.

Effect of chemical chaperone treatment on the changes in gene expression and insulin secretion in islets of db/db mice. A: Islets isolated from diabetic db/db and age-matched nondiabetic control [cont] mice (12–14 weeks of age) were cultured in the absence (control, white bars; db/db, black bars) or presence (db/db+P, striped bars) of the chemical chaperone PBA (2.5 mmol/L) for 24 h. Total RNA was extracted, reverse-transcribed, and analyzed by real-time RT-PCR. mRNA levels were expressed as fold change of the level in control islets. n = 7 in each group. B: Batches of islets were cultured in Krebs-Ringer HEPES buffer containing 0.1% BSA and 2.8 mmol/L (white bars) or 16.7 mmol/L glucose (black bars) for 1 h. Insulin was measured in an aliquot of the buffer by radioimmunoassay. Insulin secretion was expressed as fold change of the level in control islets cultured in 16.7 mmol/L glucose. n = 3 in each group. All results are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 genotype effect; †P < 0.05, ††P < 0.01 PBA treatment effect in db/db mouse islets.
FIG. 7.

Effect of chemical chaperone treatment on the expression of adaptive UPR genes in islets of db/db mice. Islets isolated from diabetic db/db and age-matched nondiabetic control (cont) mice (12–14 weeks of age) were cultured in the absence (control, white bars; db/db, black bars) or presence (db/db, striped bars) of the chemical chaperone PBA (2.5 mmol/L) for 24 h. Total RNA was extracted, reverse-transcribed, and analyzed by real-time RT-PCR. mRNA levels were expressed as fold-change of the level in control islets. Xbp1 splicing was analyzed and expressed as described in Fig. 3. All results are mean ± SEM. n = 7 in each group. *P < 0.05, **P < 0.01, ***P < 0.001 genotype effect; †P < 0.05, ††P < 0.01 PBA treatment effect in db/db mouse islets.
We examined the effects of another chemical chaperone, TMAO. No significant differences were observed in islet-associated gene expression or insulin secretion in TMAO-treated db/db islets compared with untreated db/db islets (Supplementary Fig. 6). In association with this, TMAO treatment had no effect on adaptive UPR gene expression in db/db islets (Supplementary Fig. 7). These findings suggest that the attenuation of ER stress alone is insufficient to induce improvements in gene expression and insulin secretion in db/db islets. Taken together, the data suggest that suppression of the adaptive UPR makes a necessary contribution to abnormalities of the β-cell phenotype in diabetes-prone mice.
We also tested the effects of PBA treatment on the changes in antioxidant and inflammatory stress gene expression in db/db islets (Supplementary Fig. 8). The higher levels of HO-1 and GPx mRNA in db/db islets were not affected by PBA treatment. Similarly, the improvement in chaperone activity was without effect on inflammatory cytokines or chemokines expression; the higher mRNA levels of IL-6, IL-1β, Cxcl1, and TNF-α were not different between untreated and PBA-treated db/db islets (Supplementary Fig. 8).
DISCUSSION
Here, we have used mouse models of obesity with opposing disposition to develop diabetes to study the mechanisms of β-cell compensation and failure. The data demonstrate that “robust” β-cells of diabetes-resistant ob/ob mice and “susceptible” β-cells of diabetes-prone db/db mice display several striking differences with time. Robust β-cells display a sustained adaptive UPR, a preserved β-cell phenotype, and an absence of apoptotic, inflammatory, or oxidative stress gene expression. In contrast, susceptible β-cells display a decline in the adaptive UPR, a progressive loss of β-cell differentiation, and the presence of apoptotic, inflammatory, and oxidative stress gene expression. The change in phenotype of susceptible β-cells is partially reversed by PBA treatment, suggesting that suppression of the adaptive UPR contributes to the loss of β-cell differentiation. These findings raise the possibility that the maintenance of the adaptive UPR provides a molecular link between obesity-associated insulin resistance and β-cell compensation and, conversely, that suppression of the adaptive UPR, together with inflammation, oxidative stress, and the loss of β-cell differentiation, underlies β-cell failure and progression to diabetes (Fig. 8).
FIG. 8.

Proposed mechanisms contributing to β-cell compensation and failure during progression to type 2 diabetes. Obesity and its associated metabolic changes including hyperlipidemia, glucose intolerance, and increased insulin demand lead to ER stress in pancreatic β-cells. In normal β-cells, upregulation of the adaptive UPR facilitates the enhancement of ER capacity, maintenance of β-cell compensation, and prevention of diabetes. In genetically susceptible β-cells, suppression of ER adaptation together with oxidative stress and inflammation leads to the loss of β-cell phenotype and increased β-cell death that ultimately results in type 2 diabetes.
Mechanisms of β-cell compensation.
β-cell compensation for obesity-induced insulin resistance likely involves both increased β-cell mass and enhanced insulin secretion per β-cell (2,3). Interestingly, a partial loss of β-cell differentiation was observed in islets from 6-week-old ob/ob mice as well as in prediabetic db/db mice. This may be reflective of increased proliferation and its inverse relationship with differentiation (28,29). However, the β-cell phenotype in ob/ob islets recovers rather than deteriorates with time. Perhaps β-cell mass eventually equilibrates with increased insulin demand as a result of enhanced proliferation in the early stages combined with an absence of apoptotic signaling. GSIS was enhanced in ob/ob and db/db mice at 6 weeks of age, and this was unexpectedly associated with increased expression of adaptive UPR genes. This suggests that ER stress is an early response to obesity, but also that some features of the UPR are beneficial, helping with β-cell adaptation in the compensation for obesity-associated insulin resistance. This is generally consistent with findings of UPR activation in obese nondiabetic human pancreata (30) and of the requirement of an intact UPR for normal β-cell function and glucose homeostasis in humans and mice (11,31). The maintenance of the adaptive UPR under conditions of obesity may facilitate the ongoing requirement for enhanced rates of insulin processing and secretion. Our findings suggest that XBP1 activation may play a role in the regulation of the adaptive UPR under these conditions, although coordination with the other arms of the ER stress response is likely. Alteration of XBP1 activation alone has profound effects on β-cell gene expression, insulin secretion, and apoptosis (32,33).
Mechanisms of β-cell failure.
ER stress appears sufficient to cause β-cell failure (11,31,34–36), and its potential role in the pathogenesis of type 2 diabetes is well-supported (9,10,30,37). However, our findings suggest that the presence of ER stress activation is, in itself, not an indicator of β-cell failure; rather, the nature of the subsequent response is critical in the regulation of β-cell function and survival. The expression of several proapoptotic ER stress genes, including Chop (12,37,38), Atf3 (39,40), and Trib3 (41), have been implicated in β-cell dysfunction and death, whereas upregulation of the adaptive UPR may confer protection from β-cell failure and diabetes (11,42). Importantly, the regulation of this process is not solely dependent on obesity, hyperlipidemia, or increased insulin demand because these parameters were similar in db/db and ob/ob mice. Therefore, the factors leading to the decline in XBP1 activation and suppression of the adaptive UPR in diabetic db/db mice remain unknown. Hyperglycemia may play a role (43–47), although previous studies suggest that elevated glucose levels increase rather than reduce XBP1 splicing and adaptive UPR gene expression. Interestingly, our recent studies demonstrate that inflammatory cytokines exert an inhibitory effect on XBP1 and the adaptive UPR in β-cells (48). Therefore, the low-grade inflammation observed in islets of type 2 diabetes models may influence β-cell function and survival via regulation of the pattern of downstream UPR signaling. Recent studies also demonstrate complex interactions between the UPR and oxidative stress in β-cells (37,46,49).
Our studies support and extend the case for increasing chaperone capacity of the ER as a therapeutic approach for type 2 diabetes (11,50). Chaperone treatment in vivo protects against insulin resistance (27) and β-cell dysfunction (46,51). Our studies demonstrate that part of the benefit of improving the chaperone activity of the ER in diabetes includes partial recovery in the expression of genes important for the maintenance of β-cell differentiation and function. Because our studies were performed ex vivo, the effects are likely direct and not secondary to improvements in glucose tolerance (27,51).
Our studies also support the roles of inflammation and oxidative stress as potential contributors to β-cell failure. Both features were completely absent in the robust islets of diabetes-resistant mice but were progressively exacerbated in susceptible islets. Their precise contributions remain to be clarified, and complex interactions between inflammation, oxidative stress, and ER stress are probable and of likely importance (37,40,46,48,49).
Interestingly, many genes found to be regulated by the UPR previously were reported to be dependent on hyperglycemia in db/db islets (7). In particular, the islet-associated transcription factors and selective genes important for β-cell function commonly are regulated by the UPR and hyperglycemia. The divergent regulation of other genes may have been influenced by the different time course of analyses, 2-week (7) and 24-h treatments for hyperglycemia and the UPR, respectively.
In conclusion, our study demonstrates for the first time the markedly different patterns of time-dependent changes in islet gene expression in obese mouse models of β-cell compensation and β-cell failure. The unique gene expression patterns of adaptive UPR, inflammation, oxidative stress, and β-cell differentiation in these models provide important insight into the regulation of β-cell function and survival under conditions of obesity. The role of the adaptive UPR in the protection against diabetes has been largely overlooked in studies investigating the association between deleterious UPR signaling with β-cell dysfunction and apoptosis. Thus, our study is the first to link failure of the adaptive UPR in islets of obese mice with abnormalities in β-cell gene expression and progression to diabetes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the National Health and Medical Research Council (NHMRC) of Australia. M.B. was supported by a Post-doctoral Fellowship from the Société Francophone du Diabète (SFD), Paris, France.
No potential conflicts of interest relevant to this article were reported.
J.Y.C. designed and performed experiments and wrote the manuscript. J.L. and M.B. performed experiments and reviewed the manuscript. T.J.B. contributed to discussion and reviewed the manuscript. D.R.L. conceived, designed and performed experiments and wrote the manuscript. D.R.L. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-0701/-/DC1.
REFERENCES
- 1.Kahn SE, Zraika S, Utzschneider KM, Hull RL. The beta cell lesion in type 2 diabetes: there has to be a primary functional abnormality. Diabetologia 2009;52:1003–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest 2006;116:1802–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weir GC, Marselli L, Marchetti P, Katsuta H, Jung MH, Bonner-Weir S. Towards better understanding of the contributions of overwork and glucotoxicity to the beta-cell inadequacy of type 2 diabetes. Diabetes Obes Metab 2009;11(Suppl 4):82–90 [DOI] [PubMed] [Google Scholar]
- 4.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003;52:102–110 [DOI] [PubMed] [Google Scholar]
- 5.Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 2008;10(Suppl 4):32–42 [DOI] [PubMed] [Google Scholar]
- 6.Shafrir E, Ziv E, Mosthaf L. Nutritionally induced insulin resistance and receptor defect leading to beta-cell failure in animal models. Ann N Y Acad Sci 1999;892:223–246 [DOI] [PubMed] [Google Scholar]
- 7.Kjørholt C, Akerfeldt MC, Biden TJ, Laybutt DR. Chronic hyperglycemia, independent of plasma lipid levels, is sufficient for the loss of beta-cell differentiation and secretory function in the db/db mouse model of diabetes. Diabetes 2005;54:2755–2763 [DOI] [PubMed] [Google Scholar]
- 8.Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem 2012;81:767–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 2008;29:42–61 [DOI] [PubMed] [Google Scholar]
- 10.Laybutt DR, Preston AM, Akerfeldt MC, et al. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007;50:752–763 [DOI] [PubMed] [Google Scholar]
- 11.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev 2008;29:317–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akerfeldt MC, Howes J, Chan JY, et al. Cytokine-induced beta-cell death is independent of endoplasmic reticulum stress signaling. Diabetes 2008;57:3034–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akerfeldt MC, Laybutt DR. Inhibition of Id1 augments insulin secretion and protects against high-fat diet-induced glucose intolerance. Diabetes 2011;60:2506–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kebede M, Alquier T, Latour MG, Semache M, Tremblay C, Poitout V. The fatty acid receptor GPR40 plays a role in insulin secretion in vivo after high-fat feeding. Diabetes 2008;57:2432–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kebede MA, Alquier T, Latour MG, Poitout V. Lipid receptors and islet function: therapeutic implications? Diabetes Obes Metab 2009;11(Suppl 4):10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alquier T, Peyot ML, Latour MG, et al. Deletion of GPR40 impairs glucose-induced insulin secretion in vivo in mice without affecting intracellular fuel metabolism in islets. Diabetes 2009;58:2607–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bensellam M, Laybutt DR, Jonas JC. The molecular mechanisms of pancreatic β-cell glucotoxicity: Recent findings and future research directions. Mol Cell Endocrinol 2012;364:1–27 [DOI] [PubMed] [Google Scholar]
- 18.Jonas JC, Sharma A, Hasenkamp W, et al. Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J Biol Chem 1999;274:14112–14121 [DOI] [PubMed] [Google Scholar]
- 19.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012;150:1223–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev 2008;29:351–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 2003;23:7448–7459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshida H, Matsui T, Hosokawa N, Kaufman RJ, Nagata K, Mori K. A time-dependent phase shift in the mammalian unfolded protein response. Dev Cell 2003;4:265–271 [DOI] [PubMed] [Google Scholar]
- 23.Laybutt DR, Kaneto H, Hasenkamp W, et al. Increased expression of antioxidant and antiapoptotic genes in islets that may contribute to beta-cell survival during chronic hyperglycemia. Diabetes 2002;51:413–423 [DOI] [PubMed] [Google Scholar]
- 24.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 2011;11:98–107 [DOI] [PubMed] [Google Scholar]
- 25.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol 2011;29:415–445 [DOI] [PubMed] [Google Scholar]
- 26.Cnop M, Ladriere L, Hekerman P, et al. Selective inhibition of eukaryotic translation initiation factor 2 alpha dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic beta-cell dysfunction and apoptosis. J Biol Chem 2007;282:3989–3997 [DOI] [PubMed] [Google Scholar]
- 27.Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006;313:1137–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fleischer N, Chen C, Surana M, et al. Functional analysis of a conditionally transformed pancreatic beta-cell line. Diabetes 1998;47:1419–1425 [DOI] [PubMed] [Google Scholar]
- 29.Laybutt DR, Weir GC, Kaneto H, et al. Overexpression of c-Myc in beta-cells of transgenic mice causes proliferation and apoptosis, downregulation of insulin gene expression, and diabetes. Diabetes 2002;51:1793–1804 [DOI] [PubMed] [Google Scholar]
- 30.Huang CJ, Lin CY, Haataja L, et al. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007;56:2016–2027 [DOI] [PubMed] [Google Scholar]
- 31.Delépine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet 2000;25:406–409 [DOI] [PubMed] [Google Scholar]
- 32.Allagnat F, Christulia F, Ortis F, et al. Sustained production of spliced X-box binding protein 1 (XBP1) induces pancreatic beta cell dysfunction and apoptosis. Diabetologia 2010;53:1120–1130 [DOI] [PubMed] [Google Scholar]
- 33.Lee AH, Heidtman K, Hotamisligil GS, Glimcher LH. Dual and opposing roles of the unfolded protein response regulated by IRE1alpha and XBP1 in proinsulin processing and insulin secretion. Proc Natl Acad Sci USA 2011;108:8885–8890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scheuner D, Song B, McEwen E, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 2001;7:1165–1176 [DOI] [PubMed] [Google Scholar]
- 35.Kaufman RJ, Back SH, Song B, Han J, Hassler J. The unfolded protein response is required to maintain the integrity of the endoplasmic reticulum, prevent oxidative stress and preserve differentiation in β-cells. Diabetes Obes Metab 2010;12(Suppl 2):99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fonseca SG, Ishigaki S, Oslowski CM, et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J Clin Invest 2010;120:744–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest 2008;118:3378–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cunha DA, Hekerman P, Ladrière L, et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J Cell Sci 2008;121:2308–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanabe K, Liu Y, Hasan SD, et al. Glucose and fatty acids synergize to promote B-cell apoptosis through activation of glycogen synthase kinase 3β independent of JNK activation. PLoS ONE 2011;6:e18146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zmuda EJ, Viapiano M, Grey ST, Hadley G, Garcia-Ocaña A, Hai T. Deficiency of Atf3, an adaptive-response gene, protects islets and ameliorates inflammation in a syngeneic mouse transplantation model. Diabetologia 2010;53:1438–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liew CW, Bochenski J, Kawamori D, et al. The pseudokinase tribbles homolog 3 interacts with ATF4 to negatively regulate insulin exocytosis in human and mouse beta cells. J Clin Invest 2010;120:2876–2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quan W, Hur KY, Lim Y, et al. Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia 2012;55:392–403 [DOI] [PubMed] [Google Scholar]
- 43.Greenman IC, Gomez E, Moore CE, Herbert TP. Distinct glucose-dependent stress responses revealed by translational profiling in pancreatic beta-cells. J Endocrinol 2007;192:179–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elouil H, Bensellam M, Guiot Y, et al. Acute nutrient regulation of the unfolded protein response and integrated stress response in cultured rat pancreatic islets. Diabetologia 2007;50:1442–1452 [DOI] [PubMed] [Google Scholar]
- 45.Lipson KL, Fonseca SG, Ishigaki S, et al. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab 2006;4:245–254 [DOI] [PubMed] [Google Scholar]
- 46.Tang C, Koulajian K, Schuiki I, et al. Glucose-induced beta cell dysfunction in vivo in rats: link between oxidative stress and endoplasmic reticulum stress. Diabetologia 2012;55:1366–1379 [DOI] [PubMed] [Google Scholar]
- 47.Zhang L, Lai E, Teodoro T, Volchuk A. GRP78, but not protein-disulfide isomerase, partially reverses hyperglycemia-induced inhibition of insulin synthesis and secretion in pancreatic beta-cells. J Biol Chem 2009;284:5289–5298 [DOI] [PubMed] [Google Scholar]
- 48.Chan JY, Cooney GJ, Biden TJ, Laybutt DR. Differential regulation of adaptive and apoptotic unfolded protein response signalling by cytokine-induced nitric oxide production in mouse pancreatic beta cells. Diabetologia 2011;54:1766–1776 [DOI] [PubMed] [Google Scholar]
- 49.Back SH, Scheuner D, Han J, et al. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab 2009;10:13–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Engin F, Hotamisligil GS. Restoring endoplasmic reticulum function by chemical chaperones: an emerging therapeutic approach for metabolic diseases. Diabetes Obes Metab 2010;12(Suppl 2):108–115 [DOI] [PubMed] [Google Scholar]
- 51.Xiao C, Giacca A, Lewis GF. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans. Diabetes 2011;60:918–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.