

Abstract
Insulin and proinsulin are normally produced only by the pancreas and thymus. We detected in diabetic rodents the presence of extra pancreatic proinsulin-producing bone marrow-derived cells (PI-BMDCs) in the BM, liver, and fat. In mice and rats with diabetic neuropathy, we also found proinsulin-producing cells in the sciatic nerve and neurons of the dorsal root ganglion (DRG). BM transplantation experiments using genetically marked donor and recipient mice showed that the proinsulin-producing cells in the DRG, which morphologically resemble neurons, are actually polyploid proinsulin-producing fusion cells formed between neurons and PI-BMDCs. Additional experiments indicate that diabetic neuropathy is not simply the result of nerve cells being damaged directly by hyperglycemia. Rather, hyperglycemia induces fusogenic PI-BMDCs that travel to the peripheral nervous system, where they fuse with Schwann cells and DRG neurons, causing neuronal dysfunction and death, the sine qua non for diabetic neuropathy. Poorly controlled diabetes is indeed bad to the bone.
Keywords: diabetic complications, proinsulin, bone marrow-derived cells, cell fusion
Introduction
The worldwide diabetes epidemic has shown no signs of abatement. Its prevalence has more than doubled since 1980, increasing from an estimated 153 million, three decades ago to about 347 million in 2008.1 Diabetic people suffer much morbidity and premature mortality because of chronic diabetic complications, which include cardiovascular and cerebrovascular disease, nephropathy, retinopathy, and neuropathy. Multiple pathogenic effectors downstream of hyperglycemia contribute to chronic diabetic complications.2–14 Diabetic neuropathy is the most common diabetic complication, afflicting over 50% of all diabetics, and is the leading cause of nontraumatic limb amputations. The biochemical perturbations that underlie diabetic neuropathy are very similar to those of other complications; they include oxidative stress,4,15 activation of the polyol pathway,7 increased advanced glycation end products and their receptors,15 activation of protein kinase C (PKC)9 and mitogen-activated protein kinases (MAPK),10 and inducible nitric oxide synthase.16 Furthermore, hypoxia and ischemia,6 elevated cytokines, such as tumor necrosis factor (TNF)-α 17 and IL-6,18 and nerve growth factor (NGF) deficiency19 also play important etiologic roles in diabetic neuropathy.
The consensus in the field is that in diabetes these dysregulated metabolic pathways produce malfunction, injury, and death of cells that are intrinsic to the peripheral nervous system, that is, Schwann cells, vasa nervorum, and dorsal root ganglia (DRG) neurons. Recent studies in our laboratory have revealed that a specific cell type that is extrinsic to the nervous system seems to also play a causative role in diabetic neuropathy. These diabetes-specific cells are a subpopulation of circulating bone marrow (BM)-derived cells that travel to the peripheral nervous system where they produce major tissue damage by a novel mechanism.
A few years ago, when we were working on a gene therapy for diabetes in streptozotocin (STZ)-induced diabetic mice, we made a surprising observation. Our new gene therapy formulation reversed diabetes by inducing periportal insulin-producing beta-like cells in the liver that exhibit robust glucose-stimulated insulin secretion.20 Insulin transcripts were undetectable in the liver of nondiabetic mice. Unexpectedly, we detected tiny amounts of insulin transcripts in the liver of untreated and empty vector-treated STZ-diabetic mice.20 Subsequently, we discovered that diabetes leads to the appearance of proinsulin (PI)-producing cells in multiple organs and tissues in rodents. These abnormal cells are present in STZ-induced insulin-deficient diabetes and in obese animals that have developed hyperinsulinemia and type 2 diabetes induced by high fat-diet feeding. These unique cells also occur in ob/ob mice, which have low level hyperglycemia for months before analysis.21 Interestingly, the same cells also express the proinflammatory cytokine TNF-α.22 Further examination revealed that these cells first show up in the BM, when they travel to different peripheral organs and tissues by the circulation. The proinsulin-producing bone marrow-derived cells (PI-BMDCs) appear within one to three days of intermittent hyperglycemia induced by repeated glucose injections. Our initial conjecture was that one could use such PI-BMDCs for diabetes cell therapy. Shortly after our discovery, Oh et al. reported that incubation in high-glucose medium induces insulin-gene transcription in BM cells in vitro.23 They transferred these cells to the kidney capsule of diabetic rodents and reported that the maneuver partially ameliorated the hyperglycemia, although the authors did not report the serum insulin level. When we investigated the possible utility of these cells for diabetes cell therapy, we found that the tiny amounts of PI produced by the PI-BMDCs was not detectably secreted into the culture medium, and thus, PI-BMDCs induced by exposure to a high-glucose medium have no therapeutic potential for diabetes-cell therapy.
The PI-BMDCs are F4/80+ cells, and morphological examination indicates that they are mostly macrophage-like cells. We used immunohistochemistry to localize PI-producing cells in other tissues of rodents and found PI+ in relative abundance in the peripheral nervous system of long-term diabetic mice and rats. By light microscopy, the PI+ cells appear like normal nerve cells; PI+ signals are readily detected in sciatic nerves and neurons in the DRG, but are seen only in rodents that have diabetes for over a month, when the animals have developed impaired nerve function, that is, the PI-producing neurons appear at the same time that the animals develop diabetic neuropathy. We incubated isolated DRG neurons in high-glucose medium in vitro and found that, unlike BM cells, neurons exposed to a high-glucose medium fail to turn on PI production. We hypothesized that these PI-producing neuron-like cells represent PI-BMD–neuron fusion cells.
The fusion of BMDCs with cells in different tissues and organs, for example, skeletal muscle, cardiomyocytes, hepatocytes, brain, and intestine, has been reported by different laboratories.24–29 The fusion of hematopoietic cells with Purkinje neurons has been found to occur in rodents and humans;24,30,31 the frequency of these fusion cells was increased in the presence of inflammation and after X-radiation.32,33 These were tantalizing reports, but the pathophysiological significance of fusion cells in the central nervous system remained unclear. These reports of the occurrence of fusion of BMDCs and Purkinje cells prompted us to reexamine the PI+ nerve cells that appear in rodents with diabetic neuropathy in a new light. We reasoned that the diabetes-induced PI-producing neurons might represent fusion cells formed between neurons and PI-BMDCs, with the capacity for PI production coming from the BMDC fusion partner.
PI-BMDCs in multiple tissues of diabetic rodents
We observed the presence of immunoreactive PI+ cells in both paraffin sections and frozen liver sections of diabetic mice (Fig. 1A).21 Further, PI+ cells were also observed in histological sections from adipose tissue and BM of diabetic mice and rats. The presence of insulin mRNA in the liver was corroborated by in situ nucleic acid hybridization.21 The co-occurrence of insulin transcripts and immunoreactive PI indicates that the transcripts were translated into protein. To confirm that insulingene transcription occurs in the same cells that contain immunoreactive PI, we induced STZ-diabetes in mouse insulin promoter-green fluorescent protein (MIP-GFP) transgenic mice, which express GFP driven by the insulin promoter,34 and found that diabetes led to the expression of GFP in the same cells that harbor immunoreactive PI (Fig. 1B). Immunohistochemical analysis of the liver, fat, and BM of ob/ob mice as well as high fat-diet fed mice revealed PI+ cells also occur in these type 2 diabetes models (Fig. 1A).21
Figure 1.
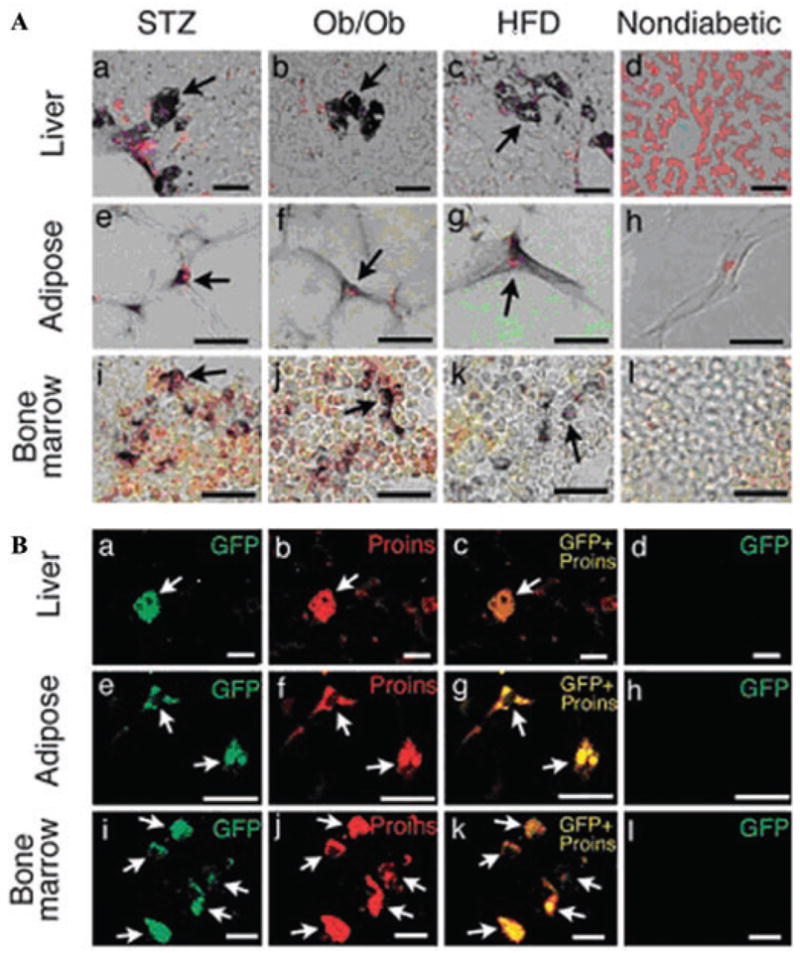
(A) Proinsulin+ cells (arrows) in the liver (a–d), abdominal adipose tissue (e–h), and bone marrow (i–l) of STZ (a, e, and i), ob/ob (b, f, and j), and high-fat diet (HFD; c, g, and k) diabetic mice and nondiabetic mice (d, h, and l). In nondiabetic mice, proinsulin+ cells were not found in the liver (d), adipose tissue (h), or bone marrow (l). (Scale bars, 25 μm.) (B) Overlap images of GFP/proinsulin in the liver (a–d), adipose tissue (e–h), and bone marrow (i–l) from STZ-induced diabetic (a–c, e–g, and i–k) and nondiabetic (d, h, and l) MIP-GFP mice. GFP and proinsulin signals completely overlap (arrows). (Scale bars: 25 μm, a–d; 20 μm, e–h; 10 μm, i–l). From Ref. 21 (with permission).
PI-BMDCs fuse with DRG neurons in mice with diabetic neuropathy
We produced diabetes by STZ treatment in eight-week-old rats and mice. Eight to twelve weeks later, all animals had developed diabetic neuropathy as evidenced by abnormal nerve function tests (impaired motor nerve conduction velocity, compound muscle action potential, sensory nerve conduction velocity, and sensory nerve action potential). By immunohistochemistry, we detected PI+ cells in the DRG and sciatic nerve of diabetic rats but not nondiabetic animals. Approximately, 10% of DRG neurons expressed PI 12 weeks after STZ-induced diabetes. The presence of insulin transcripts in DRG neurons was corroborated by in situ hybridization.35
We performed a bone marrow transplantation (BMT) experiment, transferring BM cells from transgenic mice that constitutively expressed β-gal to wild-type C57BL/6 mice, and used STZ to induce diabetes in half of the BMT recipients. By immunostaining, we found PI+ cells in the DRG and sciatic nerve (Fig. 2A) of diabetic mice; these cells displayed overlapping PI and β-gal immunostaining, indicating that they originated from the BM. Neither PI nor β-gal was detected in sections from nondiabetic BMT recipients (Fig. 2A). These observations were corroborated by data obtained in β-gal transgenic mice that have received BMT from MIP-GFP transgenic mice,34 which express GFP driven by the mouse insulin promoter (Fig. 2B).35 Additional experiments indicate that in the peripheral nervous system of diabetic animals, some of the fusion cells coexpress PI and neurofilament, a neuron-specific protein, and others coexpress PI and S100, a Schwann cell protein, indicating that both neurons and Schwann cells are individually involved in fusing with PI-BMDCs (see Fig. 1D in Terashima et al.35). Finally, when we performed BMT from male BM donors to female recipients, we detected Y chromosome-positive fusion cells in the DRG neurons of recipient mice with diabetic neuropathy,36 lending further support for a BM-neuron cell fusion event.
Figure 2.
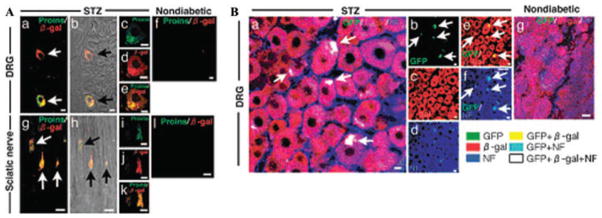
(A) Wild-type mice received BMT from β-gal donors. Immunofluorescence double staining of Proins/β-gal of DRG and sciatic nerves at eight weeks after STZ (a–e, g–k) or in nondiabetic recipients (f and l). Bright images are shown for comparison. (B) BMT of MIP-GFP donors34 to β-gal recipients. Sections taken from DRG eight weeks after STZ-induced diabetes in half the recipients. Immunofluorescence triple staining of GFP/β-gal/NF. From Ref. 35 (with permission).
To estimate the frequency of PI-BMDC–neuron fusion, we analyzed individual neurons isolated from the DRG of nondiabetic and diabetic animals and found that in nondiabetic rats,35 99.1% of the DRG neurons were diploid (2n); tetraploid (4n) cells constituted only 0.9%, and no cells were higher than tetraploid. In contrast, among neurons isolated from the DRG of diabetic rats, a substantially lower proportion, 86.6%, were diploid (2n), and there was a much higher proportion of polyploid cells (i.e., 12.5% were tetraploid, and 0.9% contained 6n or 8n [0.7% 6n and 0.2% 8n]).35 When we analyzed the distribution of PI immunostaining among the isolated DRG neurons in diabetic rats, we found that polyploidy occurs exclusively in PI-expressing cells, further indicating that PI expression is a marker for fusion cells. Findings very similar to those in diabetic rats have been reported recently in mice with diabetic neuropathy.36
Fusion of PI-BMDCs and DRG neurons in vitro
We incubated DRG neurons from Rose26-flox-stop-GFP transgenic mice (which have a loxP-flanked STOP sequence in front of the green fluorescent protein gene knocked into the Rosa26 locus in all tissues) with RIP-Cre BMDCs (from transgenic mice that express Cre recombinase driven by the rat insulin promoter) in the presence of high- and low-glucose medium, and found PI-BMDC–neuron fusion cell formation in vitro only in the presence of high glucose, but not low glucose medium. These fusion cells were marked by GFP expression. When the PI-BMDCs fuse with the neurons, the high glucose-induced Cre recombinase produced by the RIP-Cre BMDCs will access and delete the STOP signal in front of the GFP in the Rosa26-floz-stop-GFP neuron fusion partner, allowing the GFP to be expressed in the fusion cell. Therefore, the role of a high-glucose environment in the induction of PI-BMDCs and in facilitating PI-BMDC–neuron cell fusion can be reproduced under in vitro incubation conditions.36
Discussion
With the advances in glucose monitoring and individualized insulin therapy in the last two decades, it is now rare for diabetics to die of ketoacidosis. The much longer life span of diabetics in the 21st century translates into many more patients who suffer from chronic diabetic complications, including a large proportion of patients who die from them. Hyperglycemia is known to underlie most, if not all, diabetic complications. According to one scenario:
Intracellular hyperglycemia … causes increased mitochondrial production of ROS (reactive oxygen species). The ROS causes strand breaks in nuclear DNA, which activate PARP (poly[ADP-ribose] polymerase). PARP then modifies GAPDH (glyceraldehydes-3 phosphate dehydrogenase), thereby reducing its activity. Finally, decreased GADPH activates the polyol pathway, increases intracellular AGE (advanced glycation end product) formation, activates PKC (protein kinase C) and subsequently NF-κB, and activates hexosamine pathway flux” and “subsequent modification of proteins by N-acetylglucosamine” [notations in italics added by authors].37
Similar mechanisms, with minor variations in emphasis, have been proposed by others.2–14 Most of these mechanisms have also been documented to operate in diabetic neuropathy.38–40 In all cases, the culprit cells that underlie the neuropathy are assumed to be local cells in the different target organ system affected by the particular complication.
Different groups have described the fusion of hematopoietic cells with Purkinje neurons in rodents and humans.24,30,31 Hematopoietic cells derived from the BM are often thought to function as “healers,” playing a positive role in neural regeneration in neuropathy. Despite the absence of functional experiments, there is widespread speculation that BMDC–Purkinje fusion represents a mechanism that the body uses to repair damaged tissues.32,33,41–43 Experiments in our laboratory showed that PI-BMDC–neuron fusion is far from being a beneficial healing event. PI-BMDCs are a subpopulation of highly proinflammatory BMDCs specifically induced by diabetes that have fusogenic properties. They seem to play a central role in the pathogenesis of diabetic neuropathy.
Hyperglycemia induces ROS, which in turn activates poly(ADP-ribose) polymerase (PARP), which plays a central role in the pathogenesis of multiple diabetic complications. Neuronal PARP activation has been shown to occur in the spinal cord, peripheral nerves, and DRG of rodents with diabetic neuropathy. PARP inhibition was found to protect against diabetic neuropathy.39,44–50 Furthermore, PARP-knockout mice with a mixed genetic background51 or C57BL/6 background36 are resistant to diabetic neuropathy. The dogma among researchers that PARP activation within peripheral nerves, including Schwann cells, vasa nervorum, and DRG neurons is “an early and fundamental mechanism of peripheral diabetic neuropathy.”52 We note that major inflammatory pathways, for example, PKC and NF-κB, are directly downstream of PARP and much of the inflammation in diabetic complication is mediated by proinflammatory hematopoietic cells. PI-BMDCs are a diabetes-specific cell type that mediates much of the inflammation-related pathology of diabetic complications. Importantly, these same cells have great fusogenic potential that can be demonstrated under in vitro conditions.36 They directly cause tissue damage by fusing with both neurons and Schwann cells and are thus a major culprit in diabetic neuropathy.35 To further dissect the role of PARP in diabetic neuropathy, we recently genetically inactivated PARP globally, and in a BM-specific manner.36 We found that BMT of PARP-knockout BM cells to wild-type mice protects against, and conversely, BMT of wild-type cells to PARP-knockout mice confers susceptibility to, diabetic neuropathy. In other words, the major determinant in diabetic neuropathy in the BMT model is the PARP genotype of the BM cells. In vitro incubation experiments further indicate that in high-glucose PARP+ (wild-type) PI-BMDCs are fusogenic. Loss of PARP in BMDCs makes them resistant to high-glucose–induced PI expression; it also makes them lose most of their fusogenic potential despite the presence of high glucose.36
In summary, the hyperglycemia in diabetes induces the formation of fusogenic PI-BMDCs, proinflammatory cells that coexpress TNF-α. The high-glucose environment further facilitates the fusion of these cells with DRG neurons, causing nerve dysfunction and premature apoptosis, events that lead to diabetic neuropathy. Over the last decade, research in the area of skeletal biology has revealed that, in addition to controlling calcium homeostasis, skeletal cells and secretions also play key roles in energy and glucose metabolism.53 Conversely, here we show that diabetes causes BM cells to misbehave and become promiscuous, wreaking havoc as they travel to different tissues and organs by fusing with local cells. In addition to fusing with neurons and Schwann cells, we showed that PI-BMDCs also fuse with hepatocytes in diabetic animals.22 Interestingly, the pathological consequence of PI-BMDC–hepatocyte fusion in the liver is not readily apparent because of the large functional reserve of the liver. To date, diabetic neuropathy is the only chronic diabetic complication proven to be a direct victim of PI-BMDCs’ misadventures. Given the ruinous potential of this unique cell population in diabetic animals, we will be surprised if these messengers of destruction are not directly involved in other diabetic complications. Poorly controlled diabetes is bad to the bone.
Acknowledgments
This study was supported by NIH Grants HL-51586 (to LC) and P30DK079638 for the Diabetes and Endocrinology Research Center at Baylor College of Medicine. L.C. was also supported by the Betty Rutherford Chair for Diabetes Research from St. Luke’s Episcopal Hospital (Houston, Texas) and the T.T. and W.F. Chao Global Foundation.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Danaei G, Finucane MM, Lu Y, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet. 2011;378:31–40. doi: 10.1016/S0140-6736(11)60679-X. [DOI] [PubMed] [Google Scholar]
- 2.Srinivasan S, Stevens M, Wiley JW. Diabetic peripheral neuropathy. Evidence for apptosis and associated mitochondrial dysfunction. Diabetes. 2000;49:1932–1938. doi: 10.2337/diabetes.49.11.1932. [DOI] [PubMed] [Google Scholar]
- 3.Schmeichel AM, Schmelzer JD, Low PA. Oxidative injury and apoptosis of dorsal root ganglion neurons in chronic experimental diabetic neuropathy. Diabetes. 2003;52:165–171. doi: 10.2337/diabetes.52.1.165. [DOI] [PubMed] [Google Scholar]
- 4.Vincent AM, Russell JW, Low P, Feldman EL. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr Rev. 2004;25:612–628. doi: 10.1210/er.2003-0019. [DOI] [PubMed] [Google Scholar]
- 5.Leinninger GM, Edwards JL, Lipshaw MJ, Feldman EL. Mechanisms of disease: mitochondria as new therapeutic targets in diabetic neuropathy. Nat Clin Pract Neurol. 2006;2:620–628. doi: 10.1038/ncpneuro0320. [DOI] [PubMed] [Google Scholar]
- 6.Low PA, Lagerlund TD, McManis PG. Nerve blood flow and oxygen delivery in normal, diabetic, and ischemic neuropathy. Int Rev Neurobiol. 1989;31:355–438. doi: 10.1016/s0074-7742(08)60283-4. [DOI] [PubMed] [Google Scholar]
- 7.Oates PJ. Polyol pathway and diabetic peripheral neuropathy. Int Rev Neurobiol. 2002;50:325–392. doi: 10.1016/s0074-7742(02)50082-9. [DOI] [PubMed] [Google Scholar]
- 8.Thornalley PJ. Glycation in diabetic neuropathy: characteristics, consequences, causes, and therapeutic options. Int Rev Neurobiol. 2002;50:37–57. doi: 10.1016/s0074-7742(02)50072-6. [DOI] [PubMed] [Google Scholar]
- 9.Xia P, Kramer RM, King GL. Identification of the mechanism for the inhibition of Na+, K(+)-adenosine triphosphatase by hyperglycemia involving activation of protein kinase C and cytosolic phospholipase A2. J Clin Invest. 1995;96:733–740. doi: 10.1172/JCI118117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomlinson DR. Mitogen-actiavated protein kinases as glucose transducers for diabetic complications. Diabetologia. 1999;42:1271–1281. doi: 10.1007/s001250051439. [DOI] [PubMed] [Google Scholar]
- 11.Russell JW, Golovoy D, Vincent AM, et al. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J. 2002;16:1738–1748. doi: 10.1096/fj.01-1027com. [DOI] [PubMed] [Google Scholar]
- 12.King GL, Loeken MR. Hyperglycemia-induced oxidative stress in diabetic complications. Histochem Cell Biol. 2004;122:333–338. doi: 10.1007/s00418-004-0678-9. [DOI] [PubMed] [Google Scholar]
- 13.Vincent AM, Brownlee M, Russell JW. Oxidative stress and programmed cell death in diabetic neuropathy. Ann NY Acad Sci. 2002;959:368–383. doi: 10.1111/j.1749-6632.2002.tb02108.x. [DOI] [PubMed] [Google Scholar]
- 14.Groop PH, Forsblom C, Thomas MC. Mechanisms of disease: pathway-selective insulin resistance and microvascular complications of diabetes. Nat Clin Pract Endocrinol Metab. 2005;1:100–110. doi: 10.1038/ncpendmet0046. [DOI] [PubMed] [Google Scholar]
- 15.Toth C, Rong LL, Yang C, et al. Receptor for advanced glycation end products (RAGEs) and experimental diabetic neuropathy. Diabetes. 2008;57:1002–1017. doi: 10.2337/db07-0339. [DOI] [PubMed] [Google Scholar]
- 16.Vareniuk I, Pavlov IA, Obrosova IG. Inducible nitric oxide synthase gene deficiency counteracts multiple manifestations of peripheral neuropathy in a streptozotocin-induced mouse model of diabetes. Diabetologia. 2008;51:2126–2133. doi: 10.1007/s00125-008-1136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamakawa I, Kojima H, Terashima T, et al. Inactivation of TNFá ameliorates diabetic neuropathy in mice. Am J Physiol Endocrinol Metab. 2011;301:E844–E852. doi: 10.1152/ajpendo.00029.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cameron NE, Cotter MA. Proinflammatory mechanisms in diabetic neuropathy: focus on the nuclear factor kappa B pathway. Curr Drug Targets. 2008;9:60–67. doi: 10.2174/138945008783431718. [DOI] [PubMed] [Google Scholar]
- 19.Leinninger GM, Vincent AM, Feldman EL. The role of growth factors in diabetic peripheral neuropathy. J Peripher Nerv Syst. 2004;9:26–53. doi: 10.1111/j.1085-9489.2004.09105.x. [DOI] [PubMed] [Google Scholar]
- 20.Kojima H, Fujimiya M, Matsumura K, et al. NeuroD-betacellulin gene therapy induces islet neogenesis in the liver and reverses diabetes in mice. Nat Med. 2003;9:596–603. doi: 10.1038/nm867. [DOI] [PubMed] [Google Scholar]
- 21.Kojima H, Fujimiya M, Matsumura K, et al. Extrapancreatic insulin-producing cells in multiple organs in diabetes. Proc Natl Acad Sci USA. 2004;101:2458–2463. doi: 10.1073/pnas.0308690100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fujimiya M, Kojima H, Ichinose M, et al. Fusion of proinsulin-producing bone marrow-derived cells with hepatocytes in diabetes. Proc Natl Acad Sci USA. 2007;104:4030–4035. doi: 10.1073/pnas.0700220104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oh SH, Muzzonigro TM, Bae SH, et al. Adult bone marrow-derived cells trans-differentiating into insulin-producing cells for the treatment of type I diabetes. Lab Invest. 2004;84:607–617. doi: 10.1038/labinvest.3700074. [DOI] [PubMed] [Google Scholar]
- 24.Alvarez-Dolado M, Pardal R, Garcia-Verdugo JM, et al. Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature. 2003;425:968–973. doi: 10.1038/nature02069. [DOI] [PubMed] [Google Scholar]
- 25.Vassilopoulos G, Wang PR, Russell DW. Transplanted bone marrow regenerates liver by cell fusion. Nature. 2003;422:901–904. doi: 10.1038/nature01539. [DOI] [PubMed] [Google Scholar]
- 26.Wang X, Willenbring H, Akkari Y, et al. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature. 2003;422:897–900. doi: 10.1038/nature01531. [DOI] [PubMed] [Google Scholar]
- 27.Nygren JM, Jovinge S, Breitbach M, et al. Bone marrow-derived hematopoietic cells generate cardiomyocytes at a low frequency through cell fusion, but not trans-differentiation. Nat Med. 2004;10:494–501. doi: 10.1038/nm1040. [DOI] [PubMed] [Google Scholar]
- 28.Rizvi AZ, Swain JR, Davies PS, et al. Bone marrow-derived cells fuse with normal and transformed intestinal stem cells. Proc Natl Acad Sci USA. 2006;103:6321–6325. doi: 10.1073/pnas.0508593103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davies PS, Powell AE, Swain JR, Wong MH. Inflammation and proliferation act together to mediate intestinal cell fusion. PLoS One. 2009;4:e6530. doi: 10.1371/journal.pone.0006530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weimann JM, Jahansson CB, Trejo A, Blau HM. Stable reprogrammed heterokaryons form spontaneously in Purkinje Neurons after bone marrow transplant. Nat Cell Biol. 2003;5:959–966. doi: 10.1038/ncb1053. [DOI] [PubMed] [Google Scholar]
- 31.Magrassi L, Grimaldi P, Ibatici A, et al. Induction and survival of binucleated Purkinje neurons by selective damage and aging. J Neurosci. 2007;27:9885–9892. doi: 10.1523/JNEUROSCI.2539-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johansson CB, Youssef S, Koleckar K, et al. Extensive fusion of haematopoietic cells with Purkinje neurons in response to chronic inflammation. Nat Cell Biol. 2008;10:575–583. doi: 10.1038/ncb1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nygren JM, Liuba K, Breitbach M, et al. Myeloid and lymphoid contribution to non-haematopoietic lineages through irradiation-induced heterotypic cell fusion. Nat Cell Biol. 2008;10:584–592. doi: 10.1038/ncb1721. [DOI] [PubMed] [Google Scholar]
- 34.Hara M, Wang X, Kawamura T, et al. Transgenic mice with green fluorescent protein-labeled pancreatic b-cells. Am J Physiol Endocrinol Metab. 2003;284:e177–e183. doi: 10.1152/ajpendo.00321.2002. [DOI] [PubMed] [Google Scholar]
- 35.Terashima T, Kojima H, Fujimiya M, et al. The fusion of bone-marrow-derived proinsulin-expressing cells with nerve cells underlies diabetic neuropathy. Proc Natl Acad Sci USA. 2005;102:12525–12530. doi: 10.1073/pnas.0505717102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Terashima T, Kojima H, Chan L. Bone marrow expression of poly(ADP-ribose) polymerase underlies diabetic neuropathy via hematopoietic-neuronal cell fusion. FASEB J. 2011 doi: 10.1096/fj.11-186262. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brownlee M. The pathobiology of diabetic complications—a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 38.Edwards JL, Vincent AM, Cheng HT, Feldman EL. Diabetic neuropathy: mechanisms to management. Pharmacol Ther. 2008;120:1–34. doi: 10.1016/j.pharmthera.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zochodne DW. Diabetic polyneuropathy: an update. Curr Opin Neurol. 2008;21:527–533. doi: 10.1097/WCO.0b013e32830b84cb. [DOI] [PubMed] [Google Scholar]
- 40.Yasuda H, Terada M, Maeda K, et al. Diabetic neuropathy and nerve regeneration. Prog Neurobiol. 2003;69:229–285. doi: 10.1016/s0301-0082(03)00034-0. [DOI] [PubMed] [Google Scholar]
- 41.Singec I, Snyder EY. Inflammation as a matchmaker: revisiting cell fusion. Nat Cell Biol. 2008;10:503–505. doi: 10.1038/ncb0508-503. [DOI] [PubMed] [Google Scholar]
- 42.Larsson LI, Bjerregaard B, Talts JF. Cell fusions in mammals. Histochem Cell Biol. 2008;129:551–561. doi: 10.1007/s00418-008-0411-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lluis F, Cosma MP. Cell-fusion-mediated somatic-cell reprogramming: a mechanism for tissue regeneration. J Cell Physiol. 2010;223:6–13. doi: 10.1002/jcp.22003. [DOI] [PubMed] [Google Scholar]
- 44.Obrosova IG, Drel VR, Pacher P, et al. Oxidative-nitrosative stress and poly(ADP-Ribose) polymerase (PARP) activation in experimental diabetic neuropathy: the relation is revisited. Diabetes. 2005;54:3435–3441. doi: 10.2337/diabetes.54.12.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szabo C. Roles of poly(ADP-ribose) polymerase activation in the pathogenesis of diabetes mellitus and its complications. Pharmacol Res. 2005;52:60–71. doi: 10.1016/j.phrs.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 46.Southan GJ, Szabo C. Poly(ADP-ribose) polymerase inhibitors. Curr Med Chem. 2003;10:321–340. doi: 10.2174/0929867033368376. [DOI] [PubMed] [Google Scholar]
- 47.Figueroa-Romero C, Sadidi M, Feldman EL. Mechanisms of disease: the oxidative stress theory of diabetic neuropathy. Rev Endocr Metab Disord. 2008;9:301–314. doi: 10.1007/s11154-008-9104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li F, Drel VR, Szabo C, et al. Low-dose poly(ADP-Ribose) polymerase inhibitor-containing combination therapies reverse early peripheral diabetic neuropathy. Diabetes. 2005;54:1514–1522. doi: 10.2337/diabetes.54.5.1514. [DOI] [PubMed] [Google Scholar]
- 49.Ilnytska O, Lyzogubov VV, Stevens MJ, et al. Poly(ADP-ribose) polymerase inhibition alleviates experimental diabetic sensory neuropathy. Diabetes. 2006;55:1686–1694. doi: 10.2337/db06-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Drel VR, Lupachyk S, Shevalye H, et al. New therapeutic and biomarker discovery for peripheral diabetic neuropathy: PARP inhibitor, nitrotyrosine, and tumor necrosis factor-{alpha} Endocrinology. 2010;151:2547–2555. doi: 10.1210/en.2009-1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Obrosova IG, Li F, Abatan OI, et al. Role of poly(ADP-ribose) polymerase activation in diabetic neuropathy. Diabetes. 2004;53:711–720. doi: 10.2337/diabetes.53.3.711. [DOI] [PubMed] [Google Scholar]
- 52.Obrosova IG. Diabetes and the peripheral nerve. Biochim Biophys Acta. 2009;1792:931–940. doi: 10.1016/j.bbadis.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 53.Clemens TL, Karsenty G. The osteoblast: an insulin target cell controlling glucose homeostasis. J Bone Miner Res. 2011;26:677–680. doi: 10.1002/jbmr.321. [DOI] [PubMed] [Google Scholar]