

Innate immunity recognition relies on a diverse set of germ line encoded receptors, termed pattern recognition receptors (PRR), which recognize broad classes of molecular structures common to groups of microorganisms. One of the largest and best studied families of PRR are the Toll family of receptors (Toll-like receptors, TLRs) that detect microbial components with high sensitivity and selectivity[1]. Among TLRs, TLR4 selectively responds to bacterial endotoxin (E) (Gram-negative bacterial lipopolysaccharides (LPS) or lipooligosaccharides (LOS)),[2] resulting in the rapid triggering of pro-inflammatory processes necessary for optimal host immune responses to invading Gram-negative bacteria (GNB). TLR4 does not bind directly to endotoxin: LBP,[3] CD14,[4] MD-2[5] are required for efficient extraction and transfer of endotoxin monomers from the GNB outer membrane or aggregates of purified endotoxin to MD-2. The resulting monomeric E·MD-2 complex is the ligand that, depending on the structural properties of E and MD-2, specifies TLR4 activation or antagonism.[6] Although TLR4 plays a key physiologic role in host response to Gram-negative bacterial infection, an excessively potent and/or prolonged TLR4 response can promote life-threatening pathology such as septic shock.[7] TLR4 activation has also been associated with certain autoimmune diseases, non-infectious inflammatory disorders, and neuropathic pain, suggesting a wide range of possible clinical settings for application of TLR4 antagonists.[8] Conversely, agonists of TLR4 can be useful as adjuvants in vaccine development and in cancer immunotherapy [9]. Lipid A[10] (Scheme 1), the hydrophobic part of LPS, is responsible for TLR4-dependent proinflammatory activity.[11] Underacylated lipid A variants, such as tetraacylated lipid IVa[12] and E5564 (Eritoran)[13] are potent LPS antagonists (Scheme 1). The β(1→6) diglucosamine backbone of lipid A can be replaced by an aminoalkyl glucosamine moiety in aminoalkyl glucosaminide 4-phosphates (AGPs)[14] or by other non-carbohydrate structures[15] and the lipid A analogue retains TLR4 agonist or antagonist activity. One or two phosphates are typically present in synthetic lipid A mimics, but these groups could be, in principle, substituted by negatively charged isosteres. A carboxylic acid group replaces the C-1 phosphate in AGP derivatives,[14a] while a sulfate group is present in the monosaccharide lipid A mimic ONO-4007 (Scheme 1) developed by Ono Pharmaceutical Co (Osaka, Japan).[16] This compound showed TLR4 agonist activity inducing TNF-α production in tumour cells, but further clinical development was precluded by the compound's limited water solubility.
Scheme 1.
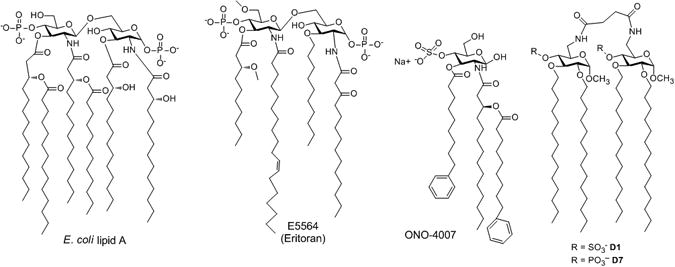
Chemical structures of E. coli lipid A, synthetic antagonist E 5564 (Eritoran), synthetic agonist ONO-4007 and synthetic compounds D1 and D7.
Here we present two innovative lipid A analogues (synthetic compounds D1 and D7) (Scheme 1), in which two methyl α-D-glucopyranoside units are bridged through a (6→6′) succinic diamide linker. In compound D7 two phosphates in C-4 and C-4′ positions mimic the phosphates in the C-1 and C-4′ position of natural lipid A, while in 1 two sulfates replace phosphates. The sulfate group, negatively charged at neutral pH, is a bioisoster of phosphate that has rarely been exploited in the design of lipid A mimetics.[16] In contrast with natural lipid A, compounds D1 and D7 are symmetric molecules (2-fold rotational symmetry C2). Four linear ether chains (C14H29) replace acyl esters (COC13H27 or COC11H23) found in lipid A and synthetic antagonists. Ether chains are more resistant to enzyme hydrolysis than esters and improve pharmacokinetic properties of the molecules and, for this reason, have been used in TLR4 antagonists previously developed by our group that target the CD14 receptor.[17]
Both lipid A mimetics D1 and D7 were rationally designed according to the recently resolved X-ray structures of lipid A[18], and antagonists lipid IVa[19] and Eritoran[20] bound to MD-2. Eritoran and lipid IVa bind to the hydrophobic pocket in human MD-2 and there is no direct interaction between both antagonists and TLR4. The structure formed by the four acyl chains of eritoran and lipid IVa complement the shape of hydrophobic cavity, occupying almost 90% of the solvent-accessible volume of the pocket (Figure 1). According to preliminary simulations, both molecules D1 and D7 can bind to MD-2 in a manner similar to that of lipid IVa,[12] with the four fatty acid chains deeply confined in the MD-2 cavity and the phosphate and sugar groups interacting with conserved residues at the cavity rim (Figure 1).
Figure 1.
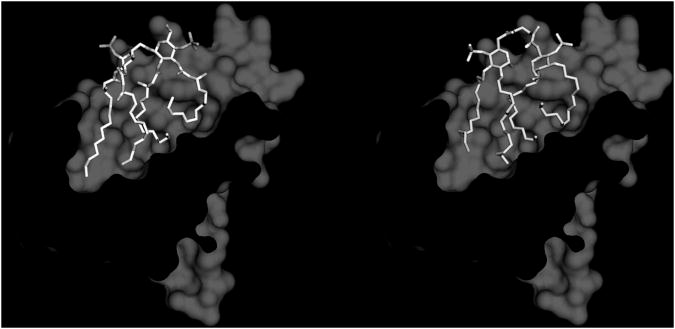
Lipid IVa (right) and molecule D1 (left) bound to the MD-2 pocket.
In both D1 and D7, the conformational flexibility of the succinic diamide linker allows the two negatively charged groups (phosphates or sulfates) to be placed at a distance of about 12 Å, similar to the distance between the phosphates of MD-2-bound lipid IVa[12] (Figure 1).
Because of their symmetric structures, both molecules D1 and D7 can be prepared through the convergent and efficient synthesis shown in Scheme 2, starting from the common precursor 3.
Scheme 2.
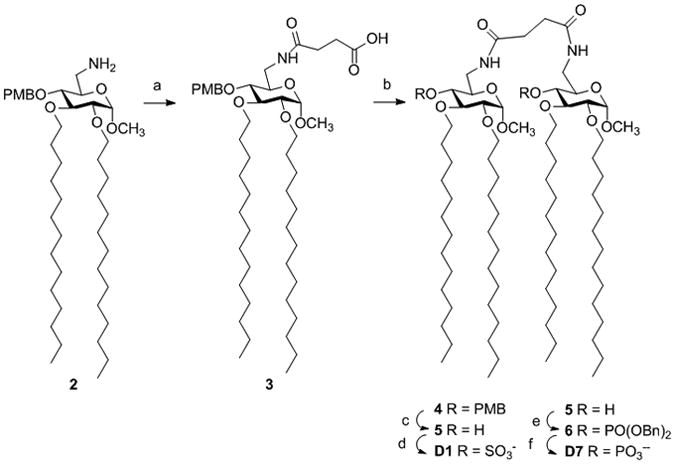
Synthesis of lipid A mimetics D1 and D7: a) succinic anhydride, dry py, RT, 97%; b) compound 2, HOBt, DIC, DIPEA, DMF, 0°C→RT, 70%; c) 1:1 TFA in CH2Cl2, RT, 86%; d) SO3.py, CH2Cl2 dry, 50% e) (iPr)2NP(OBn)2, imidazolium trifluorocaetate, then oxidation with m-chloroperbenzoic acid, 43%; f) H2/Pd-C in MeOH-THF-AcOH (71%).
The 6-deoxy-6-amino-4-O-(4′-methoxybenzyl)-2,3-di-O-tetradecyl-α-D-glucopyranoside 2 was prepared in gram quantities according to published procedures.[17b] Monosaccharide 2 with a primary amino group on C-6 was reacted with succinic anhydride to produce compound 3 (97% yield), that was condensed with 2 by treatment with hydroxybenzotriazole (HOBt), O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU), N,N′-diisopropylcarbodiimide (DIC) in the presence of the Hunig's base N,N-diisopropylethylamine (DIPEA) resulting in production of disaccharide 4 in 70% yield. The p-metoxybenzyl ethers in positions C-4 and C-4′ were cleaved by treatment with trifluoroacetic acid (TFA) solution 1:1 in CH2Cl2 affording 5 in 86% yield. To obtain the sulfated disaccharide D1, the free hydroxyl in C-4 and C-4′ position were sulfated by reaction with the SO3.pyridine complex (50% yield). Alternatively, disaccharide 5 was reacted with (iPr)2NP(OBn)2 in the presence of imidazolium trifluoroacetate and the di-benzyl phosphite was oxidized in situ to the corresponding phosphate 6 using m-chloroperbenzoic acid (43%). Treatment of compound 6 with H2 and Pd-C as catalysts followed by neutralization with Et3N[21] gave 7 as the triethylammonium salt in 71% yield. The solubility in water of D1 and D7 was surprisingly different: D1 was soluble in the aqueous buffers used for biological characterization up to a concentration of about 50 μM, while diphosphate D7 was insoluble under these conditions, precluding its biological characterization.
The ability of D1 to inhibit endotoxin-stimulated TLR4 activation was tested using stable transformants of HEK293 cells expressing TLR4 (HEK-TLR4 cells). These cells express fully functional transmembrane TLR4 but do not produce MD-2. Supplementation of the cell incubation mixture with sMD-2 as well as LBP and sCD14 was required for these cells to respond to pM amounts of added endotoxin (e.g., purified aggregates of N. meningitidis lipooligosaccharides (LOSagg)).[22] As shown in Figure 2, added D1 produced dose-dependent inhibition of cell activation (i.e., extracellular accumulation of IL-8) induced by 200 pM LOSagg.[22] Maximum inhibition was seen at 5-10 μM 1 but was incomplete, raising the possibility that D1 was itself a partial agonist of TLR4. Indeed, in the absence of added LOSagg, D1 caused dose-dependent activation of HEK-TLR4 cells (Figure 2), but not of parental HEK293 cells (Figure S1). TLR4-dependent cell activation by D1 was not due to endotoxin contamination; LAL testing (Lonza Bio-Whittaker, Walkersville, MD) with a detection limit of 10 pg endotoxin/mL, did not detect LPS contamination in the preparations of D1 used in our experiments. In sum, these data strongly suggest that molecule D1 interacts with MD-2-TLR4 and acts as a weak TLR4 agonist/partial antagonist. The limited TLR4-dependent cell activation observed when HEK-TLR4 cells were exposed to 0.2 nM LOS + 10 μM D1 most likely reflects occupation of MD-2·TLR4 by D1 rather than LOS.
Figure 2.
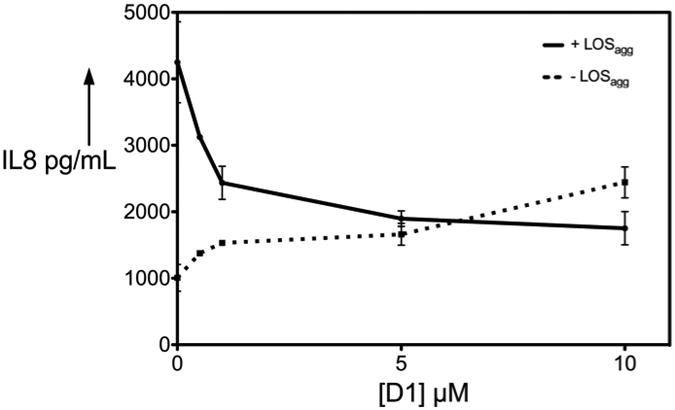
Compound D1 acts as weak TLR4 agonist, partial TLR4 antagonist. Results shown represent mean ± SEM of at least three determinations.
To test more directly the ability of D1 to inhibit LBP/sCD14-dependent extraction and transfer of endotoxin (LOS) monomers to MD-2.TLR4, the effects of 10 μM D1 were examined using uniformly radiolabeled LOS aggregates ([3H]LOSagg) plus purified recombinant LBP, sCD14, and conditioned medium containing MD-2.TLR4ecd. Molar concentrations of added MD-2·TLR4ecd, relative to LOS, were limited so as to permit detection of effects of D1 on both LBP-catalyzed extraction and transfer of LOS monomers to sCD14 (yielding monomeric [3H]LOS·sCD14; Mr ∼60,000) and transfer of [3H]LOS from [3H]LOS·sCD14 to MD-2·TLR4ecd (yielding ([3H]LOS·MD-2·TLR4ecd)2; Mr ∼190,000). Figure 3 shows that under these experimental conditions addition of 10 μM D1 partially inhibited formation of [3H]LOS·sCD14 and nearly completely inhibited transfer of [3H]LOS from [3H]LOS·sCD14 to MD-2·TLR4ecd (i.e., formation of ([3H]LOS·MD-2·TLR4ecd)2). In support of this view, D1 inhibited transfer of [3H]LOS from pre-formed [3H]LOS·sCD14 to His6-MD-2 as shown by reduced co-capture of [3H]LOS (as [3H]LOS·MD-2) to the Ni2+ HISLINK resin (Figure 4A). In contrast, at the same concentrations, 1 did not promote displacement of LOS from MD-2 (Figure 4B) nor inhibited HEK-TLR4 cells activation by pre-formed LOS·MD-2 (Figure 5).
Figure 3.
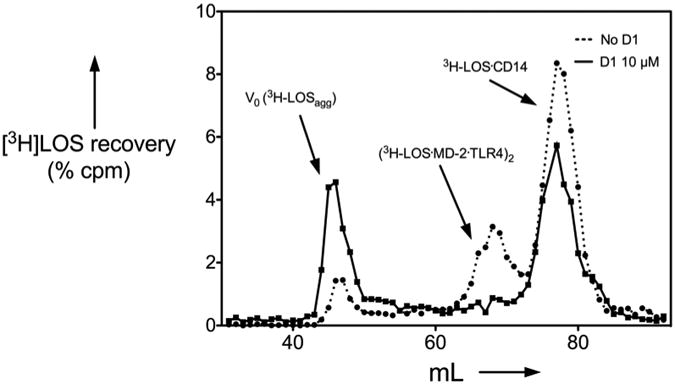
Effect of D1 (10μM) on LBP/sCD14-dependent extraction and transfer of [3H]LOS monomers from [3H]LOS aggregates to sCD14 and to MD-2·TLR4ecd (denoted as MD-2·TLR4).
Figure 4.
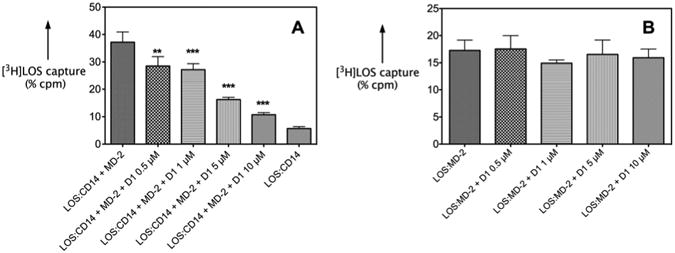
A) [3H]LOS monomer transfer from [3H]LOS·sCD14 to His6·MD-2 resulting in co-capture of [3H]LOS to Ni2+ HISLINK resin. B) [3H]LOS·MD-2 stability in presence of increasing concentration of D1. Results shown represent mean ± SEM of at least three determinations.
Figure 5.
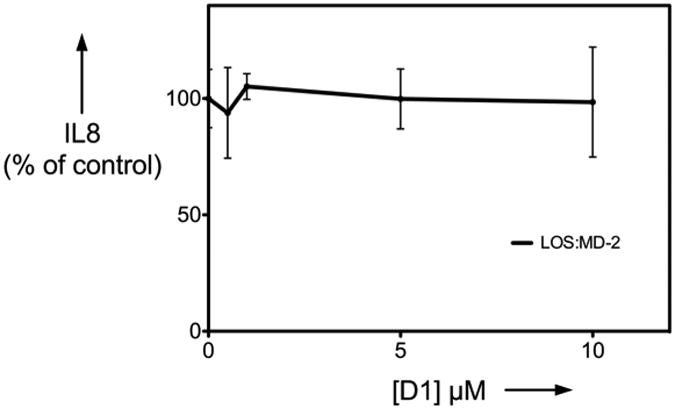
Compound D1 does not inhibit TLR4 activation by LOS·MD-2 complex. Results shown represent mean ± SEM of at least 3 determinations.
In summary, compound D1, much like other tetraacylated lipid A analogues[12-13, 23] inhibits activation of TLR4 by endotoxin by inhibiting interaction of endotoxin with both CD14 and MD-2(·TLR4). By analogy to the described interactions of lipid IVA, eritoran, and tetraacylated LPS,[11,12,19] D1 presumably acts by competitively occupying CD14 and MD-2(·TLR4). Lipid IVA has weak TLR4 agonist properties toward mouse MD-2·TLR4.[24] However, the weak TLR4 agonist properties toward human MD-2·TLR4 observed for D1 (Figure 1) is not shared by lipid IVA[20][24a], implying unique interactions of D1 with human MD-2·TLR4. Whether or not this is a consequence of the substitution of the phosphates typically present in lipid A with sulfates or other unique structural features of D1 awaits further study. Whatever the precise structural basis of the unique functional properties of D1, these properties may make these and related compounds valuable new immuno-pharmacologic agents. Under-acylated and under-phosphorylated, derivatives of lipid A that have partial TLR4 agonist properties are currently lead compounds as vaccine adjuvants, providing apparently sufficient TLR4-dependent immune boosting while tempering potential TLR4-mediated toxicity.[25] In sepsis, where immune dysregulation may be manifest as a systemic inflammatory syndrome and/or subsequent immune paralysis,[26] a compound that has both partial agonist and antagonist properties may be more advantageous than the more pure TLR4 antagonists that have been developed and tested to date. Work is in progress to characterize the pharmacodynamic and pharmacokinetic properties of D1 and to study molecular details of its interaction with the TLR4-MD-2 complex.
Supplementary Material
Acknowledgments
This work was supported by NIH/NIAID, grant number 1R01AI059372 “Regulation of MD-2 function and expression” and by the fund of Finlombarda, Regione Lombardia, “Network Enabled Drug Design” (NEDD), grant number 14546.
Contributor Information
Prof. Theresa Gioannini, Email: theresa-gioannini@uiowa.edu.
Prof. Jerrold Weiss, Email: jerrold-weiss@uiowa.edu.
Prof. Francesco Peri, Email: francesco.peri@unimib.it.
References
- 1.Miyake K. Semin Immunol. 2007;19:3–10. doi: 10.1016/j.smim.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 2.a) Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]; b) Beutler B, Du X, Poltorak A. J Endotoxin Res. 2001;7:277–280. [PubMed] [Google Scholar]; c) Beutler B. Curr Top Microbiol Immunol. 2002;270:109–120. doi: 10.1007/978-3-642-59430-4_7. [DOI] [PubMed] [Google Scholar]
- 3.Schumann RR, Leong SR, Flaggs GW, Gray PW, Wright SD, Mathison JC, Tobias PS, Ulevitch RJ. Science. 1990;249:1429–1431. doi: 10.1126/science.2402637. [DOI] [PubMed] [Google Scholar]
- 4.Wright S, Ramos R, Tobias P, Ulevitch R, Mathison J. Science. 1990;249:1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 5.a) Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gioannini TL, Teghanemt A, Zhang D, Coussens NP, Dockstader W, Ramaswamy S, Weiss JP. Proc Natl Acad Sci U S A. 2004;101:4186–4191. doi: 10.1073/pnas.0306906101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jerala R. Int J Med Microbiol. 2007;297:353–363. doi: 10.1016/j.ijmm.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 7.a) Martin GS, Mannino DM, Eaton S, Moss M. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]; b) Wafaisade A, Lefering R, Bouillon B, Sakka SG, Thamm OC, Paffrath T, Neugebauer E, Maegele M. Crit Care Med. 2011;39:621–628. doi: 10.1097/CCM.0b013e318206d3df. [DOI] [PubMed] [Google Scholar]; c) Silva E, Pedro Mde A, Sogayar AC, Mohovic T, Silva CL, Janiszewski M, Cal RG, de Sousa EF, Abe TP, de Andrade J, de Matos JD, Rezende E, Assuncao M, Avezum A, Rocha PC, de Matos GF, Bento AM, Correa AD, Vieira PC, Knobel E. Crit Care. 2004;8:R251–260. doi: 10.1186/cc2892. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cribbs SK, Martin GS. Crit Care Med. 2007;35:2646–2648. doi: 10.1097/01.CCM.0000288082.99980.90. [DOI] [PubMed] [Google Scholar]
- 8.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Nat Med. 2007;13:552–559. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 9.Hedayat M, Netea MG, Rezaei N. Lancet Infect Dis. 2011 doi: 10.1016/S1473-3099(11)70099-8. [DOI] [PubMed] [Google Scholar]
- 10.Raetz CR, Whitfield C. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rietschel ET, Wollenweber HW, Zahringer U, Luderitz O. Klin Wochenschr. 1982;60:705–709. doi: 10.1007/BF01716559. [DOI] [PubMed] [Google Scholar]
- 12.Ohto U, Fukase K, Miyake K, Satow Y. Science. 2007;316:1632–1634. doi: 10.1126/science.1139111. [DOI] [PubMed] [Google Scholar]
- 13.Rossignol DP, Lynn M. J Endotoxin Res. 2002;8:483–488. doi: 10.1179/096805102125001127. [DOI] [PubMed] [Google Scholar]
- 14.a) Johnson DA, Keegan DS, Sowell CG, Livesay MT, Johnson CL, Taubner LM, Harris A, Myers KR, Thompson JD, Gustafson GL, Rhodes MJ, Ulrich JT, Ward JR, Yorgensen YM, Cantrell JL, Brookshire VG. J Med Chem. 1999;42:4640–4649. doi: 10.1021/jm990222b. [DOI] [PubMed] [Google Scholar]; b) Johnson DA. Curr Top Med Chem. 2008;8:64–79. doi: 10.2174/156802608783378882. [DOI] [PubMed] [Google Scholar]
- 15.Lien E, Chow JC, Hawkins LD, McGuinness PD, Miyake K, Espevik T, Gusovsky F, Golenbock DT. J Biol Chem. 2001;276:1873–1880. doi: 10.1074/jbc.M009040200. [DOI] [PubMed] [Google Scholar]
- 16.Yang D, Satoh M, Ueda H, Tsukagoshi S, Yamazaki M. Cancer Immunol Immunother. 1994;38:287–293. doi: 10.1007/BF01525505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Peri F, Marinzi C, Barath M, Granucci F, Urbano M, Nicotra F. Bioorg Med Chem. 2006;14:190–199. doi: 10.1016/j.bmc.2005.08.047. [DOI] [PubMed] [Google Scholar]; b) Peri F, Granucci F, Costa B, Zanoni I, Marinzi C, Nicotra F. Angew Chem. 2007;46:3308–3312. doi: 10.1002/anie.200604932. [DOI] [PubMed] [Google Scholar]; Peri F, Granucci F, Costa B, Zanoni I, Marinzi C, Nicotra F. Angew Chem Int Ed Engl. 2007;46:3308–3312. doi: 10.1002/anie.200604932. [DOI] [PubMed] [Google Scholar]; c) Piazza M, Rossini C, Della Fiorentina S, Pozzi C, Comelli F, Bettoni I, Fusi P, Costa B, Peri F. J Med Chem. 2009;52:1209–1213. doi: 10.1021/jm801333m. [DOI] [PubMed] [Google Scholar]; d) Piazza M, Calabrese V, Baruffa C, Gioannini T, Weiss J, Peri F. Biochem Pharmacol. 2010;80:2050–2056. doi: 10.1016/j.bcp.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Piazza M, Yu L, Teghanemt A, Gioannini T, Weiss J, Peri F. Biochemistry. 2009;48:12337–12344. doi: 10.1021/bi901601b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park B, Song D, Kim H, Choi B, Lee H, Lee J. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 19.Ohto U, Fukase K, Miyake K, Satow Y. Science. 2007;316:1632–1634. doi: 10.1126/science.1139111. [DOI] [PubMed] [Google Scholar]
- 20.Kim H, Park B, Kim J, Kim S, Lee J, Oh S, Enkhbayar P, Matsushima N, Lee H, Yoo O, Lee J. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 21.Maiti KK, Decastro M, El-Sayed AB, Foote MI, Wolfert MA, Boons GJ. Eur J Org Chem. 2010;2010:80–91. doi: 10.1002/ejoc.200900973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piazza M, Damore G, Costa B, Gioannini TL, Weiss JP, Peri F. Innate immun. 2011;17:293–301. doi: 10.1177/1753425910369020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitchens RL, Munford RS. J Biol Chem. 1995;270:9904–9910. doi: 10.1074/jbc.270.17.9904. [DOI] [PubMed] [Google Scholar]
- 24.a) Muroi M, Tanamoto K. J Biol Chem. 2006;281:5484–5491. doi: 10.1074/jbc.M509193200. [DOI] [PubMed] [Google Scholar]; b) Walsh C, Gangloff M, Monie T, Smyth T, Wei B, McKinley TJ, Maskell D, Gay N, Bryant C. J Immunol. 2008;181:1245–1254. doi: 10.4049/jimmunol.181.2.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Meng J, Drolet JR, Monks BG, Golenbock DT. J Biol Chem. 2010;285:27935–27943. doi: 10.1074/jbc.M110.134668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.a) Qureshi N, Takayama K, Ribi E. J Biol Chem. 1982;257:11808–11815. [PubMed] [Google Scholar]; b) Evans JT, Cluff CW, Johnson DA, Lacy MJ, Persing DH, Baldridge JR. Exp Rev Vaccines. 2003;2:219–229. doi: 10.1586/14760584.2.2.219. [DOI] [PubMed] [Google Scholar]; c) van der Ley P, Steeghs L, Hamstra HJ, ten Hove J, Zomer B, van Alphen L. Infect Immun. 2001;69:5981–5990. doi: 10.1128/IAI.69.10.5981-5990.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiersinga WJ. Curr Opin Crit Care. 2011;17:480–486. doi: 10.1097/MCC.0b013e32834a4aeb. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.