

Abstract
Preparative scale synthesis of fourteen new N2-modified mononucleotide 5’ mRNA cap analogues was achieved. The key step involved use of an SNAr reaction with protected 2-fluoro inosine and various primary and secondary amines. The derivatives were tested in a parasitic nematode, Ascaris suum, cell-free system as translation inhibitors. The most effective compound with IC50 ~ 0.9 μM was a N2-p-metoxybenzyl-7-methylguanosine-5’-monophosphate 35.
Keywords: cap analogue, mRNA, translation inhibitor, nematode, Ascaris suum
1. Introduction
Methylation of the exocyclic amino group of guanosine is an important biological modification of RNAs. Modified nucleosides occur frequently in RNAs including N2-methyl- (m2G) and N2,N2-dimethylguanosine (m22,2G) within ribosomal (rRNA)1 and transfer (tRNA)2 RNAs, respectively. N2,N2-dimethylG as a part of the RNA cap structure is also found at the 5’ end of small nuclear RNAs (U snRNA) and some messenger RNAs (mRNA). Eukaryotic mRNAs possess a 5’ terminal cap structure consisting of a 7-methylguanosine linked via a 5’,5’-triphosphate bridge to the first transcribed nucleotide (m7GpppN, where N=G, A, C or U; MMG cap).3 In nematodes, however, two different mRNAs co-exist in cells. In addition to mRNAs with the common MMG cap, about 70% of the mRNAs possess an atypical, hypermethylated cap. This cap (TMG-cap, m32,2,7GpppG) has the N7-methyl as well as N2,N2-dimethyl in its structure and is added along with a 22 nt spliced leader sequence during trans-splicing.4
The cap plays an essential role in several processes during gene expression5 by interacting with proteins that specifically recognize its structure.6 The best-characterized cap-binding protein is the translation initiation factor eIF4E. Recognition of the mRNA cap by eIF4E is the critical, rate limiting step for efficient translation initiation, and it is a major target for translational control.7 It has been shown that eIF4E is a potent oncogene and its overexpression is associated with a variety of human cancers.8 Consequently, cap analogs as specific inhibitors to counteract elevated eIF4E level in tumor cells have been explored.9
Many nematodes, including Ascaris suum, are parasitic, infect over 2 billion people, and remain a significant health problem. The role of trans-splicing as a mechanism of nematode gene expression and the effect of the TMG cap and a spliced leader addition on mRNA metabolism have been intensively investigated.10 Ascaris cap-binding proteins must deal with two distinct populations of mRNA suggesting these proteins are unique and may be targets for the discovery of novel cap analogs that can specifically block parasite gene expression. We recently prepared and analyzed a guanosine derivative with a benzyl at N2 position (bn2m7GMP, 32) to examine the intermolecular interaction of MMG/TMG caps with Ascaris eIF4E-3.11 The addition of an N2-benzyl substituent on a monophosphate led to significant translation inhibition in an Ascaris suum embryo cell-free system. Therefore, we chose to extend these analyses to several other N2 modified derivatives.
In the present study we designed and synthesized a series of N2-substituted 7-methylguanosine 5’-monophosphates. This type of cap analog has not been widely studied due to difficulties with the development of a good method to efficiently introduce substituents at the N2 position of guanosine. To achieve our goal we used a six step strategy starting from guanosine using an SNAr reaction with fully protected 2-fluoro inosine and various primary and secondary amines. The synthesized mononucleotide TMG cap analogues were tested in a parasitic nematode, Ascaris suum, cell-free system as translation inhibitors.
2. Results and discussion
2.1 Chemistry
In order to synthesize N2-substituted mononucleotide cap analogues, we adopted and modified a method for the preparation of the N2,N2-dimethylguanosine by Eritja et al.12 The synthesis was carried out in six steps using inexpensive, commercially available guanosine as the starting material. The synthesis began with the triacetylation of guanosine using acetic anhydride (Ac2O) in the presence of triethylamine (TEA) and N,N-(dimethylamino)pyridine (DMAP) using a procedure modified from Nair et al.13 By carrying out the addition of the Ac2O at 0°C, N-acetylation was suppressed and 2’,3’,5’-tri-O-acetylguanosine (1) was obtained in a near quantitative yield (98%). In the next step, protection of the O6 group of guanosine was carried out with a p-nitrophenylethyl (NPE) group as it is believed to be stable under mild acid and base hydrolyses, and it can be easily cleaved by DBU or DBN in a β-elimination reaction. The reaction was carried out via the Mitsunobu reaction with p-nitrophenylethanol, diisopropyl azodicarboxylate (DIAD), and triphenylphosphine (TPP) in anhydrous toluene at room temperature.14 A long standing problem with the Mitsunobu reaction is the purification of the desired product from the reaction mixture. We tried replacing triphenylphosphine and diisopropyl azodicarboxylate (which are converted during the reaction into triphenylphosphine oxide and related dialkylhydrazinodicarboxylate, respectively) by other reagents to facilitate the isolation and purification processes.15 Our observations showed that obtaining pure product was possible using standard reagents but the purification required a very long chromatography process. In order to shorten the isolation of the 2’,3’,5’-tri-O-acetyl-O6-[2-(4-nitrophenyl)ethyl] guanosine (2), we partially purified the final product and then used 2 with some impurities (mainly triphenylphosphine oxide) in the next step. By-products remaining after the Mitsunobu reaction were easily removed during the purification of 3. This faster procedure allowed us to obtain a pure fluorinated derivative with good yield (85%).
The main route to N2-substituted guanosine analogues is via nucleophilic displacement of a halogen at the 2 position of inosine. Since fluoro derivatives are more reactive toward nucleophilic displacement in a SNAr reaction than other halogen derivatives,16 the fully protected 2’,3’,5’-tri-O-acetyl-O6-[2-(4-nitrophenyl)ethyl] guanosine (2) was transformed into N2-fluoro-2’,3’,5’-O-triacetyl-O6-[2-(4-nitrophenyl)ethyl] inosine (3). We tested three methods for diazotiation and fluorination of nucleosides. The first was to prepare the 2-fluoro derivative using aqueous diazotization of guanosine in the presence of fluoroboric acid.17 These conditions, however, were too harsh and led to depurination. A second approach was the introduction of the fluorine atom under anhydrous conditions with t-butyl nitrite as the diazotizing agent and HF in pyridine as the fluoride source.18 The reaction proceeded smoothly in very good yield (85%), and the main product remained fully protected. However, without careful monitoring, this reaction led to depurination or even failure to obtain product unless very high quality HF/pyridine reagent was used and a low temperature was maintained during the course of the reaction. Consequently, we also explored milder fluorination conditions using polyvinylpyridinium polyhydrogenfluoride (PVPHF) reagent developed by Olah et al.19 The advantages of this method were easy handling and an extremely convenient work-up, however, yields were limiting. Consequently, the most efficient way to prepare N2-fluoro-2’,3’,5’-O-triacetyl-O6-[2-(4-nitrophenyl)ethyl]inosine (3) is the procedure that involves using HF/pyridine for the substitution step.
The 2-fluoro intermediate was used directly in a SNAr reaction by treating it with two-fold excess of primary or secondary, aliphatic or aromatic amines varying in size and steric branching in anhydrous DMSO (Scheme 2). Incorporated amines were chosen to produce several N2 substituents (aliphatic, cyclic, or aromatic) in the cap to vary the size and steric branching of aliphatic N2 substituents (compounds 11-17) and to influence the electron density of the aromatic ring through the introduction of various substituents, such as metoxy (7), chloro (8), nitro (9), or phenyl (10), into the benzene ring. In addition to the mono substituted analogues, we also used some secondary amines and synthesized compounds bearing additional methyl (5, 11, 13, 16), ethyl (15) or benzyl (6) groups at the N2 position to check the effectiveness of these substituents on the cap as translation inhibitors. The nature of the nucleophile is the most critical parameter in the SNAr reaction. Therefore, for the simple primary amines, the substitution reaction occurred rapidly and the fluorine derivative was used up in less than 30 min at temperature not higher than 50°C. The reaction with sterically hindered and secondary amines such as dibenzylamine required longer time (in some cases even 2 days) and a higher temperature (65°C). After completion of the substitution reaction 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was added to the reaction mixture to remove the NPE group followed by the addition of a mixture of THF/MeOH/NaOHaq in order to complete acetyl deprotection. Chromatographic purification on silica gel yielded pure nucleoside derivatives 4-17 in good yield. It is worth mentioning, that in course of the reaction progress, we observed that all protecting groups (acetyl and NPE) were slowly lost if the substitution reaction was carried out under prolonged heating in the presence of excess amine. This observation led us to a simpler purification procedure in which full deprotection took place without any DBU and final N2-substituted guanosine derivatives were easily crystallized from the reaction mixture by the addition of CH2Cl2.
Scheme 2.
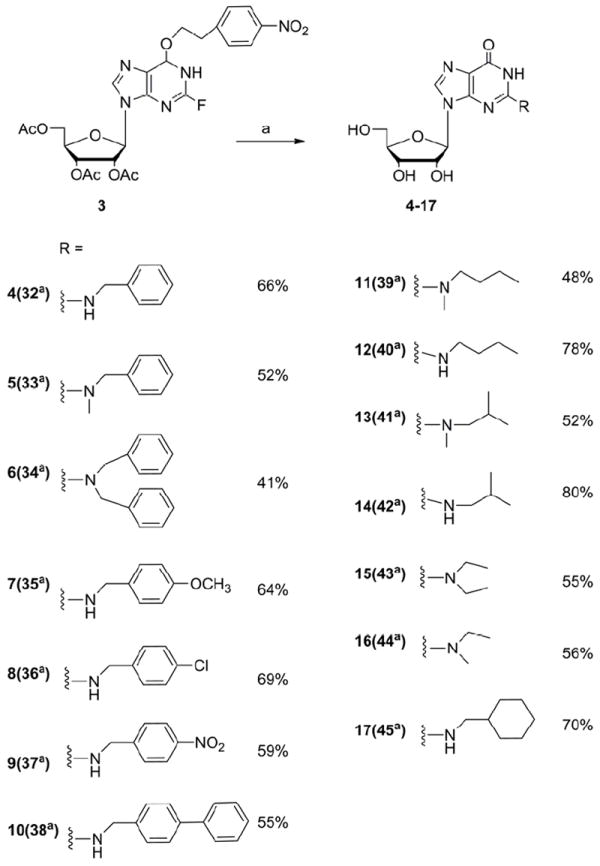
Preparation of N2-modified guanosine derivatives via a SNAr reaction; (a) amine, DMSO, 50°C to 65°C.
a numbers in brackets correspond to compounds on Scheme 3
All the N2-modified guanosine analogues were further 5’ phosphorylated using the Yoshikawa method20 with phosphorus oxide trichloride in trimethyl phosphate at 4°C and subsequently methylated at the N7 position of the guanine ring with CH3I in DMSO at RT21 (Scheme 3). The reaction progress of the last step was monitored by HPLC to avoid over methylation. Purification of the analogues was done using ion-exchange chromatography on DEAE–Sephadex A-25 (HCO3- form) and the derivatives were subsequently converted into the sodium salt using Dowex 50WX8. The structure and homogeneity of final products were confirmed by HPLC, mass spectrometry, 1H NMR and 31P NMR.
Scheme 3.
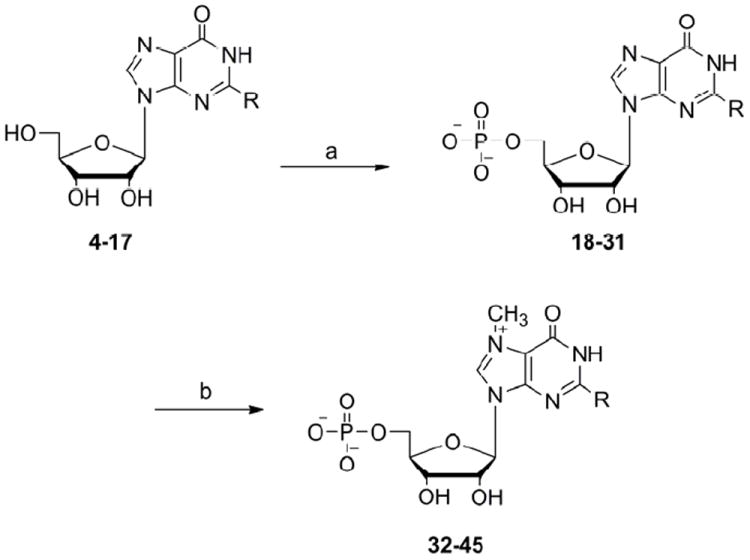
Preparation of N2-modified mononucleotide cap analogues (a) POCl3, trimethyl phosphate, 4°C (b) methyl iodide, DMSO, RT.
aR corresponds to various substituents at N2 position of guanosine (see Scheme 2, numbers in brackets)
2.2 Biology
One of the synthesized compounds, bz2m7GMP (32), has been recently used in Ascaris suum eIF4E-3/m7G- and m32,2,7G- cap binding studies.11 It was shown that bz2m7GMP binds to the protein with a 10-fold higher affinity compared with m7GMP and is a strong inhibitor of either a m7G- or m32,2,7G- capped mRNA translation in a cell-free Ascaris suum translation system. To evaluate the ability of newly synthesized N2-modified 7-methylguanosine 5’-monophosphates to inhibit cap-dependent translation in a cell-free translation system where naturally occurring mono- and trimethylated mRNAs are translated, we tested fourteen cap analogues (32-45) in the same manner as previously reported.10c,11 Two standard cap analogues were used as internal controls in the experiments: m7GTP as a positive control as it is an effective translation inhibitor in various cell-free systems (rabbit reticulocyte,9a wheat germ,23 Ascaris suum10c) and ApppG as a negative control that have very little or no inhibitory efficacy due to lack of the positive charge on guanine that is generated by the N7 methyl group. As previously shown, the inhibitory potency of cap analogues generally increases with the length of polyphosphate bridge.9a Therefore, in order to determine the strength of our new mononucleotide cap analogues we also compared them to m7GMP which is known as a weak translation inhibitor. The results (Figure 1, Table 1) indicated that any mono substitution at the N2 position of guanine (alkyl – aliphatic, cyclic, or aryl) produced inhibitory compounds with IC50 similar to m7GTP. All alkyl substituents led to similar levels of inhibition regardless of their size and steric branching. Aryl substituted compounds were more effective as translation inhibitors. The strongest inhibitor, about 7 times stronger than m7GTP, was derivative 35 with an IC50 of 0.9 μM. The electron withdrawing properties of para substituents (NO2 > Cl > Phe > OCH3) were roughly correlated with the IC50 of the compounds (12.6 μM, 3.8 μM, 4.3 μM, 0.9 μM, respectively). The enhanced π electron density of the benzene ring increases the effectiveness of cap analogues as translational inhibitors, but other factors such as the size of substituent may also play an important role. Recent studies on the co-crystal structure of m32,2,7GTP with Ascaris eIF4E-311 indicated that the two methyl groups at the N2 position of the base are solvent exposed and do not interact directly with the protein. The N2 substituents may form additional interactions with eIF4E influencing conformational changes known to form the binding pocket24 or may change the thermodynamic aspects of the protein binding to the 5’ cap.
Figure 1. Translation inhibition assay in A. suum extract.

The synthesized cap analogues were assayed for their ability to inhibit cap-dependent translation of an m7GpppG-capped Renilla luciferase mRNA in an Ascaris suum embryo cell-free translation system. The measurements were carried out as previously described and the % translation activity plotted against the inhibitor concentration.10c All measurements were made in triplicate in several preparations of extracts. Data presented are representative experiments.
Table 1.
Inhibition of translation in A. suum extract by N2-modified cap analogues
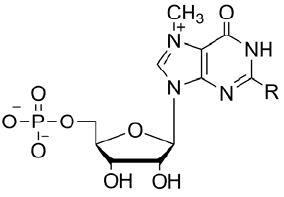
| |||||
|---|---|---|---|---|---|
| R = | IC50 | R = | IC50 | ||
| 32 |
|
2.9 μM | 39 |
|
>50 μM |
| 33 |
|
>50 μM | 40 |
|
3.9 μM |
| 34 |
|
>50 μM | 41 |
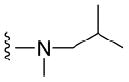
|
>50 μM |
| 42 |
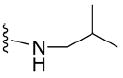
|
4.5 μM | |||
| 35 |
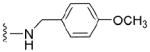
|
0.9 μM | 43 |
|
>50 μM |
| 36 |
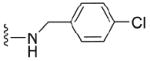
|
3.8 μM | 44 |
|
13.9 μM |
| 37 |
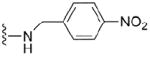
|
12.6 μM | 45 |
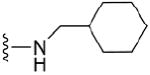
|
5.7 μM |
| 38 |
|
4.3 μM | m7GTP | 6.1 μM | |
| m7GMP | >50μM | ||||
IC50 values were extracted directly from the plotted of % translation activity values against compound concentration. IC50 values > 50 μM - no measurable effect.
Our results also showed that the addition of a second substituent at the N2 position of guanosine decreased the level of inhibition significantly (e. g compounds 32 versus 33 or 39 versus 40). These data are consistent with a comparison of the crystal structures of Ascaris eIF4E-3 with m7GTP or m32,2,7GTP.11 These studies demonstrated that the addition of the second substituent at the N2 position of guanosine leads to the loss of one hydrogen bond between the amine group of guanine and the carboxyl group of Glu116. This loss of a hydrogen bond likely influences the effectiveness of analogues as translation inhibitors.
3. Conclusions
We have prepared fourteen novel N2 modified trimethylguanosine cap analogues via a six-step synthesis from guanosine. This general strategy provides a new approach to scale-up the synthesis of a large number of modified cap analogues that should be useful in studying eIF4E function and cap-dependent translation. In addition the idea of exploring cap analogues that possess only one phosphate that typically have little inhibitory activity offers an opportunity to explore compounds that are not highly charged and can be good candidates for new drug development.
4. Materials and methods
All reagents were the highest available purity and purchased from Sigma-Aldrich Chemical Co. Triethylammonium bicarbonate (TEAB) buffer was prepared by bubbling CO2 through an ice-cold aqueous solution of redistilled triethylamine. Intermediate nucleotides were separated by ion-exchange chromatography on a DEAE-Sephadex A-25 (HCO3- form) using a linear gradient of TEAB buffer, pH 7.6. Fractions containing products were combined and evaporated under reduced pressure with several additions of ethanol and isolated as triethylammonium salts (TEA salts) and subsequently converted into the sodium salt using Dowex 50WX8 (Na+ form). Homogeneity of the final analogues was checked by reversed-phase analytical HPLC. HPLC was performed using a Supelcosil LC-18-T RP column (4.6 × 250 mm, flow rate 1.0 mL/min) with: Method A – a linear gradient of methanol from 0 to 50% (v/v) in 0.05 M ammonium acetate (pH 5.9) in 20 minutes, an isocratic elution of 50% methanol (v/v) in 0.05 M ammonium acetate (pH 5.9) till 30 minutes, Method B – a linear gradient of methanol from 0 to 50% (v/v) in 0.05 M ammonium acetate (pH 5.9) in 10 minutes and then an isocratic elution of 50% methanol (v/v) in 0.05 M ammonium acetate (pH 5.9) till 30 minutes, Method C – an isocratic elution of 50% methanol (v/v) in 0.05 M ammonium acetate (pH 5.9), on a Knauer instrument, with UV detection at 254 nm. MS spectra were acquired using Waters Micromass Q-TOF Premier spectrometer with positive electrospray ionization source. 1H and 13C NMR spectra of intermediate derivatives were obtained with a Varian UnityPlus 200 MHz spectrometer. 1H and 31P NMR spectra of the final compounds were recorded on a Varian INOVA 700 MHz spectrometer.
4.1 Experimental
4.1.1 2’,3’,5’-tri-O-acetylguanosine (1)
2.2 mL of acetic anhydride (22 mmol) was added dropwise to a suspension of guanosine (2 g, 7.0 mmol) (dried for 2 days over P4O10 in high vacuum), triethylamine (7.7 mL, 55.2 mmol) and N,N-(dimethylamino)pyridine (92 mg, 0.75 mmol) in 27 mL of acetonitrile at 0°C. The mixture was stirred until it became homogeneous and kept an additional 3 hr at room temperature. The reaction was quenched with methanol (2.3 mL). The volume was reduced to 1/3 using a rotary evaporator and diethyl ether was added dropwise to induce precipitation of a fine white powder. The product was collected by filtration, washed with diethyl ether, and then stirred for 2 hr with acetone (30 mL) at 50°C. The filtrate produced 2.8 g (98%) of a fine white powder. 1H NMR (200 MHz, DMSO) δ 10.54 (s, 1H, NH) 7.93 (s, 1H, H-8), 6.34 (br, 2H, NH2), 5.98 (d, J = 6.1 Hz, 1H, H-1’), 5.79 (t, J = 5.8 Hz, 1H, H-2’), 5.51 – 5.48 (m, 1H, H-3’), 4.40 – 4.37 (m, 1H, H-4’), 4.33 – 4.31 (m, 1H, H-5’), 4.23 – 4.28 (m, 1H, H-5”), 2.11 (s, 3H, CH3, acetyl), 2.05 (s, 3H, CH3, acetyl), 2.04 (s, 3H, CH3, acetyl); 13C NMR (200 MHz, CDCl3) δ 172.1 (C=O), 171.0 (C=O), 169.9 (C=O), 156.3 (C-6), 152.8 (C-2), 151.2 (C-4), 137.6 (C-8), 119.6 (C-5), 86.1 (C-1’), 82.3 (C-4’), 73.6 (C-2’), 72.3 (C-3’), 61.8 (C-5’), 21.1 (C, acetyl), 20.6 (C, acetyl), 20.2 (C, acetyl); m/z: calcd for C18H21N5O5 (M + H)+: 410.1306, found: 410.1305
4.1.2 2’,3’,5’-tri-O-acetyl-O6-[2-(4-nitrophenyl)ethyl]guanosine (2)
A suspension of 2’,3’,5’-tri-O-acetylguanosine (2.37 g, 5.8 mmol), triphenylphosphine (2.28 g, 8.7 mmol) and 2-(4-nitrophenyl)ethanol (1.45 g, 8.7 mmol) in anhydrous toluene was stirred for 30 min and diisopropyl azodicarboxylate (1.4 mL) was added dropwise over a period of 45 min. The reaction mixture was kept for 12 hours at RT. Then the solvent was evaporated and the residual oil was purified by column chromatography on silica gel with chloroform to produce a pure product of yellowish crystals, 2.26 g (70%). 1H NMR (200 MHz, CDCl3) δ 8.17 (d, 2H, Ph), 7.72 (s, 1H, H-8), 7.49 (d, 2H, Ph), 6.05 – 5.91 (m, 2H, H-1’, H-2’), 5.87 – 5.75 (m, 1H, H-3’), 4.73 (t, J = 6.7 Hz, 2H, O-CH2, NPE), 4.50 – 4.36 (m, 3H, H-4’, H-5’, H-5”), 3.28 (t, 6.7 2H, CH2Ph, NPE), 2.14 (s, 3H, CH3, acetyl), 2.09 (s, 3H, CH3, acetyl), 2.08 (s, 3H, CH3, acetyl); 13C NMR (200 MHz, CDCl3) δ 171.7 (C=O), 171.0 (C=O), 169.7 (C=O), 161.9, 155.5 (C-6), 154.5 (C-2), 148.8, 148.6, 138.9 (C-8), 129.3 (C, Ph), 124.8 (C, Ph), 113.6 (C-5), 86.8 (C-1’), 82.3 (C-4’), 73.4 (C-2’), 70.6 (C-3’), 67.1 (OCH2, NPE), 62.5 (C-5’), 35.5 (CH2Ph), 21.2 (C, acetyl), 20.6 (C, acetyl), 20.2 (C, acetyl); m/z: calcd for C24H26N6O10 (M + H)+: 559.1783, found: 559.1784
4.1.3 N2-fluoro-2’,3’,5’-O-triacetyl-O6-[2-(4-nitrophenyl)ethyl]inosine (3)
Dry 2’,3’,5’-tri-O-acetyl-O6-[2-(4-nitrophenyl)ethyl]guanosine (1 g, 1.79 mmol) in polypropylene tube under nitrogen was dissolved in anhydrous pyridine (6.75 mL, 0.082 mol). The tube was placed in a dry ice/acetonitrile cooling bath (-35 to -45°C) and 70% HF/pyridine solution (12 mL, 0.42 mol) was added dropwise over a period of 5 min to 45% final HF. The reaction mixture was stirred for 15 min and t-butyl nitrite (0.54 mL, 4.5 mmol) added. After 1hr, the reaction was quenched at 0°C by slowly pouring the reaction mixture into an aqueous K2CO3 solution (28.5 g in 25 mL of water) and then extracted three times with ethyl acetate. The organic layers were collected, dried over anhydrous Na2SO4 and evaporated to dryness. Purification by column chromatography using as eluate 60:1 CH2Cl2 : MeOH gave 0.85 g (85%) of product; TLC silica gel, CH2Cl2 : MeOH, 60:1 RF = 0.4; 1H NMR (700 MHz, CDCl3) δ 8.21 – 8.15 (m, 2H, Ph), 8.08 (s, 1H, H-8), 7.50 (d, 2H, Ph), 6.13 (d, J = 5.6 Hz, 1H, H-1’), 5.82 (t, J = 4.9 Hz, 1H, H-2’), 5.60 – 5.55 (m, 1H, H-3’), 4.84 (t, J = 6.7 Hz, 2H, O-CH2, NPE), 4.46 – 4.42 (m, 2H, H-4’, H-5’), 4.38 – 4.36 (m, 1H, H-5”), 3.32 (t, J = 6.7 Hz, 2H, CH2Ph, NPE), 2.15 (s, 6H, CH3, acetyl), 2.08 (s, 3H, CH3, acetyl). 13C NMR (200 MHz, CDCl3) δ 170.2 (C=O), 169.5 (C=O), 169.3 (C=O), 160.3, 159.3 (C-6), 154.2 (C-2), 147.1, 145.1, 140.8, 129.9 (Ph), 123.8 (Ph), 86.4 (C-1’), 80.5 (C-4’), 73.0 (C-2’), 72.4 (C-3’), 67.7 (OCH2, NPE), 62.8 (C-5’), 34.9 (CH2Ph), 20.7 (C, acetyl), 20.5 (C, acetyl), 20.3 (C, acetyl); m/z: calcd for C24H24FN5O10 (M + H)+: 562.1579, found: 562.1581
4.1.4 General procedure for the synthesis of N2-substitutiuted derivatives 4-17
N2-fluoro-2’,3’,5’-O-triacetyl-O6-[2-(4-nitrophenyl)ethyl]inosine (250 mg, 0.45 mmol) was dissolved in 2 mL of anhydrous dimethylsulfoxide (DMSO) and then an amine (0.9 mmol) was added. The reaction mixture was stirred at 60°C from 30 minutes to 2 days until the fluoronucleoside completely disappeared (based on TLC) followed by addition of 0.5M NaOH in THF/MeOH/H2O (5/4/2). The solvent was removed under high vacuum and the resulting oily residue was treated with CH2Cl2 to induce precipitation of a fine white powder. The product was collected by filtration and washed several times with methanol and dried over P4O10 to yield:
4.1.4.1 N2-benzylguanosine (4)
134 mg, 66%; 1H NMR (200 MHz, CDCl3) δ 7.69 (s, 1H, H-8), 7.38 – 7.19 (m, 5H, Ph), 5.66 (d, J=5.6 Hz, 1H, H-1’), 4.64 – 4.62 (m, 1H, H-2’), 4.55 – 4.47 (m, 2H, CH2Ph), 4.12 – 4.10 (m, 1H, H-3’), 3.89 – 3.84 (m, 1H, H-4’), 3.69 – 3.57 (m, 2H, H-5’); 13C NMR (50 MHz, CDCl3) δ 157.4 (C-6), 153.8 (C-2), 150.6 (C-4), 140.4 (Ph) 134.6 (C-8), 128.0 (Ph), 127.2 (Ph), 126.2 (Ph), 117.1 (C-5), 87.5 (C-1’), 85.1 (C-4’), 73.0 (C-2’), 70.6 (C-3’), 61.7 (C-5’), 44.2 (N-CH2Ph); m/z: calcd for C17H19N5O5 (M + H)+: 374.1458, found: 374.1453;
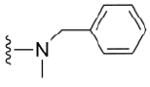
4.1.4.2 N2-benzyl, N2-methylguanosine (5)
109 mg, 52%; 1H NMR (200 MHz, CDCl3) δ 7.66 (s, 1H, H-8), 7.26 – 7.14 (m, 5H, Ph), 5.64 (d, J=5.5 Hz, 1H, H-1’), 4.62 – 4.60 (m, 1H, H-2’), 4.50 – 4.42 (m, 2H, CH2Ph), 4.13 – 4.11 (m, 1H, H-3’), 3.81 – 3.74 (m, 1H, H-4’), 3.62 – 3.51 (m, 2H, H-5’), 2.88 (s, 3H, N7-CH3); 13C NMR (50 MHz, CDCl3) δ 157.6 (C-6), 151.1 (C-2), 149.4 (C-4), 140.7, 137.2, 128.6 (Ph), 128.5 (Ph), 127.2 (Ph), 119.3 (C-5), 87.9 (C-1’), 84.6 (C-4’), 73.2 (C-2’), 71.9 (C-3’), 62.3 (C-5’), 44.8 (N-CH2Ph), 37.0 (N-CH3); m/z: calcd for C18H21N5O5 (M + H)+: 388.1615, found: 388.1613;
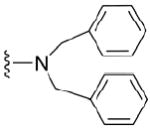
4.1.4.3 N2, N2-dibenzylguanosine (6)
103 mg, 41%, 1H NMR (200 MHz, CDCl3) δ 7.59 (s, 1H, H-8), 7.36 – 7.29 (m, 10H, Ph), 5.67 (d, J=5.6 Hz, 1H, H-1’), 4.68 (s, 4H, CH2Ph), 4.61 (t, J=5.0 Hz, 1H, H-2’), 4.16 – 4.13 (m, 1H, H-3’), 3.87 – 3.83 (m, 1H, H-4’), 3.60 – 3.43 (m, 2H, H-5’); 13C NMR (50 MHz, CDCl3) δ 155.8 (C-6), 152.2 (C-2), 150.3 (C-4), 139.8, 137.3, 128.5 (Ph), 128.2 (Ph), 127.1 (Ph), 119.6 (C-5), 88.9 (C-1’), 85.8 (C-4’), 74.4 (C-2’), 72.0 (C-3’), 62.3 (C-5’), 54.3 (N-CH2Ph). m/z: calcd for C24H25N5O5 (M + H)+: 464.1928, found: 464.1918;
4.1.4.4 N2-p-metoxybenzylguanosine (7)
140 mg, 64%, 1H NMR (200 MHz, CDCl3) δ 8.02 (s, 1H, H-8), 7.27 (d, 2H, Ph), 6,85 (d, 2H, Ph), 5.92 (d, J=5.4 Hz, 1H, H-1’), 4.73 (t, J=4.9 Hz, 1H, H-2’), 4.48 (s, 2H, CH2Ph) 4.21 - 4.18 (m, 1H, H-3’), 3.93 – 3.90 (m, 1H, H-4’), 3.72 – 3.68 (m, 2H, H-5’), 3,59 (s, 3H, O-CH3); 13C NMR (50 MHz, CDCl3) δ 158.9 (C-6), 155.9, 154.1, 150.2, 137.8, 132.8, 127.9, 118.9, 113.9, 88.9 (C-1’), 86.1 (C-4’), 74.3 (C-2’), 72.0 (C-3’), 62.7 (C-5’), 56.0 (OCH3), 44.6 (N-CH2Ph). m/z: calcd for C18H21N5O6 (M + H)+: 404.1564, found: 404.1560;
4.1.4.5 N2-p-chlorobenzylguanosine (8)
153 mg, 69%, 1H NMR (200 MHz, CDCl3) δ 8.04 (s, 1H, H-8), 7.36 – 7.30 (m, 4H, Ph), 5.90 (d, J=5.3 Hz, 1H, H-1’), 4.65 (t, J=4.8 Hz, 1H, H-2’), 4.35 (s, 2H, CH2Ph), 4.18 – 4.15 (m, 1H, H-3’), 3.91 – 3.88 (m, 1H, H-4’), 3.70 – 3.67 (m, 2H, H-5’); 13C NMR (50 MHz, CDCl3) δ 157.2 (C-6), 154.8 (C-2), 150.4 (C-4), 137.9, 136.7, 131.3, 129.2 (Ph), 128.3 (Ph), 119.8 (C-5), 88.6 (C-1’), 85.5 (C-4’), 74.3 (C-2’), 71.7 (C-3’), 62.4 (C-5’), 44.8 (N-CH2Ph). m/z: calcd for C17H18ClN5O5 (M + H)+: 408.1069, found: 408.1073;
4.1.4.6 N2-p-nitrobenzylguanosine (9)
134 mg, 59%, 1H NMR (200 MHz, CDCl3) δ 8.04 (s, 1H, H-8), 8.22 (d, 2H, Ph), 7.63 (d, 2H, Ph), 5.84 (d, J=5.2 Hz, 1H, H-1’), 4.63 (s, 2H, CH2Ph) 4.66 – 4.64 (m, 1H, H-2’), 4.18 – 4.15 (m, 1H, H-3’), 3.98 – 3.95 (m, 1H, H-4’), 3.75 – 3.71 (m, 2H, H-5’); 13C NMR (50 MHz, CDCl3) δ 156.6 (C-6), 153.7 (C-2), 150.9, 147.8, 146.4, 136.9 (C-8), 128.2 (Ph), 123.9 (Ph), 118.8 (C-5), 88.4 (C-1’), 86.8 (C-4’), 72.7 (C-2’), 71.2 (C-3’), 62.2 (C-5’), 43.8 (N-CH2Ph). m/z: calcd for C17H18N6O7 (M + H)+: 419. 1309, found: 419.1312;
4.1.4.7 N2-p-phenylbenzylguanosine (10)
134 mg, 55%, 1H NMR (200 MHz, CDCl3) δ 7.98 (s, 1H, H-8), 7.75 – 7.71 (m, 9H, Ph), 5.89 (d, J=5.3 Hz, 1H, H-1’), 4.69 – 4.65 (m, 2H, CH2Ph), 4.61 (t, J=4.9 Hz, 1H, H-2’), 4.16 – 4.14 (m, 1H, H-3’), 3.89 – 3.81 (m, 1H, H-4’), 3.64 – 3.41 (m, 2H, H-5’); 13C NMR (50 MHz, CDCl3), δ 158.7 (C-6), 153.2 (C-2), 151.8 (C-4), 140.9, 140.8, 138.1, 137.5, 129.1 (Ph), 128.5 (Ph), 127.7 (Ph), 127.6 (Ph), 127.3 (Ph), 117.4 (C-5), 88.3 (C-1’), 87.5 (C-4’), 75.6 (C-2’), 72.8 (C-3’), 63.7 (C-5’), 43.7 (N-CH2Ph); m/z: calcd for C23H23N5O5 (M + H)+: 450.1771, found: 450.1773;
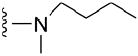
4.1.4.8 N2-butyl-N2-methylguanosine (11)
92 mg, 48%, 1H NMR (200 MHz, CDCl3) δ 8.11 (s, 1H, H-8), 5.72 (d, J=4.9 Hz, 1H, H-1’), 4.69 (t, J=5.3 Hz, 1H, H-2’), 4.13 (t, J=3.9 Hz, 1H, H-3’), 3.76 – 3.73 (m, 1H, H-4’), 3.63 – 3.61 (m, 2H, N-CH2), 3.58 – 3.46 (m, 2H, H-5’), 3.09 (s, 3H, N-CH3), 1.60 – 1.52 (m, 2H, CH2 butyl), 1.38 – 1.31 (m, 2H, CH2CH3 butyl), 0.90 (t, J = 7.3 Hz, 3H, CH3 butyl); 13C NMR (50 MHz, CDCl3), δ 158.2 (C-6), 152.3 (C-2), 140.3 (C-4), 136.9 (C-8), 118.5 (C-5), 87.9 (C-1’), 85.6 (C-4’), 74.3 (C-2’), 71.4 (C-3’), 62.4 (C-5’), 51.2 (N-CH2 butyl), 38.5 (N-CH3), 27.8 (CH2 butyl), 20.3 (CH2 butyl), 13.7 (CH3 butyl). m/z: calcd for C15H23N5O5 (M + H)+: 354. 1771, found: 354.1768;
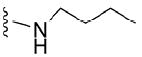
4.1.4.9 N2-butylguanosine (12)
144 mg, 78%, 1H NMR (200 MHz, CDCl3) δ 8.09 (s, 1H, H-8), 5.68 (d, J=4.8 Hz, 1H, H-1’), 4.64 – 4.59 (m, 1H, H-2’), 4.11 – 4.08 (m, 1H, H-3’), 3.76 – 3.73 (m, 1H, H-4’), 3.63 (t, J=6.2, 2H, N-CH2), 3.58 – 3.46 (m, 2H, H-5’), 1.63 – 1.54 (m, 2H, CH2 butyl), 1.28 – 1.25 (m, 2H, CH2CH3 butyl), 0.93 (t, J = 7.3 Hz, 3H, CH3 butyl); 13C NMR (50 MHz, CDCl3) δ 157.6 (C-6), 154.4 (C-2), 150.3 (C-4), 137.8 (C-8), 117.8 (C-5), 86.8 (C-1’), 84.9 (C-4’), 74.0 (C-2’), 72.3 (C-3’), 62.1 (C- 5’), 42.3 (N-CH2 butyl), 30.7 (CH2 butyl), 20.4 (CH2 butyl), 14.3 (CH3 butyl). m/z: calcd for C14H21N5O5 (M + H)+: 340. 1615, found: 340.1619;
4.1.4.10 N2-isobutyl-N2-methylguanosine (13)
100 mg, 52%, 1H NMR (200 MHz, CDCl3) δ 7.55 (s, 1H, H-8), 5.64 (d, J=5.2 Hz, 1H, H-1’), 4.64 (t, J=5.0 Hz, 1H, H-2’), 4.16 (t, J=3.8 Hz, 1H, H-3’), 3.86 – 3.81 (m, 1H, H-4’), 3.74 – 3.71 (m, 2H, N-CH2), 3.60 – 3.45 (m, 2H, H-5’), 2.97 (s, 3H, N-CH3), 1.89 – 1.85 (m, 1H, CH isobutyl), 0.94 (d, J=6.9, 6H, CH3 isobutyl); 13C NMR (50 MHz, CDCl3) δ 156.8 (C-6), 152.4 (C-2), 141.9 (C-4), 136.7 (C-8), 119.8 (C-5), 89.3 (C-1’), 85.1 (C-4’), 74.0 (C-2’), 72.3 (C-3’), 62.8 (C-5’), 52.5 (N-CH2 isobutyl), 38.6 (N-CH3), 25.2 (CH isobutyl), 20.4 (CH3 isobutyl). m/z: calcd for C15H23N5O5 (M + H)+: 354. 1771, found: 354.1777;
4.1.4.11 N2-isobutylguanosine (14)
147 mg, 80%,1H NMR (200 MHz, CDCl3) δ 7.64 (s, 1H, h-8), 5.61 (d, J=5.5 Hz, 1H, H-1’), 4.64 – 4.61 (m, 1H, H-2’), 4.15 (t, J=3.9 Hz, 1H, H-3’), 3.87 – 3.83 (m, 1H, H-4’), 3.61 – 3.58 (m, 2H, N-CH2), 3.58 – 3.47 (m, 2H, H-5’), 1.90 – 1.86 (m, 1H, CH isobutyl), 0.92 (d, J=6.5, 6H, CH3 isobutyl); 13C NMR (50 MHz, CDCl3) δ 155.9 (C-6), 151.9 (C-2), 149.2 (C-4), 137.5 (C-8), 119.8 (C-5), 87.7 (C-1’), 85.6 (C-4’), 74.6 (C-2’), 72.8 (C-3’), 62.5 (C-5’), 50.3 (N-CH2 isobutyl), 28.7 (CH isobutyl), 19.2 (CH3 isobutyl). m/z: calcd for C14H21N5O5 (M + H)+: 340.1615, found: 340.1615;
4.1.4.12 N2, N2-diethylguanosine (15)
101 mg, 55%,1H NMR (200 MHz, CDCl3) δ 7.58 (s, 1H, H-8), 5.65 (d, J=5.0 Hz, 1H, H-1’), 4.63 (br, 1H, H-2’), 4.15 (br, 1H, H-3’), 3.83 – 3.78 (m, 3H, H-4’, H-5’), 3.69 – 3.57 (m, 4H, CH2 ethyl), 1.06 (t, 6H, CH3 ethyl); 13C NMR (50 MHz, CDCl3) δ 156.8 (C-6), 151.8 (C-2), 141.6 (C-4), 136.9 (C-8), 117.3 (C-5), 87.5 (C-1’), 86.2 (C-4’), 74.2 (C-2’), 71.9 (C-3’), 61.8 (C-5’), 42.6 (N-CH2 ethyl), 13.1 (CH3 ethyl). m/z: calcd for C14H21N5O5 (M + H)+: 340.1615, found: 340.1610;
4.1.4.13 N2-ethyl-N2-methylguanosine (16)
100 mg, 56%,1H NMR (200 MHz, CDCl3) 7.79 (s, 1H, H-8), 5.67 (d, J=5.1 Hz, 1H, H-1’), 4.72 – 4.69 (t, J=5.2, 1H, H-2’), 4.16 (br, 1H, H-3’), 3.83 – 3.78 (m, 1H, H-4’), 3.69 – 3.57 (m, 2H, H-5’), 3.62 (q, J = 6.8 Hz, 2H, CH2 ethyl), 3.12 (s, 3H, N-CH3), 1.22 (t, J = 7.1 Hz, 3H, CH3 ethyl); δ 13C NMR (50 MHz, CDCl3) δ 157.7 (C-6), 152.4 (C-2), 140.2 (C-4), 137.5 (C-8), 119.2 (C-5), 88.8 (C-1’), 85.9 (C-4’), 75.0 (C-2’), 71.3 (C-3’), 63.4 (C-5’), 44.4 (N-CH2 ethyl), 37.5 (N-CH3), 11.9 (CH3 ethyl). m/z: calcd for C13H19N5O5 (M + H)+: 326.1458, found: 326.1463;
4.1.4.14 N2-cyclohexylmethylguanosine (17)
144 mg, 70%,1H NMR (200 MHz, CDCl3) δ 7.82 (s, 1H, H-8),5.68 (d, J = 3.8 Hz, 1H, H-1’), 4.72 (t, J = 4.9 Hz, 1H, H-2’), 4.15 (t, J = 5.7 Hz, 1H, H-3’), 3.85 – 3.80 (m, 1H, H-4’), 3.61 – 3.52 (m, 2H, H-5’), 3.32 – 3.28 (m, 2H, N-CH2), 1.82 – 0.98 (m, 11H, cyclohexyl); 13C NMR (50 MHz, CDCl3) δ 156.1 (C-6), 153.2 (C-2), 150.4 (C-4), 137.9 (C-8), 120.1 (C-5), 87.9 (C-1’), 86.6 (C-4’), 73.9 (C-2’), 71.9 (C-3’), 62.3 (C-5’), 44.9 (N-CH2), 35.7 (cyclohexyl), 29.9 (cyclohexyl), 25.8 (cyclohexyl), 25.1 (cyclohexyl). m/z: calcd for C17H25N5O5 (M + H)+: 380.1928, found: 380.1925;
4.1.5 General procedure for the synthesis of N2-substitutiuted 5’- monophosphates 32-45
Phosphorus oxide trichloride (POCl3) (3.5 eq) in trimethyl phosphate (0.05 eq) was cooled to 0°C and added to the dried over P4O10 compound 4-17 (1 eq). The reaction mixture was stirred at 0°C. After 2 hr, 1 M aqueous TEAB was added to neutralize the pH of the reaction mixture. The product was purified by ion-exchange chromatography on a DEAE-Sephadex A-25 column using a linear 0–1 M TEAB gradient to produce products 18-31 as TEA salts. Methyl iodide (7 eq) was added to a suspension of mononucleotide derivative (18-31) in 1.5 mL anhydrous dimethylsulfoxide (DMSO) and stirred at room temperature for 2 hr. The mixture was poured into water and extracted three times with diethyl ether. The aqueous phase was purified on DEAE–Sephadex using a linear 0 - 0.8 M gradient of TEAB.
4.1.5.1 N2-benzyl-7-methylguanosine-5’-monophosphate (32)
129 mg, 65%, sodium salt; 1H NMR (700 MHz, D2O) δ 7.46 – 7.32 (m, 5H, Ph), 6.07 (d, J = 3.4 Hz, 1H, H-1’), 4.61 (s, 2H, CH2Ph), 4.58 (t, J = 4.7 Hz, 1H, H-2’), 4.43 (t, J =5.7 Hz, 1H, H-3’), 4.35 – 4.33 (m, 1H, H-4’), 4.15 – 4.11 (m, 1H, H-5’), 4.09 (s, 3H, N7-CH3), 4.00 – 3.97 (m, 1H, H-5”); 31P NMR (283 MHz, D2O) 3.234; m/z: calcd for C18H23N5O8P: 468.1127, found: 468.1131; HPLC (Method B) tR 9 min.
4.1.5.2 N2-benzyl-N2,7-dimethylguanosine-5’-monophosphate (33)
110 mg, 68%, sodium salt; 1H NMR (700 MHz, D2O) δ 7.45 – 7.31 (m, 5H, Ph), 6.11 (d, J = 3.5 Hz, 1H, H-1’), 4.68 (s, 2H,CH2Ph), 4.62 (t, J = 4.7 Hz, 1H, H-2’), 4.44 (t, J = 5.4 Hz, 1H, H-3’), 4.35 – 4.32 (m, 1H, H-4’), 4.15 – 4.11 (m, 1H, H-5’), 4.10 (s, 3H, N7-CH3), 4.01 – 3.97 (m, 1H, H-5”), 3.17 (s, 3H, N2-CH3); 31P NMR (283 MHz, D2O) 3.139; m/z: calcd for C19H25N5O8P : 482.1284, found: 482.1287; HPLC (Method B) tR 13 min.
4.1.5.3 N2,N2-dibenzyl-7-dimethylguanosine-5’-monophosphate (34)
84 mg, 58%, sodium salt; 1H NMR (700 MHz, D2O) δ 7.42 – 7.28 (m, 10H, 2x Ph), 6.07 (d, J = 3.6 Hz, 1H, H-1’), 4.82 (s, 4H, CH2Ph), 4.56 (t, J = 4.9 Hz, 1H, H-2’), 4.42 (t, J = 5.6 Hz, 1H, H-3’), 4.31 – 4.28 (m, 1H, H-4’), 4.09 (s, 3H, N7-CH3), 4.08 – 4.06 (m, 1H, H-5’), 3.97 – 3.93 (m, 1H, H-5”); 31P NMR (283 MHz, D2O) 3.085; m/z: calcd for C25H29N5O8P: 558.1597, found: 558.1592; HPLC (Method B) tR 20.5 min.
4.1.5.4 N2-p-metoxybenzyl-7-methylguanosine-5’-monophosphate (35)
134 mg, 66%, sodium salt; 1H NMR (700 MHz, D2O) δ 7.39 (d, 2H, Ph), 6.95 (d, 2H, Ph), 6.08 (d, J = 3.4 Hz, 1H, H-1’), 4.60 (t, J = 4.7 Hz, 1H, H-2’), 4.51 (s, 2H, CH2Ph), 4.44 (t, J = 5.7 Hz, 1H, H-3’), 4.36 – 4.34 (m, 1H, H-4’), 4.17 – 4.13 (m, 1H, H-5’), 4.07 (s, 3H, N7-CH3), 4.02 – 3.98 (m, 1H, H-5”), 3.79 (s, 3H, OCH3); 31P NMR (283 MHz, D2O) 3.011; m/z: calcd for C19H25N5O9P: 498.1233, found: 498.1237; HPLC (Method B) tR 12.25 min.
4.1.5.5 N2-p-chlorobenzyl-7-methylguanosine-5’-monophosphate (36)
119 mg, 53 %, sodium salt; 1H NMR (700 MHz, D2O) δ 7.40 (s, 4H, Ph), 5.90 (d, J=3.6, 1H, H-1’), 4.66 – 4.62 (m, 1H, H-2’), 4.50 (s, 2H, CH2Ph), 4.35 - 4.32 (m, 1H, H-3’), 4.29 – 4.26 (m, 1H, H-4’), 4.09 (s, 3H, N7-CH3), 4.07 – 3.97 (m, 2H, H-5’, H-5”); 31P NMR (283 MHz, D2O) 2.590; m/z: calcd for C18H22N5O8PCl: 502.0894, found: 502.0897; HPLC (Method B) tR 15 min.
4.1.5.6 N2-p-nitrobenzyl-7-methylguanosine-5’-monophosphate (37)
96 mg, 49%, sodium salt; 1H NMR (700 MHz, D2O) δ 8.25 (d, 2H, Ph), 7.62 (d, 2H, Ph), 6.01 (d, J = 3.5 Hz, 1H, H-1’), 4.68 (s, 2H, CH2Ph), 4.52 – 4.50 (m, 1H, H-2’), 4.41 – 4.39 (m, 1H, H-3’), 4.32 – 4.29 (m, 1H, H-4’), 4.10 – 4.09 (m, 1H, H-5’), 4.07 (s, 3H, N7-CH3), 3.98 – 3.94 (m, 1H, H-5”); 31P NMR (283 MHz, D2O) 3.339; m/z: calcd for C18H22N6O10P: 513.0978, found: 513.0976; HPLC (Method C) tR 4.5 min.
4.1.5.7 N2-p-phenylbenzyl-7-methylguanosine-5’-monophosphate (38)
114 mg, 60 %, sodium salt; 1H NMR (700 MHz, D2O) δ 7.84 – 7.41 (m, 9H, Ph-Ph), 6.09 (d, J = 3.5 Hz, 1H, H-1’), 4.69 (s, 2H, CH2Ph), 4.64 (t, J = 4.7 Hz, 1H, H-2’), 4.46 – 4.44 (m, 1H, H-3’), 4.39 – 4.32 (m, 1H, H-4’), 4.19 – 4.16 (m, 1H, H-5’), 4.08 (s, 3H, N7-CH3), 4.05 – 4.01 (m, 1H, H-5”); 31P NMR (283 MHz, D2O) 3.121; m/z: calcd for C24H25N5O8P: 542.1440, found: 542.1443; HPLC (Method C) tR 15.4 min.
4.1.5.8 N2-butyl-N2,7-dimethylguanosine-5’-monophosphate (39)
78 mg, 55%, sodium salt; 1H NMR (700 MHz, D2O) δ 6.11 (d, J = 3.2 Hz, 1H, H-1’), 4.69 (t, J = 4.7 Hz, 1H, H-2’), 4.46 (t, J = 5.9 Hz, 1H, H-3’), 4.38 – 4.35 (m, 1H, H-4’), 4.22 – 4.18 (m, 1H, H-5’), 4.09 (s, 3H, N7-CH3), 4.06 – 4.03 (m, 1H, H-5”), 3.68 – 3.60 (m, 2H, CH2 butyl), 3.15 (s, 3H, N2-CH3), 1.66 – 1.60 (m, 2H, CH2 butyl), 1.37 – 1.28 (m, 2H, CH2CH3 butyl), 0.92 (t, J = 7.4 Hz, 3H, CH3 butyl). 31P NMR (283 MHz, D2O)1.548; m/z: calcd for C16H27N5O8P : 448.1440, found: 448.1445; HPLC (Method A) tR 18.7 min.
4.1.5.9 N2-butyl-7-methylguanosine-5’-monophosphate (40)
134 mg, 60%, sodium salt; 1H NMR (700 MHz, D2O) δ 6.11 (d, J = 3.0 Hz, 1H, 1H, H-1’), 4.69 (t, J = 4.8 Hz, 1H, H-2’), 4.46 (t, J = 5.8 Hz, 1H, 1H, H-3’), 4.40 – 4.36 (m, 1H, H-4’), 4.24 – 4.20 (m, 1H, H-5’), 4.08 (s, 3H, N7-CH3), 4.07 – 4.05 (m, 1H, H-5”), 3.45 –3.40 (m, 2H, CH2 butyl), 1.64 – 1.55 (m, 2H, CH2 butyl), 1.42 – 1.32 (m, 2H, CH2CH3 butyl), 0.91 (t, J = 7.4 Hz, 3H, CH3 butyl); 31P NMR (283 MHz, D2O) 0.770; m/z: calcd for C15H25N5O8P: 434.1284, found: 434.1285; HPLC (Method A) tR 13.2 min.
4.1.5.10 N2-isobutyl-N2,7-dimethylguanosine-5’-monophosphate (41)
78 mg, 51%, sodium salt; 1H NMR (700 MHz, D2O) δ 6.11 (d, J = 3.2 Hz, 1H, H-1’), 4.67 (t, J = 4.8 Hz, 1H, H-2’), 4.46 (t, J = 5.9 Hz, 1H, H-3’), 4.37 – 4.35 (m, 1H, H-4’), 4.22 – 4.18 (m, 1H, H-5’), 4.09 (s, 3H, N7-CH3), 4.07 – 4.03 (m, 1H, H-5”), 3.45 – 3.40 (m, 2H, N-CH2), 3.17 (s, 3H, N2-CH3), 2.15 – 2.05 (m, 1H, CH isobutyl), 0.92 (d, J = 6.5 Hz, 6H, CH3 isobutyl); 31P NMR (283 MHz, D2O) 1.646; m/z: calcd for C16H27N5O8P: 448.1440 found: 448.1442; HPLC (Method A) tR 21.5 min.
4.1.5.11 N2-isobutyl-7-methylguanosine-5’-monophosphate (42)
130 mg, 57 %, sodium salt; 1H NMR (700 MHz, D2O) δ 6.12 (d, J = 3.3 Hz, 1H, H-1’), 4.67 (t, J = 4.9 Hz, 1H, H-2’), 4.47 (t, J = 5.6 Hz, 1H, H-3’), 4.38 – 4.36 (m, 1H, H-4’), 4.20 – 4.16 (m, 1H, H-5’), 4.09 (s, 3H, N7-CH3), 4.06 – 4.02 (m, 1H, H-5”), 3.32 – 3.20 (m, 2H, N-CH2), 1.98 – 1.89 (m, 1H, CH isobutyl), 0.93 (d, J = 6.7 Hz, 6H, CH3 isobutyl); 31P NMR (283 MHz, D2O) 2.061; m/z: calcd for C15H25N5O8P: 434.1284, found: 434.1287; HPLC (Method A) tR 16 min.
4.1.5.12 N2,N2-diethyl-7-methylguanosine-5’-monophosphate (43)
83 mg, 52%, sodium salt; 1H NMR (700 MHz, D2O) δ 6.12 (d, J = 3.1 Hz, 1H, H-1’), 4.70 (t, J = 4.9 Hz, 1H, H-2’), 4.46 (t, J = 5.5 Hz, 1H, H-3’), 4.37 – 4.35 (m, 1H, H-4’), 4.22 – 4.18 (m, 1H, H-5’), 4.08 (s, 3H, N7-CH3), 4.07 – 4.04 (m, 1H, H-5”), 3.60 (q, J = 6.1 Hz, 4H, CH2 ethyl), 1.22 (t, J = 7.1 Hz, 6H, CH3 ethyl); 31P NMR (283 MHz, D2O) 1.299; m/z: calcd for C15H25N5O8P: 434.1284, found: 434.1291; HPLC (Method A) tR 14.2 min.
4.1.5.13 N2-ethyl-N2,7-dimethylguanosine-5’-monophosphate (44)
109 mg, 70%, sodium salt; 1H NMR (700 MHz, D2O) δ 6.12 (d, J = 3.4 Hz, 1H, H-1’), 4.70 (t, J = 4.8 Hz, 1H, H-2’), 4.46 (t, J = 5.8 Hz, 1H, H-3’), 4.38 – 4.35 (m, 1H, H-4’), 4.23 – 4.19 (m, 1H, H-5’), 4.08 (s, 3H, N7-CH3), 4.07 – 4.05 (m, 1H, H-5”), 3.64 (dd, J = 14.3, 7.1 Hz, 2H), 3.15 (s, 3H), 1.20 (t, J = 7.1 Hz, 3H); 31P NMR (283 MHz, D2O) 0.866; m/z: calcd for C14H23N5O8P: 420.1127, found: 420.1128; HPLC (Method A) tR 13.9 min.
4.1.5.14 N2-cyclohexylmethyl-7-methylguanosine-5’-monophosphate (45)
122 mg, 56%, sodium salt; 1H NMR (700 MHz, D2O) δ 6.12 (d, J = 3.6 Hz, 1H, H-1’), 4.66 (t, J = 4.8 Hz, 1H, H-2’), 4.47 (t, J = 5.7 Hz, 1H, H-3’), 4.38 – 4.35 (m, 1H, H-4’), 4.16 – 4.12 (m, 1H, H-5’), 4.10 (s, 3H, N7-CH3), 4.02 – 3.98 (m, 1H, H-5”), 3.30 – 3.27 (m, 2H, N-CH2), 1.79 – 0.95 (m, 11H, cyclohexyl); 31P NMR (283 MHz, D2O) 3.218; m/z: calcd for C18H29N5O8P: 474.1597, found: 474.1592; HPLC (Method A) tR 15.6 min.
Supplementary Material
Scheme 1.
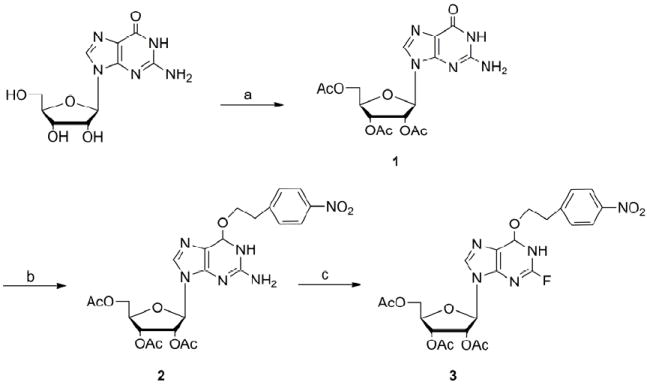
Preparation of N2-fluoro-2’,3’,5’-O-triacetyl-O6-[2-(4-nitrophenyl)ethyl] inosine; (a) acetic anhydride, DMAP, Et3N, AcCN, 4°C to RT (b) NPE, PPh3, DIAD, toluene, RT (c) HF/pyridine, tBuONO, pyridine, -40°C.
Acknowledgments
This work was partially supported by a grant N N301 096339 from the Ministry of Science and Higher Education, Poland, National Institutes of Health grants R0149558 and AI080805 (to R.E.D.) and 02/EuroNanoMed/2011. We gratefully acknowledge prof. Edward Darzynkiewicz from from the Department of Biophysics, Institute of Experimental Physics, Warsaw University for all kind of support. We also thank members of the Davis lab for their help with biological experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Andersen NM, Douthwaite S. J Mol Biol. 2006;359:777–786. doi: 10.1016/j.jmb.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 2.(a) Edqvist J, Straby KB, Grosjean H. Biochimie. 1995;77:54–61. doi: 10.1016/0300-9084(96)88104-1. [DOI] [PubMed] [Google Scholar]; (b) Grosjean H, Benne R, editors. Modification and editing of RNA. ASM Press; Washington, DC: 1998. [Google Scholar]
- 3.Furuichi Y, Shatkin A. J Adv Virus Res. 2000;55:135–184. doi: 10.1016/S0065-3527(00)55003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Thomas JD, Conrad RC, Blumenthal T. Cell. 1988;54:533–539. doi: 10.1016/0092-8674(88)90075-x. [DOI] [PubMed] [Google Scholar]; (b) Liou RF, Blumenthal T. Mol Cell Biol. 1990;10:1764–1768. doi: 10.1128/mcb.10.4.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Sharp PA. Cell. 1994;77:805–815. doi: 10.1016/0092-8674(94)90130-9. [DOI] [PubMed] [Google Scholar]; (b) Lewis JD, Izaurralde E. Eur J Biochem. 1997;247:461. doi: 10.1111/j.1432-1033.1997.00461.x. [DOI] [PubMed] [Google Scholar]; (c) Rhoads RE. Prog Mol Subcell Biol. 1985;9:104–155. [Google Scholar]; (d) Gingras AC, Raught B, Sonenberg N. Annu Rev Biochem. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- 6.(a) Sonenberg N, Hinnebusch A. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Von Der Haar T, Gross J, Wagner G, McCarthy J. Nat Struct Mol Biol. 2004;11:503–511. doi: 10.1038/nsmb779. [DOI] [PubMed] [Google Scholar]; (c) Izaurralde E, Lewis J, McGuigan C, Jankowska M, Darzynkiewicz E, Majtaj IW. Cell. 1994;78:657–668. doi: 10.1016/0092-8674(94)90530-4. [DOI] [PubMed] [Google Scholar]; (d) Calero G, Wilson KF, Ly T, Rios-Steiner JL, Clardy JC, Cenione RA. Nat Struct Biol. 2002;9:912–917. doi: 10.1038/nsb874. [DOI] [PubMed] [Google Scholar]; (e) Huber J, Cronshagen U, Kadokura M, Marshallsay C, Wada T, Sekine M, Luhrmann R. EMBO J. 1998;17:4114–4126. doi: 10.1093/emboj/17.14.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Strasser A, Dickmanns A, Luhrmann R, Ficner R. EMBO J. 2005;24:2235–2243. doi: 10.1038/sj.emboj.7600701. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Liu H, Rodgers ND, Jiao X, Kiledjian M. EMBO J. 2002;21:4699–4708. doi: 10.1093/emboj/cdf448. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) van Dijk E, Cougot N, Meyer S, Babajko S, Wahle E, Seraphin B. EMBO J. 2002;21:6915–6924. doi: 10.1093/emboj/cdf678. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Wang Z, Jiao X, Carr-Schmid A, Kiledjian M. Proc Natl Acad Sci U S A. 2002;99:12663–12668. doi: 10.1073/pnas.192445599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) McKendrick L, Pain VM, Morley SJ. Int J Biochem Cell Biol. 1999;31:31–35. doi: 10.1016/s1357-2725(98)00129-0. [DOI] [PubMed] [Google Scholar]; (b) Sonenberg N, Gingras AC. Curr Opin Cell Biol. 1998;10:268–275. doi: 10.1016/s0955-0674(98)80150-6. [DOI] [PubMed] [Google Scholar]; (c) von der Haar T, Gross JD, Wagner G, McCarthy JEG. Nat Struct Mol Biol. 2004;11:503–511. doi: 10.1038/nsmb779. [DOI] [PubMed] [Google Scholar]
- 8.(a) Clemens MJ, Bommer UA. Int J Biochem Cell Biol. 1999;31:1–23. doi: 10.1016/s1357-2725(98)00127-7. [DOI] [PubMed] [Google Scholar]; (b) De Benedetti A, Graff JR. Oncogene. 2004;23:3189–3199. doi: 10.1038/sj.onc.1207545. [DOI] [PubMed] [Google Scholar]; (c) Mamane Y, Petroulakis E, Rong L, Yoshida K, Ler LW, Sonenberg N. Oncogene. 2004;23:3172–3179. doi: 10.1038/sj.onc.1207549. [DOI] [PubMed] [Google Scholar]
- 9.(a) Cai A, Jankowska-Anyszka M, Centers A, Chlebicka L, Stepinski J, Stolarski R, Darzynkiewicz E, Rhoads RE. Biochemistry. 1999;38:8538–8547. doi: 10.1021/bi9830213. [DOI] [PubMed] [Google Scholar]; (b) Ghosh P, Park C, Peterson MS, Bitterman PB, Polunovsky VA, Wagner CR. Bioorg Med Chem Lett. 2005;15:2177–2180. doi: 10.1016/j.bmcl.2005.01.080. [DOI] [PubMed] [Google Scholar]; (c) Jia Y, Chiu T-L, Amin EA, Polunovsky V, Bitterman PB, Wagner CR. Eur J Med Chem. 2010;45:1304–1313. doi: 10.1016/j.ejmech.2009.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jemielity J, Kowalska J, Rydzik AM, Darzynkiewicz E. New J Chem. 2010;34:829–844. [Google Scholar]
- 10.(a) Jankowska-Anyszka M, Lamphear BJ, Aamondt EJ, Harrington T, Darzynkiewicz E, Stolarski R, Rhoads RE. J Biol Chem. 1998;273:10538–10542. doi: 10.1074/jbc.273.17.10538. [DOI] [PubMed] [Google Scholar]; (b) Keiper BD, Lamphear BJ, Deshpande AM, Jankowska-Anyszka M, Aamodt EJ, Blumenthal T, Rhoads RE. J Biol Chem. 2000;275:10590–10596. doi: 10.1074/jbc.275.14.10590. [DOI] [PubMed] [Google Scholar]; (c) Lall S, Friedman CC, Jankowska-Anyszka M, Stepinski J, Darzynkiewicz E, Davis RE. J Biol Chem. 2004;279:45573–45585. doi: 10.1074/jbc.M407475200. [DOI] [PubMed] [Google Scholar]; (d) Cheng G, Cohen L, Mikhli C, Jankowska-Anyszka M, Stepinski J, Darzynkiewicz E, Davis RE. Mol Biochem Parasitol. 2007;153:95–106. doi: 10.1016/j.molbiopara.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Liu W, Zhao R, McFarland C, Kieft J, Niedzwiecka A, Jankowska-Anyszka M, Stepinski J, Darzynkiewicz E, Jones DNM, Davis RE. J Biol Chem. 2009;284:31333–331349. doi: 10.1074/jbc.M109.049858. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wallace A, Filbin M, Veo B, McFarland C, Jankowska-Anyszka M, Stepinski J, Darzynkiewicz E, Davis RE. Mol Cell Biol. 2010;30:1958–1970. doi: 10.1128/MCB.01437-09. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ruszczynska-Bartnik K, Maciejczyk M, Stolarski R. J Mol Model. 2011;17:727–737. doi: 10.1007/s00894-010-0773-x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Lasda EL, Blumenthal T. Wiley Interdiscip Rev RNA. 2011;2:417–434. doi: 10.1002/wrna.71. [DOI] [PubMed] [Google Scholar]; (i) Allen MA, Hillier LW, Waterston RH, Blumenthal T. Genome Res. 2011;21:255–264. doi: 10.1101/gr.113811.110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Zaslaver A, Baugh LR, Sternberg PW. Cell. 2011;145:981–992. doi: 10.1016/j.cell.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu W, Jankowska-Anyszka M, Piecyk K, Dickson L, Wallace A, Niedzwiecka A, Stepinski J, Stolarski R, Darzynkiewicz E, Kieft J, Zhao R, Jones DNM, Davis RE. Nucleic Acids Res. 2011;20:8820–8832. doi: 10.1093/nar/gkr650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aviñó A, Mayordomo A, Espuny R, Bach M, Eritja R. Nucleosides & Nucleotides. 1995;7:1613–1617. [Google Scholar]
- 13.Nair V, Turner GA, Chamberlain SD. J Am Chem Soc. 1987;109:7223–7224. [Google Scholar]
- 14.Himmelsbach F, Schultz S, Trichtinger T, Charubala R, Pfleiderer W. Tetrahedron. 1984;40:59–72. [Google Scholar]
- 15.(a) Harned AM, Song He H, Toy PH, Flynn DL, Hanson PR. J Am Chem Soc. 2005;127:52–53. doi: 10.1021/ja045188r. [DOI] [PubMed] [Google Scholar]; (b) Lan P, Porco JA, South MS, Parlow JJ. J Com Chem. 2003;5:660–669. doi: 10.1021/jo035129g. [DOI] [PubMed] [Google Scholar]; (c) Fleckenstein CA, Plenio H. Adv Synth Catal. 2006;348:1058–1062. [Google Scholar]; (d) Dandapani S, Curran DP. Tetrahedron. 2002;58:3855–3864. [Google Scholar]
- 16.Liu J, Robins MJ. J Am Chem Soc. 2007;129:5962–5968. doi: 10.1021/ja070021u. [DOI] [PubMed] [Google Scholar]
- 17.Acedo M, Fabrega C, Aviñó A, Fagan P, Wammer D, Eritja R. Nucleic Acids Res. 1994;22:2982–2989. doi: 10.1093/nar/22.15.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Woo J, Sigurdsson T, Hopkins PB. J Am Chem Soc. 1993;115:3407–3415. [Google Scholar]; (b) Allerson CR, Chen SL, Verdine GL. J Am Chem Soc. 1997;119:7423–7433. [Google Scholar]
- 19.Olah AG, Li X. Synlett. 1990:267–269. [Google Scholar]
- 20.Yoshikawa M, Kato T, Takenishi T. Tetrahedron Lett. 1967;50:5065–5068. doi: 10.1016/s0040-4039(01)89915-9. [DOI] [PubMed] [Google Scholar]
- 21.(a) Adams BL, Morgan M, Shatkin AJ. J Biol Chem. 1978;253:2589–2595. [PubMed] [Google Scholar]; (b) Darzynkiewicz E, Dekiel I, Lassota P, Tahara SM. Biochemistry. 1987;26:4372–3480. doi: 10.1021/bi00388a028. [DOI] [PubMed] [Google Scholar]
- 22.Cohen LS, Mikhli C, Friedman C, Jankowska-Anyszka M, Stepinski J, Darzynkiewicz E, Davis RE. RNA. 2004;10:1609–1624. doi: 10.1261/rna.7690504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lax S, Fritz W, Browning K, Ravel J. Proc Natl Acad Sci USA. 1985;82:330–333. doi: 10.1073/pnas.82.2.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Volpon L, Osborne MJ, Topisirovic I, Siddiqui N, Borden KL. The EMBO J. 2006;25:5138–5149. doi: 10.1038/sj.emboj.7601380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.