

Abstract
Induced pluripotent stem cells (iPSC) are important tools in regenerative medicine. Yet, it is becoming increasingly clear that the reprogramming process, including retroviral transduction with potent oncogenes like c-Myc and long-term cultivation, may induce genetic instability. Genetically altered iPS cells can grow out and dominate the cell culture. This review intends to comprehensively summarize the current knowledge on genetic instability of embryonic and iPSCs, with an emphasis on cytogenetic alterations, and compares these data with what is known from tumorigenesis.
Keywords: Induced pluripotent stem cells, gene therapy, chromosomal instability, genetic integrity, tumor, leukemia, clonal evolution, chromosome, mutation, P53
Introduction
Induced pluripotent stem cells (iPSC) provide important tools to understand basic functions of stemness and reprogramming, to perform pharmacologic testing, to develop disease models and to perform gene correction before transfer into the patient [1,2]. They also have great potential for regenerative medicine, providing cellular sources to replace defective cells, particularly in age-related diseases. However, a major concern in applying iPS cells in the clinic is whether they maintain their genetic integrity. There is increasing evidence that iPS cells may acquire genetic and epigenetic alterations during reprogramming and cultivation [3,4]. Retroviral gene transfer to express reprogramming factors may itself result in insertional mutagenesis by activating cellular oncogenes. It may be associated with an increased genetic instability and finally lead to clonal and even malignant outgrowth [5].
Stem cells give their genetic information to millions of daughter cells. Therefore, it is of utmost importance that they protect this genetic information from deleterious damage. The genetic information is coded in about 20.000-25.000 genes that are systematically ordered in our 46 chromosomes and regulated by an epigenetic layer of information, e.g. by DNA methylation and chromatin modifications. Although epigenetic modifications control reprogramming and stemness, they are not further discussed in this review. Somatic mutations are estimated to occur at a rate of ~3.4 × 10-7 per cell division in normal humans [6]. Yet we have developed efficient repair systems and immune surveillance to eliminate cells carrying gene mutations or chromosome defects. Stem cells in particular have developed efficient mechanisms to maintain their genomic integrity (For review please refer to [7,8]).
How to define genetic instability?
In principle, there are three different modes of genetic instability: acquisition of mutations, mismatch repair deficiency and chromosomal instability. Mismatch repair deficiency is caused by mutations in DNA repair genes like MLH1 or MSH2 that are responsible for resolving replicate errors within microsatellites. In the case of mismatch repair deficiency, short repetitive sequences may show an increased frequency of length mutations, especially in highly proliferating tissues. In contrast, chromosomal instability leads to gross alterations of chromosome number or structure. Chromosomal instability refers to the cell-to-cell variability, i.e. chromosome aberrations that differ from cell to cell, that may ultimately lead to the outgrowth of chromosomally aberrant clones [9]. Frequently, chromosomal instability is used synonymously with the presence of chromosome aberrations, but this is the consequence rather than chromosomal instability per se.
Ways to decipher genetic instability
Karyotyping and fluorescence in situ hybridization (FISH)
Microscopic analyses of chromosome number and structure within single cells requires the preparation of metaphases and the application of chromosome banding techniques to construct a karyotype. This is still the gold standard to obtain an overview of the genetic integrity of reprogrammed cells. However, the resolution of this method is only about 5 to 10 Mb. Higher resolution of defined genetic regions can be achieved by FISH using fluorochrome-labeled DNA probes complementary to the regions of interest. After binding to the complementary regions, the probes produce distinct fluorescence signals within the metaphase or interphase cell that allow investigation of the number, structure and localization of the respective genetic regions with high resolution. However, only defined regions, complementary to the respective probes, can be investigated. In multicolorFISH (mFISH), a simultaneous hybridization of six fluorochromes onto metaphases, each chromosome is marked with a specific mixture of fluorochromes. With specialized software modules, each chromosome appears in a different so-called pseudo-color (Figure 1). This method is very helpful in clarifying the origin of complex chromosomal rearrangements or marker chromosomes [10]. For example, it is useful for the identification of translocations that might not be detectable by classical banding analysis due to a similar banding pattern of the aberrant chromosomes. However, only inter-, not intrachromosomal rearrangements can be detected. A disadvantage of this method is the disability to detect deletions or inversions.
Figure 1.

mFISH karyogram of a patient with myelodysplastic syndrome and a complex karyotype: by simultaneous hybridization of six fluorochromes onto metaphases, each chromosome is marked with a specific mixture of fluorochromes. A specialized software module (MetaSystems GmbH, Altlussheim, Germany) depicts the chromosomes in so-called pseudo-colors as shown here. In addition to classical banding analysis, this is a useful method to identify unknown chromosomal material or cryptic aberrations.
High-resolution array comparative genomic hybridization (aCGH)
High-resolution array comparative genomic hybridization (aCGH) or single nucleotide polymorphism (SNP) arrays are the appropriate techniques to detect small gains or losses up to single exons. The methods are based on the comparison of test genomes (e.g. cultured iPSC or initial fibroblast cells) to a reference genome. Fluorescently labeled probes are hybridized to oligonucleotide probes immobilized to a microarray (up to several million per array) slides. Probe design is very flexible in array-CGH, facilitating dense coverage of nonpolymorphic unique sequence probes of genomic regions, whereas SNP arrays depend on the genomic location of SNPs. Copy-number differences are calculated from the fluorescence intensities of the fluorochromes. Significant deviation from unity in the ratios of the fluorescence intensity values is indicative of a deletion or gain in the test compared to the reference DNA (Figure 2).
Figure 2.
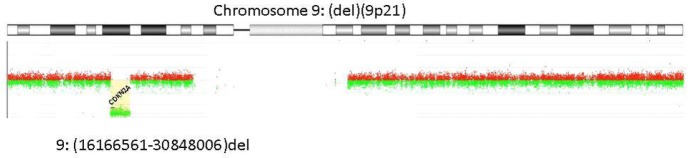
Array-CGH Result: Genomic profile of chromosome 9 as determined by Human Genome 400k Microarray (Agilent Technologies, Santa Clara, CA, USA). Below the ideogram of chromosome 9, its profile resulting from Genomic Workbench (Agilent) is given: log2 ratios of array probes are plotted against the chromosomal localization indicating a homozygous deletion of around 14.7 Mb affecting the region from 16.166561-30.848006 Mb.
Nowadays, next generation sequencing is introduced to detect mutations in small cell populations with a very high sensitivity, given that coverage is at least 1000x. Meanwhile, whole-genome sequencing has identified more than one thousand heterozygous single-nucleotide variants (SNVs) in human iPS cell lines [11].
Genetic instability in reprogrammed cells
Recently, several reports brought attention to the presence of genetic alterations in embryonic stem (ES) and iPS cells [12-14]. Human iPS cell lines from different laboratories, independent of the reprogramming methods used, were found to contain an average of five protein-coding mutations. Some mutations preexisted in small subclones, the others occurred during the reprogramming process [12]. Significantly more copy number variants (CNV), i.e. gains or losses of genomic regions, are present in early-passage human iPS cells than intermediate-passage human iPS cells, fibroblasts or human ES cells [13]. Most CNVs are formed de novo and render the affected cells at a selective disadvantage. Therefore, genetically aberrant cells are rapidly counter-selected. Thus, it seems possible to generate iPS cell lines without copy-number changes, if appropriate conditions for reprogramming and non-integrating vectors are used [11]. However, copy-number variations frequently affect tumor suppressor genes and oncogenes [13,15] and include homogeneously staining regions (HSR), cytogenetically detectable gene amplifications that are considered a hallmark of cancer cells [16]. Actually, many genes of ES cells deregulated due to copy-number variations are functionally linked to cancer [17].
Investigating different sources of mouse (induced) pluripotent stem cells, four hotspots of chromosomal aberrations were detected: full trisomy 11 (with a minimally recurrent gain in 11qE2), full trisomy 8, and deletions in chromosomes 10qB and 14qC-14qE. The most recurrent aberration in mouse PSCs, gain 11qE2, turned out to be fully syntenic to the common aberration 17q25 in human PSCs, while other recurrent aberrations were found to be species-specific. Analysis of rhesus macaque PSCs revealed a gain in chromosome 16q, syntenic to the hotspot in human 17q [18]. Therefore, Ben-David and Benvenisty strongly emphasize that attention should be paid to the thorough cytogenetic characterization of ESC and iPSC. This is also recommended to exclude chromosome aberrations in cell lines used as control, which may lead to false conclusions. Chromosome aberrations may interfere with germline transmission of ESC, as described by Liu et al. for trisomy 8 [19]. During the last few years, comprehensive studies have been performed to genetically characterize large numbers of murine and human ES and iPS cell lines. The findings of these studies are summarized in Tables 1 and 2.
Table 1.
Genomic alterations of embryonic stem (ES) cells
| Cell type | Conditions | No of aberrant/analysed clones | Karyotype | Literature |
|---|---|---|---|---|
| mES cell lines | Culture in medium containing leukemia inhibitory factor | 22 of 29 clones from 9 cell lines | Trisomy 8 in 21 clones; | Liu X et al, Dev Dyn (1997) [19] |
| Trisomy 15 in 2 clones | ||||
| T(Y;12)(Tel;C1), Inv(3) | ||||
| mES cell lines | Cell lines from different institutions | 35 of 88 cell lines | Trisomy 8, loss of sex chromosome, trisomy 11 | Sugawara A et al, Comp Med (2006) [56] |
| mES cell lines | Various conditions | 49 of 129 cell lines with gene expression profiles obtained from the GEO database* | Trisomy 8, trisomy 11, deletion 10qB and 14C-14E | Ben-David U & Benvenisty N, Stem Cells (2012) [18] |
| hES cell lines | Prolonged culture | 13 of 13 cell lines | Gains of chromosomes 12, 17 and X; i(12)(p10); | Baker DE et al, Nat Biotechnol (2007) [16] and review therein |
| 18 of 18 cell lines | Smallest region of amplification: 17q11-q12, 17q25-ter; | |||
| other aberrations | ||||
| hES cell lines (HS181, SHEF-3, SHEF-1) | Transfer to a feeder-free culture system | 2 of 3 cell lines | 47,XY,+14 | Catalina P et al, Mol Cancer (2008) [22] |
| 47,XX,+12/48,idem,+der(20) | ||||
| hES cell lines (HUES1, HUES3, HUES4) | Prolonged culture | 2 of 17 cell lines | Trisomy 12 der(2) | Cowan C et al, N Engl J Med (2004) [57] |
| hESC | Long-term culture | 4 of 5 cell lines analysed by FISH and aCGH | Amplification of 20q11.21 | Lefort N et al, Nat Biotechnol (2008) [25] |
| hES cell lines HS-181, HS293 | Long-term culture on human feeder-layers | 0 of 2 cell lines | Genetically stable | Catalina P et al, Leuk Res (2009) [58] |
| hESC | Cell lines from different institutions | 12 of 38 cell lines with gene expression profiles obtained from the GEO database* | Trisomy 12 and 17 | Mayshar Y et al, Cell Stem Cell (2010) [23] |
| hES cells | Cell lines from different institutions | 17 of 17 cell lines analysed with SNP arrays | 843 copy number variations (CNV), 50kb to 30Mb in size in karyotypically normal cell lines, | Närvä E et al, Nat Biotechnol (2010) [17] |
| Gain of 1p36, 2p11, 7q35, 14q32, 22q11, loss of 15q11 | ||||
| hESC | Cell lines from different institutions | 42 of 125 cell lines analysed with karyotyping and high-resolution SNP arrays | Extra copies of chromosomes 1, 12, 17, 2 or X, | The International Stem Cell Initiative, Nat Biotechnol (2011) [14] |
| Minimal amplicons 17q25 and 20q11.2, | ||||
| Gain of 12p, | ||||
| Loss of 10p13-pter, 18q21-qter, 22q13-qter | ||||
| hESC | Different time points during cultivation | 69 cell lines analysed with high-resolution SNP arrays | Duplications of 12p, 20q and X chromosome, trisomy 15 | Laurent LC et al, Cell Stem Cell (2011) [15] |
| hESC | Prolonged culture | 1 of 3 cell lines analysed by G-banding, aCGH, FISH | ins(2;?)(p16;?) | Dekel-Naftali M et al, Eur J Hum Genet (2012) [21] |
| ish amp(1)(q21q32) |
chromosome aberrations were detected by a methodology based on gene expression profiling (Ben-David et al., Cell Stem Cell 2011 [59]).
Table 2.
Genomic alterations of induced pluripotent stem (iPS) cells
| Cell type | Factors used for repro-gramming | No of aberrant/analysed clones | Karyotype | Literature |
|---|---|---|---|---|
| miPS cell lines | 4 factors including c-Myc | 6 of 9 cell lines | Trisomy or Robertsonian translocation of chromosome 14, trisomy 8 | Chen Q et al, Chromosome Res (2011) [29] |
| miPS cell lines | Various conditions | 27 of 127 cell lines with gene expression profiles obtained from the GEO database* | Trisomy 8, trisomy 11/gains of 11D-11E, deletions of 14C-14E | Ben-David U & Benvenisty N, Stem Cells (2012) [18] |
| hiPSC | Non-Silencing of 4 factors | 1 of 4 clones using karyotyping and mFISH | Translocation between chromosome 1 and 17, gain of 1q, trisomy 5 | Ramos-Mejia V et al, Cell Res (2010) [26] |
| hiPSC | Cell lines from different institutions | 13 of 66 cell lines with gene expression profiles obtained from the GEO database* | Trisomy 1, 9, 12 and 17, gain of 12p and 17q, Loss of 15q | Mayshar Y et al, Cell Stem Cell (2010) [23] |
| hiPS cell llines | No influence of reprogramming factors | 22 hiPS cell lines analysed with SNP arrays | Median number of of copy number variations (CNV) 109; higher number in early than in late passages | Hussein SM et al, Nature (2011) [13] |
| hiPSC | Cell lines from different institutions | 3 of 11 cell lines analysed with karyotyping and SNP arrays | Extra copies of chromosome 12, inversion of chromosome 5 | The International Stem Cell Initiative, Nat Biotechnol (2011) [14] |
| hiPS | Different time points during cultivation and differentiation | 37 cell lines analysed with high-resolution SNP arrays | Deletions of regions containing tumor suppressor genes, duplications of regions containing oncogenes | Laurent LC et al, Cell Stem Cell 2011) [15] |
| hiPSC | Reprogramming by | 4 of 8, 2 of 6, 3 of 4 clones, resp, using array containing probes for chr. 5-13 and parts of chr. 4 and 14 | Deletion of chromosome 9, 13, 14, duplication of chromosome 13, gain of chromosome 8, amplification of chromosome 7 | Pasi CE et al, Cell Death Diff (2011) [28] |
| - c-MYC | ||||
| - 3 factors | ||||
| - 4 factors | ||||
| hiPSC | Prolonged culture | 1 of 2 cell lines analysed by G-banding, aCGH, FISH | ish amp(1)(q21qter) | Dekel-Naftali et al, Eur J Hum Genet (2012) [21] |
chromosome aberrations were detected by a methodology based on gene expression profiling (Ben-David et al., Cell Stem Cell 2011 [59]).
A 3.1Mb gain in 1q found in cultured human ES cells includes JARID1B, a polycomb-related histone demethylase [14] that is overexpressed in different human tumors like breast, prostate cancer or uveal melanoma. Moreover, a jumping translocation of chromosome 1q seems to inhibit senescence during neural differentiation of hESC into a neural stem cell population that could be propagated for more than 50 passages [20]. Jumping translocations of 1q are recurrent chromosome aberrations of hematologic neoplasms and pediatric brain tumors associated with a poor prognosis. Also, in another report, recurrent gain of chromosome 1q was described in one hESC and one hiPSC line, providing clonal advantage in culture [21].
Repeatedly, trisomy 12 or an isochromosome 12p were observed in human ES and iPS cell lines (Tables 1 and 2). Catalina et al. [22] reported clonal advantage of cells with trisomy 12 in hESC lines. Mayshar and colleagues [23] report that a substantial number of human iPSC and ES cell lines carry full and partial chromosomal aberrations - again, a high incidence of chromosome 12 duplications resulting in significant enrichment for cell-cycle-related genes like NANOG. Such aneuploidy may limit the differentiation capacity and increase the tumorigenicity of human iPSCs. This result was further substantiated by the data of Moon et al. [24], who found that undifferentiated CHA3-hESCs with trisomy 12 undergo abnormal cell division with multiple spindles and that these ESC derivates are able to form a tumor-like tissue after transplantation in mice. Notably, trisomy 12 is a recurrent chromosome aberration in different malignancies like chronic lymphocytic leukemia, and isochromosome 12p is the typical change of testicular germ cell tumors [http://cgap.nci.nih.gov/Chromosomes/Mitelman].
The International Stem Cell Initiative [14] analysed 125 human ES cell lines and 11 iPS cell lines, from 38 laboratories worldwide, for genetic changes occurring during culture. Most lines remained karyotypically normal, but there was a progressive tendency to acquire changes on prolonged culture, commonly affecting chromosomes 1, 12, 17 and 20. Notably, an amplicon in chromosome 20q11.21, including three genes expressed in human ES cells, ID1, BCL2L1 and HM13, occurred in >20% of the lines. This amplification at 20q11.21 had been described previously in four cell lines of different origin and was proposed to provide a selective advantage to hES cells in culture [25]. Of the genes located within the amplified region, BCL2L1 may be a strong candidate for driving culture adaptation of ES cells.
iPSC lines that do not silence the expression of the ectopic reprogramming factors may display enhanced propensity to genomic instability [26]. To this end, excisable vectors or transgene-free reprogramming, under serum-free and feeder-layer-free conditions, may significantly decrease the risk of genetic instability and malignant transformation [27]. Comparative genomic hybridization analysis of stem cells reprogrammed by expressing c-Myc revealed the presence of genomic deletions and amplifications, whose signature was suggestive of oncogene-induced DNA replication stress. The genomic aberrations were, to a significant degree, dependent on c-Myc expression and their presence could explain why p53 inactivation facilitates stem cell reprogramming [28]. This is in line with our observations in nine independent murine iPS (miPS) cell lines from three laboratories where we found recurrent trisomy and/or translocation of chromosome 14 to be associated with the use of c-MYC for reprogramming [29]. Other key factors regulating genetic instability are ATM and P16 [30].
Yet it seems possible to generate iPSCs lacking gene-disrupting mutations using current reprogramming methods. Using whole-genome paired-end DNA sequencing and a sensitive algorithm, only few (one or two) spontaneous sequence variants per line and no evidence for endogenous retro-element transposition were identified [31]. It is also possible to produce gene-corrected disease-specific iPSC lines that do not show any chromosomal alterations if investigated by spectral karyotyping and high-resolution array CGH analysis [2].
Key regulators of genetic instability in stem cells
Except for rare mismatch repair deficiency or chromosomal breakage syndromes caused by inherited mutations of components of different DNA repair pathways, the reasons for increased genetic instability are poorly understood. Clearly, mutations may occur under physiological conditions in normal cells, mostly due to replicative errors. Accordingly, normal cells can acquire numerical chromosome aberrations during mitosis. Metabolic processes generate oxidative species, potent mutagens. Thus, at a low frequency, genetic changes are an integral part of living cells.
DNA damage may also be induced by X-rays, radio- and chemotherapy and by a plethora of mutagenic substances. As long as mutated cells are recognized and eliminated, genetic changes will cause no harm. However, if replicative errors accumulate in highly proliferative cells, DNA repair systems and immune surveillance mechanisms may be overloaded and no longer able to protect genetic stability. With regard to genetic instability of stem cells, quiescence is a highly effective safeguard mechanism.
It is difficult to decipher the mechanisms that increase the rate of genetic instability and allow the outgrowth of genetically defective clones. Oncogenes, tumor suppressor genes, telomere attrition or a combination of several factors have been discussed. Very recently, RNase H2, an enzyme required to remove endogenous ribonucleotides from DNA was shown to be essential for genomic integrity in early embryogenesis [32].
There may be various factors inducing genetic instability during the process of reprogramming: insertional mutagenesis, transcription factors like c-MYC used for reprogramming, enhanced proliferation, cell cultivation over long periods of time, etc. Extensive passaging seems to be associated with an increased rate of genetic instability. Low rates of mosaicism observed in late, but not in early passages imply a direct correlation between number of passages and aneuploidy rate [21]. Chromosome aberrations may already be present in somatic cells, particularly from aged individuals, used for reprogramming. Recently, it has been shown that cellular reprogramming is capable of reversing aging-related features in somatic cells, despite the presence of genomic alterations [33].
c-MYC-induced genetic instability and telomere attrition
There is an obvious correlation between extent of chromosomal aberrations and transcriptional factors used for their reprogramming. Particularly the oncogene c-MYC, one of originally four factors to generate induced pluripotent stem (iPS) cells [34] has been demonstrated to induce genetic instability. As mentioned above, we have observed trisomy and translocations in miPS cell lines from three laboratories, which provided a growth advantage over the miPS cells with a normal karyotype. Remarkably, there was a significantly higher frequency of chromosome aberrations in the miPS cell lines induced with c-Myc than those without c-Myc [29]. Sabine May and colleagues elegantly showed that the overexpression of c-MYC leads to telomere aggregates that reflect the onset and propagation of breakage-bridge-fusion cycles initiated by end-to-end telomeric chromosome fusions. c-Myc-dependent telomere remodeling thus precedes the onset of genomic instability and subsequently leads to chromosomal rearrangements [35]. Investigating murine hematopoietic stem cells overexpressing c-myc, we could show that loss of the histone methyltransferase suv39h1 can prevent the induction of chromosomal instability. Suv39h1-deficient (c-myc-overexpressing) cells had short, yet less critically short telomeres than wild-type (c-myc-overexpressing) cells, indicating that alternative telomere lengthening may play a role to stabilize the telomeres [36]. Here we have to bear in mind that one single critically short telomere - and not the average telomere length - may induce a chromosome segregation defect or a chromosomal rearrangement.
Telomere erosion is physiologically associated with aging. Cells from the elderly have shortened telomeres and increased genetic instability, thus possibly explaining why tumor risks rise with age and why aged somatic cells can be reprogrammed less efficiently than cells from younger individuals. Several components of the telomerase complex have been implicated in telomere maintenance and long-term genomic stability in ES cells [37]. One of them is Zscan4 regulating telomere extension in ES cells. Knockdown of Zscan4 leads to telomere shortening and to increased rate of chromosome abnormalities [38].
TP53 - the guardian of the genome
TP53 is considered to be the most important gene to protect genetic stability and is also a key regulator of stem cell maintenance and pluripotency. After genetic damage occurs in a cell, TP53 induces a halt in the cell cycle to allow repair, or alternatively induces senescence or apoptosis, in case the damage cannot be repaired properly. TP53 has a critical function in regulating stem cell quiescence in adult hematopoietic stem cells [39]. Moreover, high expression levels of TP53 promote differentiation of embryonic stem cells [40] and limit efficient reprogramming of somatic cells to iPSC [41,42]. This may be due to the critical role of TP53 in preventing the reprogramming of cells carrying various types of DNA damage, including short telomeres, DNA repair deficiencies, or exogenously inflicted DNA damage [43]. P53 deletion impairs clearance of chromosomal-instable stem cells in aging telomere-dysfunctional mice [44]. TP53 is part of a huge network. Several regulators like necdin, a growth-suppressing protein [39], aurora kinase A [45] or miR-138, directly binding to the 3’ untranslated region (UTR) of TP53, seem to control TP53 expression [46] in adult and embryonal stem cells. Two downstream targets of P53, i.e. Puma and p21, represent cooperating checkpoints limiting self-renewal and chromosomal instability of somatic stem cells in response to telomere dysfunction [47].
Lessons to be learned from genetic defects in somatic stem cells
If we wish to understand the consequences defined genetic changes may have for genetically modified stem or iPS cells, it may be worth evaluating the harm they cause in somatic stem cells. From genetic analyses of leukemias and solid tumors, we have a broad knowledge of recurrent abnormalities associated with certain tumor subtypes and with clinical outcome. It is clear that the activation of oncogenes like c-MYC and the inactivation of tumor suppressor genes cooperate with telomere attrition and epigenetic changes to transform cells. There are different modes of activation of an oncogene, like point mutations, amplification or translocation next to enhancer elements. Also, there are different modes of inactivation of a tumor suppressor gene, like inactivating or dominant-negative mutations, deletions, uniparental disomy or loss of the whole chromosome.
Not surprisingly, TP53 is the gene most frequently altered in human malignancies. In myeloid neoplasms, TP53 mutations predict high relapse rates and a very poor prognosis [48,49]. Notably, TP53 mutations have been identified in the majority of myeloid neoplasms with a complex karyotype carrying a high number of chromosome aberrations, per se the most important factor to indicate an extremely poor prognosis. The influence of TP53 on genetic stability is further highlighted by recent observations that TP53 mutations are present in nearly all pediatric brain tumors exhibiting signs of chromothripsis, a kind of “shredding,” and complex rearrangements within one or a few chromosomes [50]. TP53 mutations seem to provide a profound survival advantage, since small clones with TP53 mutations identified only by next generation sequencing can induce disease progression years later [49]. Yet, the recent advances in next generation sequencing have allowed the gain of deeper insights into clonal evolution and shown that there is a significant genetic diversity and highly dynamic growth and disappearance of genetically aberrant clones in human malignancies [51]. These studies will hopefully allow in the future key players like TP53 that drive malignant transformation and progression to be distinguished from by-stander lesions. This knowledge will certainly be extremely helpful in judging the malignant potential of defined genetic lesions in reprogrammed cells.
Malignant transformation of genetically modified stem cells after gene therapy
There have been several reports that patients in different gene therapy trials have developed leukemias. This allows us to better understand what genetic alterations provide a selective growth advantage to genetically modified hematopoietic stem cells. Unequivocally, vector integration into the host genome is the first step towards malignant transformation. As mouse transplantation models have clearly shown, even the integration of one vector upstream of a potent oncogene is sufficient for insertional mutagenesis [52]. There seems to be a limited number of oncogenes that are prone to insertional mutagenesis, particularly EVI1 inducing myeloid malignancies like myelodysplastic syndromes and acute myeloid leukemias, and LMO1 and LMO2 inducing T lymphoblastic leukemias. Notably, in addition to insertional mutagenesis into LMO2, T cell leukemias from patients having undergone gene therapy of SCID-X1 frequently contained chromosome aberrations, for example deletions of 6q and 9p affecting the tumor suppressor gene CDKN2A or a SIL-TAL1 fusion [53,54]. These changes are known as recurrently altered chromosome regions of sporadic T lymphoblastic leukemias, demonstrating that chromosome aberrations contribute to leukemogenesis of genetically modified stem cells. In a gene therapy trial for chronic granulomatous disease, two patients developed myelodysplasia (MDS) due to insertional mutagenesis into the EVI1 locus. Both patients showed monosomy 7, a typical chromosome aberration of MDS that triggered the clonal expansion of the malignant clone [55].
Conclusions
From the knowledge we have today about genetic instability of genetically modified and reprogrammed cells, we can deduce that a better understanding of the factors that induce an increased genetic instability is warranted before extensive clinical use. There is evidence that enhanced cell culturing, prolonged expression of transcription factors like c-Myc and inactivation of TP53 may play a significant role. A better understanding will also include reliable criteria to classify genetic alterations according to risk for malignant transformation [27]. It will be of the utmost importance to compare genetic alterations in hES and iPS cells with those recurrently found in malignancies. As we show, there are surprising parallels, if we base the comparison on those genetic lesions that provide a selective growth advantage in vitro. Another challenge will be to develop appropriate animal models to investigate the malignant potential of defined genetic alterations in vivo. Integrating all this knowledge will enable us to set up highly qualified laboratories dedicated to rigorous genetic screening of reprogrammed cells with state-of-the-art technologies. Careful genetic analyses will help to reduce risks of clinical use when reprogrammed cells are transferred into patients.
Acknowledgement
This work is supported by funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) for the Cluster of Excellence REBIRTH (From Regenerative Biology to Reconstructive Therapy).
Conflict of interest
None of the authors has editorial or financial conflicts of interest (e.g., consultancy, stock ownership, equity interests, patent or licensing agreements).
References
- 1.Muller LU, Milsom MD, Harris CE, Vyas R, Brumme KM, Parmar K, Moreau LA, Schambach A, Park IH, London WB, Strait K, Schlaeger T, Devine AL, Grassman E, D’Andrea A, Daley GQ, Williams DA. Overcoming reprogramming resistance of Fanconi anemia cells. Blood. 2012;119:5449–5457. doi: 10.1182/blood-2012-02-408674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu G, Liu N, Rittelmeyer I, Sharma AD, Sgodda M, Zaehres H, Bleidissel M, Greber B, Gentile L, Han DW, Rudolph C, Steinemann D, Schambach A, Ott M, Scholer HR, Cantz T. Generation of healthy mice from gene-corrected disease-specific induced pluripotent stem cells. PLoS Biol. 2011;9:e1001099. doi: 10.1371/journal.pbio.1001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nishino K, Toyoda M, Yamazaki-Inoue M, Fukawatase Y, Chikazawa E, Sakaguchi H, Akutsu H, Umezawa A. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS Genet. 2011;7:e1002085. doi: 10.1371/journal.pgen.1002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pera MF. Stem cells: The dark side of induced pluripotency. Nature. 2011;471:46–47. doi: 10.1038/471046a. [DOI] [PubMed] [Google Scholar]
- 5.Kustikova O, Fehse B, Modlich U, Yang M, Dullmann J, Kamino K, von Neuhoff N, Schlegelberger B, Li Z, Baum C. Clonal dominance of hematopoietic stem cells triggered by retroviral gene marking. Science. 2005;308:1171–1174. doi: 10.1126/science.1105063. [DOI] [PubMed] [Google Scholar]
- 6.Araten DJ, Golde DW, Zhang RH, Thaler HT, Gargiulo L, Notaro R, Luzzatto L. A quantitative measurement of the human somatic mutation rate. Cancer Res. 2005;65:8111–8117. doi: 10.1158/0008-5472.CAN-04-1198. [DOI] [PubMed] [Google Scholar]
- 7.Mandal PK, Blanpain C, Rossi DJ. DNA damage response in adult stem cells: pathways and consequences. Nat Rev Mol Cell Biol. 2011;12:198–202. doi: 10.1038/nrm3060. [DOI] [PubMed] [Google Scholar]
- 8.Tichy ED, Liang L, Deng L, Tischfield J, Schwemberger S, Babcock G, Stambrook PJ. Mismatch and base excision repair proficiency in murine embryonic stem cells. DNA Repair (Amst) 2011;10:445–451. doi: 10.1016/j.dnarep.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geigl JB, Obenauf AC, Schwarzbraun T, Speicher MR. Defining ‘chromosomal instability’. Trends Genet. 2008;24:64–69. doi: 10.1016/j.tig.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 10.Lange K, Gadzicki D, Schlegelberger B, Gohring G. Recurrent involvement of heterochromatic regions in multiple myeloma-a multicolor FISH study. Leuk Res. 2010;34:1002–1006. doi: 10.1016/j.leukres.2009.10.027. [DOI] [PubMed] [Google Scholar]
- 11.Cheng L, Hansen NF, Zhao L, Du Y, Zou C, Donovan FX, Chou BK, Zhou G, Li S, Dowey SN, Ye Z, Program NCS, Chandrasekharappa SC, Yang H, Mullikin JC, Liu PP. Low incidence of DNA sequence variation in human induced pluripotent stem cells generated by non-integrating plasmid expression. Cell Stem Cell. 2012;10:337–344. doi: 10.1016/j.stem.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gore A, Li Z, Fung HL, Young JE, Agarwal S, Antosiewicz-Bourget J, Canto I, Giorgetti A, Israel MA, Kiskinis E, Lee JH, Loh YH, Manos PD, Montserrat N, Panopoulos AD, Ruiz S, Wilbert ML, Yu J, Kirkness EF, Izpisua Belmonte JC, Rossi DJ, Thomson JA, Eggan K, Daley GQ, Goldstein LS, Zhang K. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hussein SM, Batada NN, Vuoristo S, Ching RW, Autio R, Narva E, Ng S, Sourour M, Hamalainen R, Olsson C, Lundin K, Mikkola M, Trokovic R, Peitz M, Brustle O, Bazett-Jones DP, Alitalo K, Lahesmaa R, Nagy A, Otonkoski T. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011;471:58–62. doi: 10.1038/nature09871. [DOI] [PubMed] [Google Scholar]
- 14.Amps K, Andrews PW, Anyfantis G, Armstrong L, Avery S, Baharvand H, Baker J, Baker D, Munoz MB, Beil S, Benvenisty N, Ben-Yosef D, Biancotti JC, Bosman A, Brena RM, Brison D, Caisander G, Camarasa MV, Chen J, Chiao E, Choi YM, Choo AB, Collins D, Colman A, Crook JM, Daley GQ, Dalton A, De Sousa PA, Denning C, Downie J, Dvorak P, Montgomery KD, Feki A, Ford A, Fox V, Fraga AM, Frumkin T, Ge L, Gokhale PJ, Golan-Lev T, Gourabi H, Gropp M, Lu G, Hampl A, Harron K, Healy L, Herath W, Holm F, Hovatta O, Hyllner J, Inamdar MS, Irwanto AK, Ishii T, Jaconi M, Jin Y, Kimber S, Kiselev S, Knowles BB, Kopper O, Kukharenko V, Kuliev A, Lagarkova MA, Laird PW, Lako M, Laslett AL, Lavon N, Lee DR, Lee JE, Li C, Lim LS, Ludwig TE, Ma Y, Maltby E, Mateizel I, Mayshar Y, Mileikovsky M, Minger SL, Miyazaki T, Moon SY, Moore H, Mummery C, Nagy A, Nakatsuji N, Narwani K, Oh SK, Oh SK, Olson C, Otonkoski T, Pan F, Park IH, Pells S, Pera MF, Pereira LV, Qi O, Raj GS, Reubinoff B, Robins A, Robson P, Rossant J, Salekdeh GH, Schulz TC, Sermon K, Sheik Mohamed J, Shen H, Sherrer E, Sidhu K, Sivarajah S, Skottman H, Spits C, Stacey GN, Strehl R, Strelchenko N, Suemori H, Sun B, Suuronen R, Takahashi K, Tuuri T, Venu P, Verlinsky Y, Ward-van Oostwaard D, Weisenberger DJ, Wu Y, Yamanaka S, Young L, Zhou Q International Stem Cell Initiative. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat Biotechnol. 2011;29:1132–1144. doi: 10.1038/nbt.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laurent LC, Ulitsky I, Slavin I, Tran H, Schork A, Morey R, Lynch C, Harness JV, Lee S, Barrero MJ, Ku S, Martynova M, Semechkin R, Galat V, Gottesfeld J, Izpisua Belmonte JC, Murry C, Keirstead HS, Park HS, Schmidt U, Laslett AL, Muller FJ, Nievergelt CM, Shamir R, Loring JF. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell. 2011;8:106–118. doi: 10.1016/j.stem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker DE, Harrison NJ, Maltby E, Smith K, Moore HD, Shaw PJ, Heath PR, Holden H, Andrews PW. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat Biotechnol. 2007;25:207–215. doi: 10.1038/nbt1285. [DOI] [PubMed] [Google Scholar]
- 17.Narva E, Autio R, Rahkonen N, Kong L, Harrison N, Kitsberg D, Borghese L, Itskovitz-Eldor J, Rasool O, Dvorak P, Hovatta O, Otonkoski T, Tuuri T, Cui W, Brustle O, Baker D, Maltby E, Moore HD, Benvenisty N, Andrews PW, Yli-Harja O, Lahesmaa R. High-resolution DNA analysis of human embryonic stem cell lines reveals culture-induced copy number changes and loss of heterozygosity. Nat Biotechnol. 2010;28:371–377. doi: 10.1038/nbt.1615. [DOI] [PubMed] [Google Scholar]
- 18.Ben-David U, Benvenisty N. High prevalence of evolutionarily conserved and species-specific genomic aberrations in mouse pluripotent stem cells. Stem Cells. 2012;30:612–622. doi: 10.1002/stem.1057. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, Wu H, Loring J, Hormuzdi S, Disteche CM, Bornstein P, Jaenisch R. Trisomy eight in ES cells is a common potential problem in gene targeting and interferes with germ line transmission. Dev Dyn. 1997;209:85–91. doi: 10.1002/(SICI)1097-0177(199705)209:1<85::AID-AJA8>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 20.Varela C, Denis JA, Polentes J, Feyeux M, Aubert S, Champon B, Pietu G, Peschanski M, Lefort N. Recurrent genomic instability of chromosome 1q in neural derivatives of human embryonic stem cells. J Clin Invest. 2012;122:569–574. doi: 10.1172/JCI46268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dekel-Naftali M, Aviram-Goldring A, Litmanovitch T, Shamash J, Reznik-Wolf H, Laevsky I, Amit M, Itskovitz-Eldor J, Yung Y, Hourvitz A, Schiff E, Rienstein S. Screening of human pluripotent stem cells using CGH and FISH reveals low-grade mosaic aneuploidy and a recurrent amplification of chromosome 1q. Eur J Hum Genet. 2012;20:1248–1255. doi: 10.1038/ejhg.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Catalina P, Montes R, Ligero G, Sanchez L, de la Cueva T, Bueno C, Leone PE, Menendez P. Human ESCs predisposition to karyotypic instability: Is a matter of culture adaptation or differential vulnerability among hESC lines due to inherent properties? Mol Cancer. 2008;7:76. doi: 10.1186/1476-4598-7-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayshar Y, Ben-David U, Lavon N, Biancotti JC, Yakir B, Clark AT, Plath K, Lowry WE, Benvenisty N. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell. 2010;7:521–531. doi: 10.1016/j.stem.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 24.Moon SH, Kim JS, Park SJ, Lim JJ, Lee HJ, Lee SM, Chung HM. Effect of chromosome instability on the maintenance and differentiation of human embryonic stem cells in vitro and in vivo. Stem Cell Res. 2011;6:50–59. doi: 10.1016/j.scr.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 25.Lefort N, Feyeux M, Bas C, Feraud O, Bennaceur-Griscelli A, Tachdjian G, Peschanski M, Perrier AL. Human embryonic stem cells reveal recurrent genomic instability at 20q11.21. Nat Biotechnol. 2008;26:1364–1366. doi: 10.1038/nbt.1509. [DOI] [PubMed] [Google Scholar]
- 26.Ramos-Mejia V, Munoz-Lopez M, Garcia-Perez JL, Menendez P. iPSC lines that do not silence the expression of the ectopic reprogramming factors may display enhanced propensity to genomic instability. Cell Res. 2010;20:1092–1095. doi: 10.1038/cr.2010.125. [DOI] [PubMed] [Google Scholar]
- 27.Baum C, Modlich U, Gohring G, Schlegelberger B. Concise review: managing genotoxicity in the therapeutic modification of stem cells. Stem Cells. 2011;29:1479–1484. doi: 10.1002/stem.716. [DOI] [PubMed] [Google Scholar]
- 28.Pasi CE, Dereli-Oz A, Negrini S, Friedli M, Fragola G, Lombardo A, Van Houwe G, Naldini L, Casola S, Testa G, Trono D, Pelicci PG, Halazonetis TD. Genomic instability in induced stem cells. Cell Death Differ. 2011;18:745–753. doi: 10.1038/cdd.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Q, Shi X, Rudolph C, Yu Y, Zhang D, Zhao X, Mai S, Wang G, Schlegelberger B, Shi Q. Recurrent trisomy and Robertsonian translocation of chromosome 14 in murine iPS cell lines. Chromosome Res. 2011;19:857–868. doi: 10.1007/s10577-011-9239-y. [DOI] [PubMed] [Google Scholar]
- 30.Kinoshita T, Nagamatsu G, Kosaka T, Takubo K, Hotta A, Ellis J, Suda T. Ataxia-telangiectasia mutated (ATM) deficiency decreases reprogramming efficiency and leads to genomic instability in iPS cells. Biochem Biophys Res Commun. 2011;407:321–326. doi: 10.1016/j.bbrc.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 31.Quinlan AR, Boland MJ, Leibowitz ML, Shumilina S, Pehrson SM, Baldwin KK, Hall IM. Genome sequencing of mouse induced pluripotent stem cells reveals retroelement stability and infrequent DNA rearrangement during reprogramming. Cell Stem Cell. 2011;9:366–373. doi: 10.1016/j.stem.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, Devenney PS, Sexton D, Grimes G, Holt IJ, Hill RE, Taylor MS, Lawson KA, Dorin JR, Jackson AP. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell. 2012;149:1008–1022. doi: 10.1016/j.cell.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prigione A, Hossini AM, Lichtner B, Serin A, Fauler B, Megges M, Lurz R, Lehrach H, Makrantonaki E, Zouboulis CC, Adjaye J. Mitochondrial-associated cell death mechanisms are reset to an embryonic-like state in aged donor-derived iPS cells harboring chromosomal aberrations. PLoS One. 2011;6:e27352. doi: 10.1371/journal.pone.0027352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 35.Louis SF, Vermolen BJ, Garini Y, Young IT, Guffei A, Lichtensztejn Z, Kuttler F, Chuang TC, Moshir S, Mougey V, Chuang AY, Kerr PD, Fest T, Boukamp P, Mai S. c-Myc induces chromosomal rearrangements through telomere and chromosome remodeling in the interphase nucleus. Proc Natl Acad Sci U S A. 2005;102:9613–9618. doi: 10.1073/pnas.0407512102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vajen B, Modlich U, Schienke A, Wolf S, Skawran B, Hofmann W, Buesche G, Kreipe H, Baum C, Santos-Barriopedro I, Vaquero A, Schlegelberger B, Rudolph C. Histone methyltransferase Suv39h1 deficiency prevents Myc-induced chromosomal instability in murine myeloid leukemias. Genes Chromosomes Cancer. 2013;52:423–30. doi: 10.1002/gcc.22040. [DOI] [PubMed] [Google Scholar]
- 37.Flores I, Blasco MA. The role of telomeres and telomerase in stem cell aging. FEBS Lett. 2010;584:3826–3830. doi: 10.1016/j.febslet.2010.07.042. [DOI] [PubMed] [Google Scholar]
- 38.Zalzman M, Falco G, Sharova LV, Nishiyama A, Thomas M, Lee SL, Stagg CA, Hoang HG, Yang HT, Indig FE, Wersto RP, Ko MS. Zscan4 regulates telomere elongation and genomic stability in ES cells. Nature. 2010;464:858–863. doi: 10.1038/nature08882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menendez S, Antipin J, Reva B, Koff A, Nimer SD. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4:37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jain AK, Allton K, Iacovino M, Mahen E, Milczarek RJ, Zwaka TP, Kyba M, Barton MC. p53 regulates cell cycle and microRNAs to promote differentiation of human embryonic stem cells. PLoS Biol. 2012;10:e1001268. doi: 10.1371/journal.pbio.1001268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, Okita K, Yamanaka S. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460:1132–1135. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, Wahl GM, Izpisua Belmonte JC. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460:1140–1144. doi: 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marion RM, Strati K, Li H, Murga M, Blanco R, Ortega S, Fernandez-Capetillo O, Serrano M, Blasco MA. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460:1149–1153. doi: 10.1038/nature08287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Begus-Nahrmann Y, Lechel A, Obenauf AC, Nalapareddy K, Peit E, Hoffmann E, Schlaudraff F, Liss B, Schirmacher P, Kestler H, Danenberg E, Barker N, Clevers H, Speicher MR, Rudolph KL. p53 deletion impairs clearance of chromosomal-instable stem cells in aging telomere-dysfunctional mice. Nat Genet. 2009;41:1138–1143. doi: 10.1038/ng.426. [DOI] [PubMed] [Google Scholar]
- 45.Lee DF, Su J, Ang YS, Carvajal-Vergara X, Mulero-Navarro S, Pereira CF, Gingold J, Wang HL, Zhao R, Sevilla A, Darr H, Williamson AJ, Chang B, Niu X, Aguilo F, Flores ER, Sher YP, Hung MC, Whetton AD, Gelb BD, Moore KA, Snoeck HW, Ma’ayan A, Schaniel C, Lemischka IR. Regulation of embryonic and induced pluripotency by aurora kinase-p53 signaling. Cell Stem Cell. 2012;11:179–194. doi: 10.1016/j.stem.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ye D, Wang G, Liu Y, Huang W, Wu M, Zhu S, Jia W, Deng AM, Liu H, Kang J. MiR-138 promotes induced pluripotent stem cell generation through the regulation of the p53 signaling. Stem Cells. 2012;30:1645–1654. doi: 10.1002/stem.1149. [DOI] [PubMed] [Google Scholar]
- 47.Sperka T, Song Z, Morita Y, Nalapareddy K, Guachalla LM, Lechel A, Begus-Nahrmann Y, Burkhalter MD, Mach M, Schlaudraff F, Liss B, Ju Z, Speicher MR, Rudolph KL. Puma and p21 represent cooperating checkpoints limiting self-renewal and chromosomal instability of somatic stem cells in response to telomere dysfunction. Nat Cell Biol. 2012;14:73–79. doi: 10.1038/ncb2388. [DOI] [PubMed] [Google Scholar]
- 48.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701–713. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 49.Jadersten M, Saft L, Smith A, Kulasekararaj A, Pomplun S, Gohring G, Hedlund A, Hast R, Schlegelberger B, Porwit A, Hellstrom-Lindberg E, Mufti GJ. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J. Clin. Oncol. 2011;29:1971–1979. doi: 10.1200/JCO.2010.31.8576. [DOI] [PubMed] [Google Scholar]
- 50.Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J, Jager N, Remke M, Shih D, Northcott PA, Pfaff E, Tica J, Wang Q, Massimi L, Witt H, Bender S, Pleier S, Cin H, Hawkins C, Beck C, von Deimling A, Hans V, Brors B, Eils R, Scheurlen W, Blake J, Benes V, Kulozik AE, Witt O, Martin D, Zhang C, Porat R, Merino DM, Wasserman J, Jabado N, Fontebasso A, Bullinger L, Rucker FG, Dohner K, Dohner H, Koster J, Molenaar JJ, Versteeg R, Kool M, Tabori U, Malkin D, Korshunov A, Taylor MD, Lichter P, Pfister SM, Korbel JO. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59–71. doi: 10.1016/j.cell.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, Kandoth C, Fulton RS, McLellan MD, Dooling DJ, Wallis JW, Chen K, Harris CC, Schmidt HK, Kalicki-Veizer JM, Lu C, Zhang Q, Lin L, O’Laughlin MD, McMichael JF, Delehaunty KD, Fulton LA, Magrini VJ, McGrath SD, Demeter RT, Vickery TL, Hundal J, Cook LL, Swift GW, Reed JP, Alldredge PA, Wylie TN, Walker JR, Watson MA, Heath SE, Shannon WD, Varghese N, Nagarajan R, Payton JE, Baty JD, Kulkarni S, Klco JM, Tomasson MH, Westervelt P, Walter MJ, Graubert TA, DiPersio JF, Ding L, Mardis ER, Wilson RK. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Z, Dullmann J, Schiedlmeier B, Schmidt M, von Kalle C, Meyer J, Forster M, Stocking C, Wahlers A, Frank O, Ostertag W, Kuhlcke K, Eckert HG, Fehse B, Baum C. Murine leukemia induced by retroviral gene marking. Science. 2002;296:497. doi: 10.1126/science.1068893. [DOI] [PubMed] [Google Scholar]
- 53.Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, Clappier E, Caccavelli L, Delabesse E, Beldjord K, Asnafi V, MacIntyre E, Dal Cortivo L, Radford I, Brousse N, Sigaux F, Moshous D, Hauer J, Borkhardt A, Belohradsky BH, Wintergerst U, Velez MC, Leiva L, Sorensen R, Wulffraat N, Blanche S, Bushman FD, Fischer A, Cavazzana-Calvo M. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, Brugman MH, Pike-Overzet K, Chatters SJ, de Ridder D, Gilmour KC, Adams S, Thornhill SI, Parsley KL, Staal FJ, Gale RE, Linch DC, Bayford J, Brown L, Quaye M, Kinnon C, Ancliff P, Webb DK, Schmidt M, von Kalle C, Gaspar HB, Thrasher AJ. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, Schmidt M, Kramer A, Schwable J, Glimm H, Koehl U, Preiss C, Ball C, Martin H, Gohring G, Schwarzwaelder K, Hofmann WK, Karakaya K, Tchatchou S, Yang R, Reinecke P, Kuhlcke K, Schlegelberger B, Thrasher AJ, Hoelzer D, Seger R, von Kalle C, Grez M. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 56.Sugawara A, Goto K, Sotomaru Y, Sofuni T, Ito T. Current status of chromosomal abnormalities in mouse embryonic stem cell lines used in Japan. Comp Med. 2006;56:31–34. [PubMed] [Google Scholar]
- 57.Cowan CA, Klimanskaya I, McMahon J, Atienza J, Witmyer J, Zucker JP, Wang S, Morton CC, McMahon AP, Powers D, Melton DA. Derivation of embryonic stem-cell lines from human blastocysts. N Engl J Med. 2004;350:1353–1356. doi: 10.1056/NEJMsr040330. [DOI] [PubMed] [Google Scholar]
- 58.Catalina P, Bueno C, Montes R, Nieto A, Ligero G, Sanchez L, Jara M, Rasillo A, Orfao A, Cigudosa J, Hovatta O, Greaves M, Menendez P. Genetic stability of human embryonic stem cells: A first-step toward the development of potential hESC-based systems for modeling childhood leukemia. Leuk Res. 2009;33:980–990. doi: 10.1016/j.leukres.2008.08.028. [DOI] [PubMed] [Google Scholar]
- 59.Ben-David U, Mayshar Y, Benvenisty N. Large-scale analysis reveals acquisition of lineage-specific chromosomal aberrations in human adult stem cells. Cell Stem Cell. 2011;9:97–102. doi: 10.1016/j.stem.2011.06.013. [DOI] [PubMed] [Google Scholar]