

Abstract
In bacteria, replication is a carefully orchestrated event that unfolds the same way for each bacterium and each cell division. The process of DNA replication in bacteria optimizes cell growth and coordinates high levels of simultaneous replication and transcription. In metazoans, the organization of replication is more enigmatic. The lack of a specific sequence that defines origins of replication has, until recently, severely limited our ability to define the organizing principles of DNA replication. This question is of particular importance as emerging data suggest that replication stress is an important contributor to inherited genetic damage and the genomic instability in tumors. We consider here the replication program in several different organisms including recent genome-wide analyses of replication origins in humans. We review recent studies on the role of cytosine methylation in replication origins, the role of transcriptional looping and gene gating in DNA replication, and the role of chromatin’s 3-dimensional structure in DNA replication. We use these new findings to consider several questions surrounding DNA replication in metazoans: How are origins selected? What is the relationship between replication and transcription? How do checkpoints inhibit origin firing? Why are there early and late firing origins? We then discuss whether oncogenes promote cancer through a role in DNA replication and whether errors in DNA replication are important contributors to the genomic alterations and gene fusion events observed in cancer. We conclude with some important areas for future experimentation.
Keywords: replication, origin, replicon, transcription, epigenetics, checkpoints
Introduction to DNA Replication
Making another strand of DNA that is exactly the same as the existing strand can be argued to be the single most important job of a cell. Unwinding, replicating, and rewinding 3 billion base pairs must be an incredibly challenging task. The cell accomplishes this by creating a series of sites at which replication will be initiated and by opening the double-stranded DNA at those sites to form a small bubble. DNA is replicated outward from these sites in both directions by replication “forks.” Once these forks are formed, they may encounter obstacles and roadblocks: DNA adducts, the presence of other enzymes, or a lack of dNTPs. A complex machinery exists to stabilize replication forks, to help prevent their collapse and failure, and to prevent them from firing under conditions in which they are likely to fail. Fork failure has been associated with genetic changes, and recent data indicate that DNA replication may be an important contributor to the genetic alterations observed in humans. We might imagine that there would be a strong drive to organize DNA replication in a way that optimizes the cell’s ability to faithfully create a replica of its DNA.
DNA replication is initiated when a trans-acting factor binds to a cis element to initiate replication.1 In the circular genomes of bacteria, there is only one site at which replication initiates, the origin of replication. If there were only one site at which DNA replication initiated in human cells, it would take 20 days to replicate the entire genome.2 Instead, in eukaryotes, there are many locations along chromosomes at which replication initiates.3 In humans, an estimated 30,000 DNA replication origins are activated with each cell division.4 At each of these origins, spaced approximately 100 kb apart, protein complexes form that prepare the DNA to initiate bidirectional replication, that is, fire. A set of proteins that is highly conserved from yeast to humans has been identified that form the prereplication complex. First, the origin recognition complex (ORC) protein binds, and it is followed in turn by Cdc6, Cdt1, and minichromosome maintenance 2 to 7 (MCM2-MCM7) helicase.2,5,6 Actually initiating replication, that is, “firing” the origins, occurs in response to an increase in the activity of S phase–promoting kinases Cdc7/Dbf4 and S-phase, cyclin-dependent kinases.7-9 These kinases recruit additional components including the CDC45 protein7 and the α DNA polymerase and thereby trigger the conversion of the prereplicative complex to actively firing origins.
The activity of S-phase kinases and Cdc7/Dbf4 is tightly regulated in order to ensure that DNA replication only occurs during S phase and that each portion of chromosomes is replicated only once per cell cycle. Changes in cyclin-dependent kinase activity,10 proteasome-mediated destruction of Cdt1,11 and a Cdt1 inhibitory protein called geminin prevent the assembly of new prereplication complexes until mitosis, when prereplication complexes are permitted to reassemble. By restricting the formation of the prereplication complexes to only a short window before S phase, the cell can ensure that origins fire only once during the cell cycle.12 When the cells enter the next S phase, only existing complexes are permitted to fire.13,14 After an origin fires or when it is passively replicated by a replication fork from a neighboring origin, the origin loses its initiation proteins.15 Thus, origins that have already fired are prevented from reloading a prereplication complex prior to the next cell division.16 Through these mechanisms, the cell prevents origins from firing more than once per cycle,17 which could result in re-replication, DNA damage, or cell death.
Even if DNA replication is restricted to one round in a cell cycle, it is still a process that is fraught with challenges. Replication forks must navigate challenging DNA structures such as triplet repeats, palindromic sequences, G-quartets, telomeric repeats, DNA adducts, and tRNA genes, all of which create problems that cause the replication forks to stall.18 They must also replicate genes that are actively being transcribed, which may result in a collision with an RNA polymerase. There may be low levels of dNTPs or proteins important for the creation of the replisome.19 As a result of all of these challenges, replication forks are often in danger. In budding yeast, forks stall approximately once per 10 kb.20 When DNA polymerases encounter barriers, one possibility is that the blockage is removed and the polymerase can continue.21 Other possible outcomes can result in errors in the passage of genetic information, for instance, single-stranded DNA and DNA breaks can be formed, which can lead to single-stranded 3′ tails from broken forks annealing with microhomology to single-stranded DNA nearby.22 Indeed, a pattern of mutations characterized by microhomologies is induced by treatment with the inhibitor of ribonucleotide reductase hydroxyurea or the DNA polymerase inhibitor aphidicolin.23,24
When replication problems arise, the cell can activate checkpoint mechanisms that integrate information about the presence of DNA damage and the replication status of the cell and provide the cell with multiple different types of assistance. RPA-bound, single-stranded DNA accumulates at stalled forks and acts as a signal to checkpoint kinases.25 The checkpoint kinases ATR/Mec1 and ATM/Tel1 stabilize stalled forks19 and prevent their collapse.19,26-28 In yeast with mutations in the ATR pathway, stalled replication forks rapidly collapse into a reversed-fork conformation.19,27 These structures recombine very easily and can lead to genome rearrangements including inverted repeats19,27,29 and translocations.30-35 In an analysis comparing the most important contributors to cell death in yeast, checkpoint activation of stalled replication forks was found to be central to cell viability, while other pathways such as the regulation of mitosis, gene expression, and origin firing only contributed modestly.36
Interest in replication fork–mediated genomic changes has increased recently because of deep sequencing studies of human genomes.22,28,34,37,38 The complex genome rearrangements discovered in some patients with genetic disorders suggest elaborate replication fork failure.22,28,34,37,38 One disorder that has been investigated is Pelizaeus-Merzbacher disease an X-linked myelination disorder often caused by nonrecurrent duplications of the proteolipid protein 1 gene. Analysis of this gene in patients revealed complex patterns of duplications.22,34 The types of alterations included multiple copy number changes, deletions, inverted duplications, triplications, insertion of short sequences at break points that were templated by nearby genomic regions, and microhomology at the break-point junctions. Based on comparisons with experimental observations from bacteria, yeast, and human studies,22,39 the changes were considered consistent with a replication-based model for their generation based on fork stalling and template switching.40 Further studies of the dystrophin gene and other disease-causing genes revealed a similar pattern with deletions and duplications that were interpreted as likely resulting from stalled and dislodged replication forks that re-engaged at a different template based on a limited region of homology.41-43
Because of the critical importance of performing the complex act of DNA replication, we anticipate that the cell has evolved an optimized and efficient strategy to achieve high fidelity transmission of genetic information. Yet, we still do not fully appreciate the most important principles for the organization of DNA replication in eukaryotes. The last year has seen an explosion of information. High-throughput methods for identifying origins have become possible, and several reports have emerged describing the systematic identification of a large number of origins in multiple species. Information on the timing of DNA replication and on the 3-dimensional organization of chromatin has provided insights into the dynamics of origin firing and its relationship with the spatial organization of chromatin. Other studies have highlighted the role of new pathways in preparing origins for replication, in organizing 3-dimensional replication structure, and in creating replication checkpoints.
We review the relevant literature on DNA replication in different species, focusing on recent advances. We then synthesize this information to discuss the question: what are the most central problems that a cell faces when it replicates its DNA, and has it organized the selection of origins for firing and the timing of origin firing in order to minimize these problems? We then consider how these findings relate to the formation of tumors and whether they can explain the types of mutations observed in tumors. Finally, we articulate what we believe will be the exciting areas of research that will advance this field.
Replication in Different Organisms
Replication in bacteria
In a bacterial cell cycle, chromosome replication starts at a single genomic locus, at a well-defined sequence, the origin. The replication origin in Escherichia coli, oriC, contains multiple DnaA binding sites, and replication begins when the initiator protein, DnaA, binds to these sequences and opens an adjacent AT-rich element.5,44,45 This transition remodels the replication origins for prereplication complex assembly.46 Replication proceeds in both directions away from the origin and is performed by a holoenzyme particle with 2 polymerase cores. One polymerase performs leading strand synthesis, while the other performs lagging strand synthesis through the use of Okazaki fragments. Once the polymerases complete the task of DNA replication, the chromosome dimer is resolved by site-specific recombination. The same sequence is used as an origin in every cell cycle, so the replication program unfolds in the same way in each bacterium and each cell cycle.1 During exponential growth, E. coli can have 2 to 3 simultaneous rounds of replication, which results in there being more copies of the DNA encoding genes near the origin relative to genes near the terminus of replication. Highly expressed genes cluster near the origin,47 which may be advantageous since these sequences are often single-stranded during replication.48,49 The position of genes within the genome is clearly important because if the distance of genes to the origin of replication is changed, growth rate slows.50
DNA replication in budding yeast Saccharomyces cerevisiae
Budding yeast such as S. cerevisiae differ from bacteria in that they have not one but many sequences that can serve as origins of replication. DNA replication initiates at sites that contain an AT-rich DNA motif bound by the ORC.51 These sequences can be identified functionally by their ability to support autonomous replication of bacterial plasmids in yeast backgrounds.52,53 However, of the 12,000 ARS present in the S. cerevisiae genome, only approximately 400 (3.3%) are functional.54 Origins that actually fire are often found in a broader sequence context that involves a relative depletion of nucleosomes near the origin.54-57
Since DNA is not loose within the nucleus but rather tightly wound around octamers of histones and further organized into 30-nm fibers of condensed nucleosomes, accessing a single strand of DNA for replication requires the ability to open and unravel chromatin and gain access to an individual strand. Depleting nucleosomes nearby the potential origin may facilitate formation of a prereplication complex. RNA polymerases face similar challenges as DNA polymerases58,59 and binding sites for transcription factors that regulate the accessibility of DNA to RNA polymerases can also facilitate replication initiation.59 For instance, the B3 element of the ARS1 replication origin is a recognition site for the transcription factor Abf1. Replacing the Abf1 recognition site with the recognition site for a different transcription factor, GAL4, allowed the origin to retain its capacity to nucleate DNA replication.60 Further, mutations that disrupt silent chromatin at telomeres in budding yeast activate a telomeric origin that is typically silent.61 Thus, in S. cerevisiae, origins that have ARS sequences that are accessible can be quite efficient, and consequently, replication unfolds in a fairly consistent way from one cell cycle to the next.62
Replication in fission yeast
In the fission yeast Schizosaccharomyces pombe, origins are not characterized by a specific sequence as in budding yeast but do share the presence of AT-rich islands.63,64 These sequences can be bound by an S. pombe ORC subunit that contains AT-hook domains.65,66 AT-rich sequences have been reported to exclude nucleosomes because the intrinsic stiffness of polyA is energetically unfavorable for nucleosome formation.66,67 This past year, single-molecule deep sequencing of replication origins in 3 fission yeast (S. pombe, Schizosaccharomyces octosporus, and Schizosaccharomyces japonicus) was combined with data on genome-wide nucleosome occupancy in the same species.68 In S. pombe and S. octosporus, polyA tracts and, to a lesser extent, AT richness were strong predictors of origin function. However, in S. japonicus, AT content was a negative predictor of origin function, and instead, origins had high GC content and were enriched in polyG and the binding site for a protein known to have nucleosome-excluding properties.69 The authors concluded that the sequences of the fission yeast genomes that function as origins are the nucleosome-depleted regions of the genome with the highest affinity for the ORC.
Replication in embryonic frogs and flies
Frog and fly embryos use a radically different replication model from the approach used by bacterium. Early in the development of both frog and fly embryos, when rapid chromosomal replication is needed to keep up with the fast pace of cell division, DNA replication initiates at many sites throughout the genome all at the same time.70,71 The regions of the chromosomes that are used to initiate DNA replication are not characterized by the presence of any particular sequence, and instead, origins are formed and fire from seemingly random sequences present at short, regular intervals. Any plasmid DNA injected into a Xenopus egg can replicate with origins that are not consistent from one cycle to the next.59 By firing many origins simultaneously, DNA replication is completed rapidly, and the cells divide quickly.72-74 In early Drosophila cleavage embryos, S phase lasts just a few minutes, whereas DNA replication takes 8 hours in somatic cells.75,76 Under these conditions, each dividing embryonic cell has a different and distinct replication program.
In both Xenopus and Drosophila, after the midblastula transition, there is a shift to a much longer S phase.76,77 A large number of origins are inactivated, and in Drosophila, replicon size increases from 8 kb in early embryos to 40 kb in cultured cells.5,71,78 In addition, there is no longer synchrony in origin firing, and instead, some origins fire earlier in S phase, while others fire later.76,77 This shift corresponds to the time at which the developing organism ceases to rely exclusively on maternally supplied transcripts as it does as an early zygote and activates the transcription of its own genes. After the midblastula transition, there is a site-specific preference for promoter regions and AT-rich sequences.78,79 At the DNA polymerase α gene locus, DNA replication initiates at nonspecific chromosomal sites in preblastoderm embryos, but in more differentiated cells, initiation is restricted to 2 sites, one of which coincides with the promoter region of the adjacent dE2F gene.78
To more directly test the relationship between origins of replication and transcription factor binding sites, an inducible transcription template was introduced into Xenopus eggs. When the corresponding transcription factor was introduced, a site-specific origin of replication was formed.59 Transcription itself was not required, but the presence of the transcription factor binding site resulted in changes in the acetylation state of histone lysines. The results reinforce the importance of chromatin conformation and displacement of nucleosomes by transcription factors in selecting regions of DNA to serve as origins.
Replication in metazoans
In mature cells of higher eukaryotes, there are specific sequences that identify the positions at which replication initiates. For instance, if the origin of replication in the human β-globin gene locus along with 8 kb of surrounding DNA is transferred to a different site in the genome, it can still direct site-specific initiation of replication. However, replication origins in higher eukaryotes, unlike in bacteria or budding yeast, do not share a consensus sequence, even though the proteins that are recruited to origins in higher eukaryotes are similar to those in bacteria and yeast.6,45 Unfortunately, the types of functional assays that successfully identified origins of replication in yeast based on their ability to promote the replication of bacterial plasmids, have not been successful in mammalian cells.45,80 However, enough origins have been identified that patterns have emerged. Some of the specific sequences enriched near origins in fission yeast are also important for higher eukaryotes as well. For instance, Xenopus ORC tends to bind to asymmetrical, AT-rich sequences,81 while Drosophila ORC tends to bind AT-rich sequences82 that are negatively supercoiled.81
In higher eukaryotes, origins are not utilized with the same efficiency; some initiate replication in almost every cell cycle, while others rarely fire and instead are passively duplicated by a replication fork initiated at neighboring origins. In S. cerevisiae and S. pombe, the overall efficiency is less than 50%, but some origins are used in almost every cell cycle, and others are largely inactive.83,84 In metazoans, the efficiency of specific replication origins has been estimated at 5% to 20%.85-87 Thus, in different cell cycles and in different types of cells, different origins are selected for firing,88 and the cells therefore have a different replication pattern.89,90
Many of the known replication origins map to intergenic regions close to promoters in higher eukaryotes,5,91-95 and highly expressed genes tend to cluster near origins of replication.47,48 Further, transcriptionally active regions in mammalian cells tend to replicate early in S phase.95 Origins that fire early tend to have more open, accessible chromatin,95-96 acetylated histone lysines,97 and high levels of activating histone dimethylation and trimethylation on H3K4.98-100 Modulation of histone acetylation and chromatin conformation patterns is sufficient to affect DNA replication programs.101-104 Origins of replication are also associated with nearby unmethylated CpG islands and promoters.93,105,106 Origins at unmethylated CpG islands replicate earlier than those at methylated CpG islands.107 Origins activated late in S phase are associated with nontranscribed, heterochromatic regions.95,105,108,109 Eventually, all of the genome must be replicated, so origins in heterochromatic regions do ultimately fire, but they tend to fire late in S phase.110 In humans as well as in Xenopus, while origins tend to be present near transcription start sites, actual transcription is not necessary for origin activity.111
One example of an origin of replication that demonstrates the association between transcription and replication in mammalian cells is the β-globin locus, a site that is strongly transcriptionally induced in erythroid cells that synthesize large amounts of hemoglobin.112,113 In the human erythroleukemic cell line K562, expression of the β-globin locus is strongly dependent on a locus control region (LCR) >20 kb upstream of the β-globin gene that contains binding sites for transcription factors including those of the Maf and bZip family proteins. Although the LCR is located kilobases away, it is required for replication initiation because deletion of a region containing the LCR abolishes replication initiation from the β-globin origin.114
The association between origins of replication and transcription start sites could reflect a functionally important role for transcription factors in activating replication origins. In viruses, yeast, Drosophila, and Xenopus, transcription factors play a role in activating origins.5,59 Drosophila Rb and E2F bind the chorion origin,115 the Drosophila angiotensin-converting enzyme origin binds c-Myb homologs and Rb,116 and the lamin B2 origin associates with USF and SP1.92,117 As described above, transcription factors play an important role in the remodeling of nucleosomes in order to make single strands of DNA accessible to the RNA polymerase58,59 or replication proteins.118,119 There might also be a physical interaction between the transcription factors and the DNA replication machinery. c-Jun, whose binding sites are enriched near human origins, can activate polyoma virus DNA replication by stimulating the binding of the virus-encoded initiator, large T antigen, to origins.120 In addition to c-Jun, other transcription factors such as GAL4,121 bovine papilloma virus E2,122 c-Rel,123 and the p53 tumor suppressor124 can also enhance polyoma DNA replication through their binding sites. In bovine papilloma virus, the E2-E1 interaction can result in enhanced binding of E1 to the origin with consequent stimulation of DNA unwinding and replication.125-127
The oncogenic transcription factor c-Myc has also been implicated in the control of replication origin firing,128 possibly through its role in remodeling chromatin.129 Myc forms heterodimers with members of the Max family of proteins and binds to the E-box DNA sequence CACGTG. Myc-bound E-boxes can then associate with other proteins to remodel chromatin.130 In addition to its role in promoting transcription, c-Myc was found to co-immunoprecipitate with protein components of the ORC, suggesting a role in replication. Myc overexpression resulted in DNA damage during S phase,128 which may reflect a contribution of excessive origin firing. Further, c-Myc overexpression in primary human fibroblasts accelerated S phase, while c-Myc–deficient fibroblasts exhibited a prolonged S phase.131
A recent paper closely investigated the role of c-Myc in regulating the firing of the lamin B origin,132 one of the well-mapped human replication origins (Fig. 1).133 The lamin B origin contains an E-box to which c-Myc and its partner Max bind in early G1 phase. The histone methyltransferase mixed lineage leukemia 1 (MLL1) was recruited by c-Myc to the lamin B origin and added methyl groups to lysine 4 of histone H3 in nearby nucleosomes. After MLL1 and c-Myc were released from the origin, the H3K4me3 modification persisted and served to recruit the histone acetylase HBO1, which acetylated lysines on histone H4. Hyperacetylation resulted in lower nucleosome occupancy, which facilitated the loading of MCM proteins at the origin.
Figure 1.
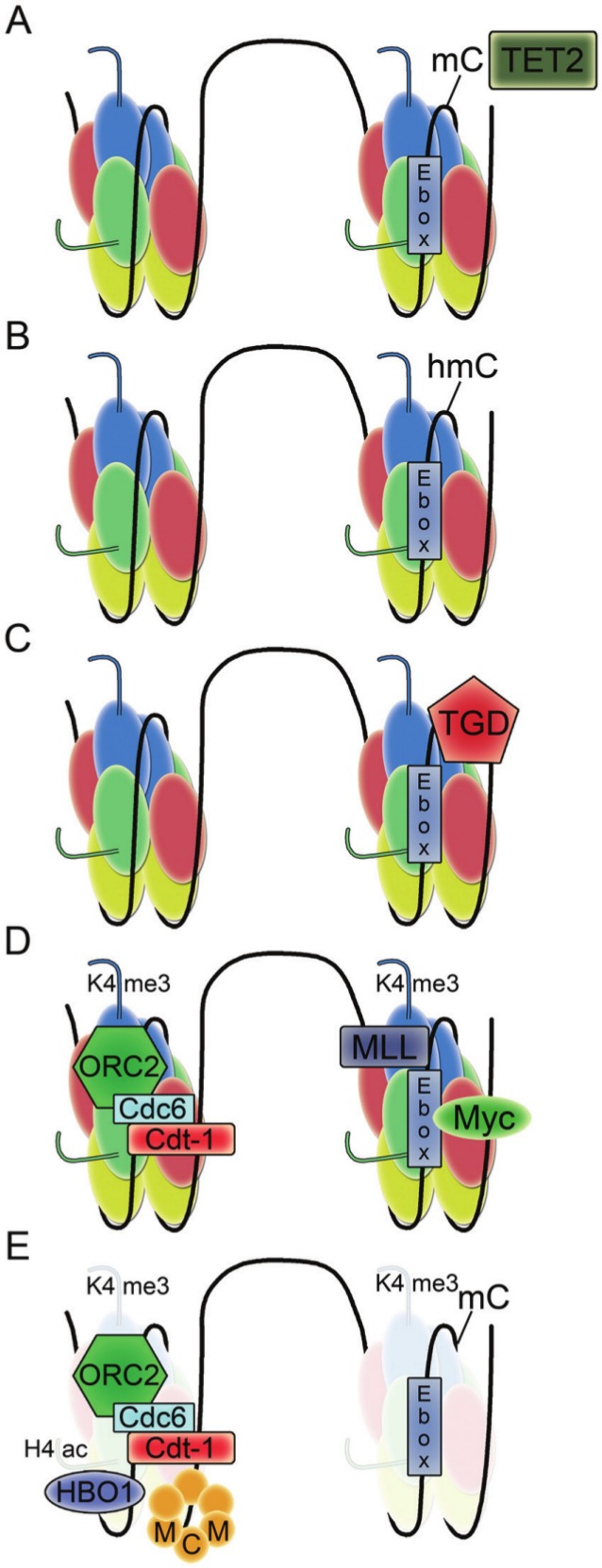
Model for the initiation of DNA replication at the lamin B2 origin. The TET2 enzyme converts methylated cytosine to hydroxymethylated cytosine. The hydroxymethylated cytosine is excised by thymine DNA glycosylase. The E-box with unmodified cytosine is recognized by Myc as origin proteins ORC2, Cdc6, and Cdt1 are bound. Myc recruits MLL, which modifies histone H3 on lysine 4 to the trimethyl form. The histone H3K4me3 mark promotes the binding of HBO1 acetylase and facilitates histone H4 hyperacetylation. The resulting hyperacetylation favors nucleosome remodeling that facilitates the loading of MCM proteins.
The authors also discovered that loading of the c-Myc protein to the origin was controlled by demethylation of CpG sites in the E-box.132 During the transition from G0 to G1, CpG sites were first converted to hydroxy-CpG by Ten-Eleven Translocation (TET) enzymes and then repaired by thymine DNA glycosylase-mediated base excision repair, which removed the modified nucleotides. Removal of the modifications prepared the CpGs for Myc binding and the rest of the remodeling required for MCM complex loading. The findings support a model in which cytosine demethylation, Myc recruitment, and histone methylation-acetylation crosstalk result in nucleosome remodeling, replication complex recruitment, and origin licensing. It will be interesting to determine whether this model is confirmed and extended to other origins. For the DHFR origin, comparison of the active and inactive X chromosomes suggests that the retention of methylation at the CpG regions is not important for origin function.107,111 Further, mouse embryonic stem cells with genetic defects in CpG methylation did not exhibit differences in origin firing for many single copy loci.134
Global origin mapping
Without a functional assay for sequences capable of initiating replication,45 the main approach to understanding DNA replication in mammalian cells until recently had been to identify and investigate individual positions at which DNA replication initiates. With this strategy, about 30 origins were identified.86,135 In the past few years, with the widespread adoption of high-throughput sequencing technologies, there has been an explosion of information as hundreds of origins have been identified and mapped. The ability to systematically define human origins has made it possible to consider anew the properties of replication origins in humans and the important principles of DNA replication.
One of the first such studies involved mapping replication origins based on the sequence composition of the surrounding bases. The leading and lagging strands of replicated DNA display different rates for each of the possible base pair substitutions.136-138 In most species, the leading strand is richer in G relative to C and, to a lesser degree, richer in T relative to A.139 The most widely held view is that the cause of this skew is deamination of C to T.140 The rate of cytosine deamination is 140-fold higher in single-stranded than double-stranded DNA,141 and the leading strand spends more time single-stranded than the lagging strand during DNA replication. In bacterial chromosomes, where there is a single origin and a single terminus, and each nucleotide is always either leading or lagging, a strong compositional bias is observed.139,142 Drawing GC skews, defined as (G – C)/(G + C), in sliding windows has become a standard method to identify the origin and terminus of replication in bacteria as the sign of the TA and GC skews changes abruptly when crossing replication origins and termination sites.143,144 In yeast chromosomes, there are many autonomously replicating sequences, only some of which are used in each replication cycle, so a particular single-stranded sequence may be replicated as the leading strand in some cell cycles and the lagging strand in others. At the very end of yeast chromosomes, however, there is only one leading/lagging option, and in these sequences, the same asymmetry present in bacterial chromosomes is observed.145
Analysis of nucleotide composition asymmetry around experimentally determined origins in mammals revealed that in 6 out of 9 cases, the skew displayed an abrupt sign switch at the origin similar to the pattern in prokaryotes.146 This observation led to the computational prediction of around 1,000 putative origins representing about 27% of the genome.146,147 Within 50 kbp of putative origins, the mean density of genes that are transcribed in the same direction as they are replicated was 8.2 times greater than genes transcribed and replicated in opposite directions.147 These findings would suggest a strong concordance between directionality of transcription and replication in mammals but, as described further below, have been reanalyzed.
Experimental analyses have also been performed to systematically capture and map large numbers of replication origins. One approach has been to isolate transitory, RNA-DNA short nascent strand molecules created at the beginning of DNA replication based on their protection from exonuclease treatment and to hybridize them to microarrays.105 Applying this approach with microarrays that cover 1% of the genome resulted in a dataset of 283 origins of replication with interorigin distances that ranged from 1 kb to 500 kb. Origin sequences were more evolutionarily conserved than expected by chance, and half of them mapped within or near CpG islands.105 Most of the origins overlapped transcriptional regulatory elements, and in this study, a significant correlation was observed between origins and c-Jun and c-Fos binding sites. The position of origins correlated strongly with DNAse-hypersensitive sites, acetylated histone H4, and H3K4 dimethyl and trimethyl marks. The findings were consistent with the replication machinery preferentially recognizing open chromatin structures near promoters of actively transcribed genes, although approximately 30% of origins did not overlap with open chromatin marks. Similar findings were reported in other studies of origins identified by nascent strands99,148 or with a protocol in which restriction fragments containing origins are trapped in gelling agarose based on their partially circular nature and sequenced.149 While the findings of enrichment near CpG islands and transcription start sites were consistent among these studies, unfortunately, the concordance between the actual sites identified as origins was low when comparing in silico predicted versus experimentally defined origins or between origins from 2 different experimental approaches.99,105,150 For the computationally predicted origins, the sense strand for transcription was very likely to also be the leading strand for replication, while this same trend was not observed in a reanalysis of the origins defined by the presence of the nascent strand.150 Further, reanalysis of the experimentally observed origins revealed that the expected lagging strand/leading strand nucleotide skew was observed around origins in intergenic regions but not in origins found in introns, raising questions as to whether identifying origins based on nucleotide skew would lead to an unbiased set.150
Topological constraints, gene gating, and higher order chromatin structure
In addition to recent information on the mapping of origins genome-wide, there have also been a recent series of papers describing DNA replication with respect to the 3-dimensional topology of chromatin. All DNA-related processes including both replication and transcription generate torsional energy that can result in negatively or positively supercoiled DNA when the double helix underwinds or overwinds.151 When 2 forks converge during replication termination or when 1 fork clashes with a transcription bubble,152,153 especially if the helix is anchored and cannot rotate, the expectation is that there will be positive supercoiling ahead of the replication fork or transcription bubble and negative supercoiling behind it (Fig. 2A).152 If the torsional stress that is created by the encounter cannot simply diffuse through the chromosome by untwisting it, then type I and type II topoisomerases can make single- or double-stranded breaks, respectively, and catalyze strand passage reactions, thus changing the linkage of DNA molecules.151,153 Both top1 (a type I topoisomerase) and top2 (a type II topoisomerase) travel with replication forks in yeast and likely cooperate in the resolution of torsion produced by replication forks.154
Figure 2.
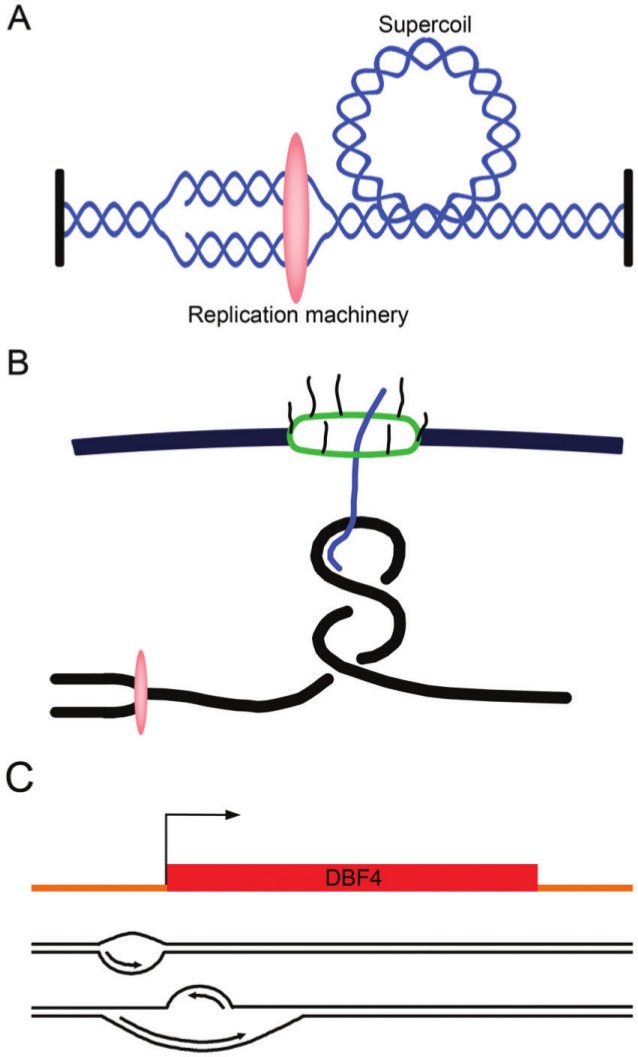
Topological problems and a potential solution associated with DNA replication. (A) Unwinding of the DNA causes positive supercoils to accumulate ahead of the advancing replication machinery (oval). (B) The topological problem associated with nuclear pore–gated transcribed genes. A model for a replication fork approaching a gene that is gated to the nuclear pore complex is shown. (C) An example of bidirectional replication initiation. The DBF4 origin contains 2 initiation zones. Replication starts at the first zone and progresses in the direction of transcription toward the second initiation zone. Replication then initiates from a second origin and proceeds on the opposite strand.
Recent studies have also shown that RNA polymerase II–transcribed units are organized into loops,155 which may facilitate RNA polymerase recycling and promote repetitive rounds of transcription of the same gene.155 Through gene gating, these loops are coupled to the nuclear pore complex, which allows for a coupling of transcription with mRNA export (Fig. 2B). The proteins that mediate this interaction are the THO/TREX and TREX-2 complexes and the nucleoporins.156-159 Gene gating might serve the purpose of decreasing the likelihood that the nascent RNA becomes tangled in the RNA polymerase bubble.160,161 However, by attaching the transcribed DNA to a fixed structure such as the nuclear pore, there may be an exacerbation of topological problems associated with transcription, especially when the replication fork arrives.154,156
The challenge faced by the replication fork when it encounters these gene loops is demonstrated by the finding that transcribed genes represent the most abundant sites of replication fork pausing in the yeast genome, and replisome pausing at transcribed genes is independent of the gene’s orientation with respect to the replication fork.162 During S phase, top2 is recruited to these loops and likely plays a role in relieving the associated torsional stress.163 Further, top2 may work together with the components of the THO/TREX/nuclear pore to alleviate torsional stress during DNA replication152 as top2 mutants are synthetic sick with mutations in the THO/TREX/nuclear pore complex.164 Consistent with a potential role for gene gating in DNA replication, THO knockdown in human cells results in defects not only in gene expression and hyperrecombination that likely result from problems during transcription165,166 but also changes in the kinetics of DNA replication.166
While the selection of individual origins suggests a randomness to the replication process in higher eukaryotes, in fact, the replication of eukaryotic chromosomes does exhibit a temporal and spatial organization within the nucleus. The characteristic timing and location of specific chromosomal regions are thought to reflect a higher order structure of the genome within the nucleus.167-169 One manifestation of the effects of a higher order structure of chromatin on replication is the presence of replication foci that are the nexus of multiple origins. Their position in the nucleus as well as temporal order of activation are inherited throughout cell cycles.170-173 Possibly as a result of this higher order structure, even though different individual origins may be selected to fire, there is a reproducible replication timing for broad regions of the genome.82,83,173-176 When followed for as many as 15 cell cycles, labeled foci that represent replication factories do not mix, separate, or change in shape.170,173 Moreover, replicon clusters that fire at different times during S phase occupy different subnuclear compartments.170
High-resolution replication timing profiles in mouse embryonic stem cells revealed multimegabase, coordinately replicated regions of the genome that are separated into distinct nuclear regions.176 The regions that act as boundaries were consistent between several embryonic stem cell lines and induced pluripotent stem cells. Upon differentiation to neural precursor cells, approximately 20% of the genome transitioned to different domains, with the predominant pattern being a consolidation into fewer, larger replication units in the differentiated cells. These studies and others have led to a model in which distant genomic regions of similar replication timing come together to form replication factories in which DNA is replicated in multiple regions simultaneously (Fig. 3).177-179
Figure 3.
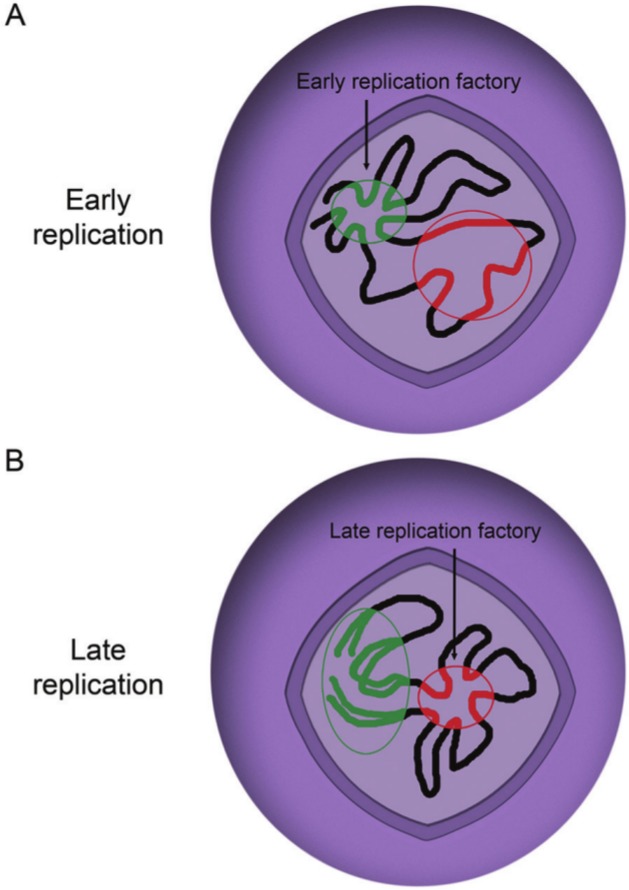
Model for the organization of genomic DNA with respect to DNA replication. Replication origins in open chromatin are grouped together into a replication factory early in S phase. Later in S phase, a different set of origins are clustered together to form a replication factory.
Proteins that regulate the formation of these replication factories have recently been identified. In mice, the absence of the nuclear matrix–localized Rif1 protein resulted in changes in the temporal order of origin firing as well as the physical definition of chromosome domains,180 implicating Rif1 as part of the machinery that establishes the accessibility of different origin clusters for replication factors. In yeast, mutations in the forkhead transcription factors resulted in extensive changes in the timing of origin firing, with early firing origins consistently firing later than normal and late firing origins shifting to an earlier firing time.181 The role of the forkhead proteins in transcription was unrelated to this phenotype, which more likely reflects the newly discovered ability of forkhead transcription factors to control the clustering of early origins and their association with the key initiation factor Cdc45 in G1.181
Understanding the Organization of DNA Replication
How does the cell organize the firing of its origins?
Bacteria exhibit a very strict replication model in which a single origin is fired at a specific time, while origin sequences in embryonic flies and frogs vary from cycle to cycle, do not share consensus motifs, but are regularly spaced. Higher eukaryotes use a different approach from either of these. There are specific sequences that function consistently as origins in higher eukaryotes, but only a fraction fire with each cell cycle. The simplest model would be one in which origins are selected at random for firing. After all, since S phase is distinct from the phase in which origins are licensed, perhaps the location of origins of replication does not matter.45 However, if potential origins were distributed randomly along the genome, one expects a geometric (exponential) distribution of separations.182 This would result in some very large interorigin distances, which could raise problems. If the gap is too large, the cell might not have time to replicate that region, and the region might be unreplicated at mitosis or delay the length of S phase.88
Recent studies employing DNA combing have suggested 2 different models to explain how the cell avoids this problem (Fig. 4). In DNA combing, sites of DNA replication are labeled at 2 different times with 2 different nucleotides and visualized with fluorescent antibodies. The data provide quantitative information on replication rates. Guilbaud and colleagues183 performed DNA combing studies and found that DNAse-hypersensitive sites and CpG islands were more abundant at early firing locations and steadily decrease as the replication forks head toward late firing regions. Replication fork velocity did not change during S phase, but the global fork density increased over the course of S phase, which was interpreted to indicate that the efficiency of firing increases through S phase (Fig. 4A).88,184 This may reflect changes in DNA supercoiling in front of the fork, other proteins being recruited to the later origins, possibly because they have been released by the early firing forks, or increased S-phase kinase activity.185-187 Combining the DNA combing data with chromosome conformation capture that provides information on long-range chromatin interactions177 revealed the presence of U-shaped domains that correspond to blocks of enriched interaction. Thus, the genome is segmented into replication timing domains that correspond to spatially compartmentalized chromatin units. These domains are insulated on either side by 2 boundaries of open, accessible, actively transcribed chromatin that are also enriched in the insulator binding protein CTCF. The authors proposed a model for U-shaped chromatin domains in which replication would first initiate in efficient zones with an open chromatin structure on the outside of the domain and then work progressively and with increasing efficiency toward origins at the base of the U with more compact chromatin.183,188 This “domino” model may help to explain why replication progresses much faster than the known speed of a single fork, how late firing origins that are in regions of inaccessible chromatin can initiate, and why adjacent origins fire synchronously. Further, under this model, even if there are long stretches of chromosomes that are origin-free, replicating them would not take excessively long. The increasing density of origin firing with S-phase progression predicted by this model has been observed in budding yeast and frog embryo extracts.62,71,184,189
Figure 4.
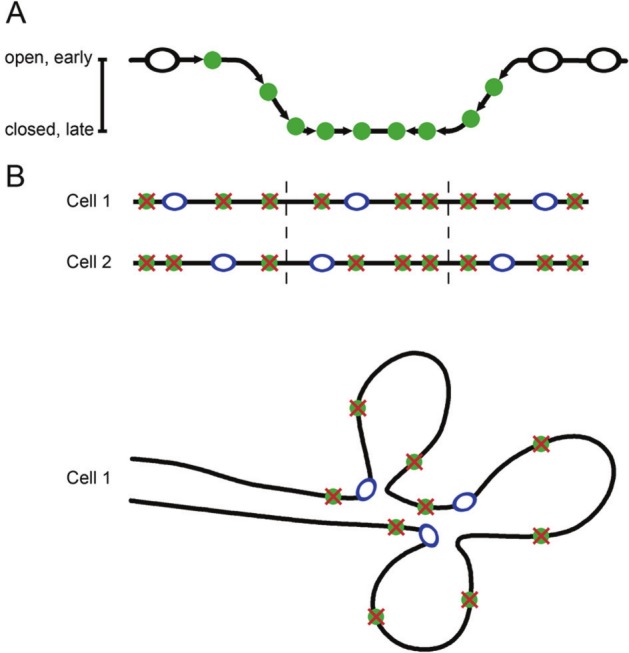
Two possible models to explain the lack of long gaps between replication origins. (A) The “domino model” postulates that replication begins at replication origins present in open chromatin. As replication progresses, replication origins in closed chromatin become more accessible as the replication fork unwinds the DNA. (B) An alternative model is that one origin within a cluster of potential origins is selected in each cell for firing. This ensures both the flexibility of origin choice and consistent spacing of origins.
Cayrou and colleagues148 also performed DNA combing experiments and arrived at a somewhat different model for DNA replication. They compared their DNA combing data in Drosophila with several models of origin firing. A random origin firing model in which origins are randomly activated did not fit the observed data. Similarly, a model of “increasing origin efficiency” that assumes that origin firing increases over the course of S phase, as described above, also did not match the data because the distribution of simulated interorigin distances was wider than that in the actual data. The best fit was for a model in which replication is organized into replicons, and within each replicon, one origin is fired, and others are silenced (Fig. 4B). In this scenario, there would be a mechanism that imposes regularity in the distribution of potential origins, thus preventing large gaps.190 Lebofsy and colleagues85 reached a similar conclusion when they mapped all detectable initiation zones throughout a 1.5-Mb region in human chromosome 14q11.2 using a single-molecule approach. The authors also found regular interorigin distances and a high potential-to-active origin ratio. They hypothesized that an excess of potential origins may provide a “safety net” in case of perturbed DNA replication. If a fork is blocked, a neighboring origin can be activated to provide a new fork. The firing of backup origins might also help to rescue a cell when a checkpoint has been activated.
Is the cell coordinating transcription and replication?
One of the clear messages from recent genome-wide analyses of origins in humans is an association with transcription start sites. As described above, in multiple species, early firing origins tend to have an open chromatin configuration and to be located near transcription start sites.5,91-94 In general, a tissue-specific gene replicates earlier in S phase in cells in which it is expressed than in cells in which it is transcriptionally silent.191 It is important to note, however, that in at least some studies, a relationship between replication timing and transcription during early S phase was not observed.192 The association between replication and transcription does not require transcription per se, as transcription from a promoter is not important for its use as a replication origin.111 Instead, the more likely model is that the most accessible origins fire early, and those near transcriptional regulators happen to be more accessible.185 From this perspective, the association between replication and transcription might be considered a consequence of both processes relying on open chromatin.
However, even if transcription is not required per se, it is also possible that there is an advantage to the cell in coordinating replication and transcription so that highly transcribed genes are replicated first. Transcriptionally active regions are more likely to cause fork stalling,162 so replicating them at the very beginning of S phase might minimize the possibility that they have failed to complete replication by the end of S phase or that they delay cell cycle progression. Firing order can also be interpreted as conveying a hierarchy to the genome. For instance, the conditions that permitted replication to start may no longer exist at the end of the process,193 and the fidelity of replication might decline if nucleotides and replication factors are depleted.110 About 50% of bacterial genomes have higher A and T content in the portions of the genome that are replicated last,142 which may reflect replication under conditions of depleted nutrients since guanine and cytosine are in more limited supply in the bacterial cytosol.194-196 Also, the rate of evolutionary sequence change is slower for genes near the origin in bacteria.195,197 In human cancers, a preference for deletions over amplifications in late-replication timing zones may indicate dNTP depletion late in replication.198 Replicating higher priority genomic regions, identified by their high transcription rate, early in S phase could help the cell to address this problem.
Another possibility is that the genome has been organized to minimize collisions between DNA and RNA polymerases. Because both forks advance and progression of the transcription bubble generates positive supercoiling, a head-on collision between replication and transcription machinery is expected to cause severe topological impediments and fork pausing.199-201 Fork stalling in response to head-on collisions with the RNA polymerase has been observed using electron microscopy to monitor fork progression after inverting the direction of ribosomal operons,202 with 2-dimensional gel electrophoresis of replication fork intermediates,203 and microarrays.204,205 Most studies indicate that replication is slowed more substantially as a result of head-on collisions with transcription units that oppose the fork compared to co-directional encounters,199,206 although co-oriented collisions can also result in RNA polymerase stalling.162,199,207,208 Replication fork arrest as a result of collisions with transcription complexes can lead to double-stranded breaks, DNA damage response, mutagenesis, and chromosomal deletions.204
Prokaryotes have only one replication origin, and they have organized their genomes in a way that has been interpreted as minimizing collisions between replication forks and transcription bubbles. For E. coli and many other bacteria, highly expressed and essential genes are transcribed in the same direction as the replication fork progresses.144,209-212 Seven rDNA operons in E. coli are all on the leading strand, resulting in co-orientation of replication and transcription.213 This orientation minimizes the stalling and potential genomic damage from head-on RNA polymerase and DNA polymerase collisions as well as loss of RNA templates.142,214
As described above, the most recent analysis suggests that human cells have not achieved co-directionality between transcription and replication. Necsulea and colleagues150 analyzed experimental and skew predictions of origins. While for the computationally predicted origins there was a strong leading strand bias, for the experimental data, the association between the direction of transcription and replication was essentially random. Further, the leading strand fraction was similar for highly and lowly expressed genes and was not different for genes expressed in S phase. There are several possible explanations. One possibility is that head-on collisions in eukaryotes are not as consequential as might be expected based on findings in bacteria. Perhaps the head-on collisions studied most closely in bacteria are particularly disastrous. The severity of replication fork arrest due to a head-on collision is correlated with the level of gene expression, and in some studies, only heavily transcribed genes significantly impeded the progression of the replication fork when inverted.204 Bacterial rRNAs, the most intensively studied genes, have very high transcription rates. Also, in bacterial rRNAs, there may be clustering of multiple RNA polymerases that work together,215 so a collision with this multi-RNA polymerase complex may lead to particularly ruinous consequences. On the other hand, data in E. coli show that genomic integrity can be affected by head-on collisions with genes that are not highly transcribed based on the rpoB gene, which is expressed at a low level but is very long.204
Even in bacteria, whether the main goal of genome organization is eliminating head-on collisions is being reconsidered.142 The frequency of co-directional genes is approximately 75% in Bacillus subtilis 216 but only 55% in E. coli. 217 Also, only a few genes are really highly expressed, making the model less likely.142 Moreover, according to the collision model, gene strand bias should be higher in fast-growing bacteria where transcription and replication are frequent, but that trend is not observed.218
Another possibility is that eukaryotes have evolved other mechanisms to prevent head-on collisions. Yeast have evolved a clever solution in which they ensure that certain highly transcribed genes are replicated only in a single direction by erecting a nucleic acid–protein barrier to forks entering from the 3′ direction opposite to the direction of transcription.219 For ribosomal DNA repeats in yeast, a replication fork barrier that consists of a cis-chromosomal motif and effector proteins blocks forks that are moving head-on toward the heavily transcribed 35S rRNA and prevents them from entering the 3′ end of the gene.220-224 The rRNA is then replicated by forks advancing co- directionally. However, these mechanisms may not be as relevant for higher eukaryotes.224,225
Another possibility is that in eukaryotes, the presence of a transcription loop is problematic, and the directionality of the replication machinery is not as important. In support of this model, the regions of the genome with the highest number of pause sites in S. cerevisiae are highly transcribed RNA polymerase II genes.162 Azvolinsky and colleagues162 found that in yeast, forks pause at transcribed genes, regardless of their relative orientation. Top2 seems to modulate DNA topology at sites of interference between replication and transcription, and it is present at the base of transcribed genes.163 In response to checkpoint arrest, the physical continuity between transcribed genes and the nuclear envelope created by the THO/TREX-2 complexes is modulated by checkpoint kinases that phosphorylate nuclear pore complex proteins.226 Checkpoint deficiency can be rescued by disrupting the tethering of transcribed genes to the nuclear pore complex or by introducing a double-stranded break between a fork and a highly transcribed gene. Thus, the topological impediments to DNA polymerase progression are reduced by the activation of checkpoints that counteract the topological tension generated at gene gates. These findings might be consistent with a model in which the navigation of gated genes, and not whether the collision is head-on or co-directional, is the most important barrier to faithful DNA replication in higher eukaryotes. From this perspective, perhaps embryonic cell cycles do not require checkpoints because the cells have not yet initiated a transcription program, and they do not need checkpoint kinases to counter the tension induced by gene gating.226 Similarly, bacteria lack a nucleus and would not be expected to face the topological challenges posed by gene gating.
A final intriguing possibility is that the genome is, in fact, organized to minimize head-on collisions, but instead of solving the problem through the orientation of genes, higher eukaryotes resolve it in the way they activate their origins. Evidence for this model comes from a report on asymmetric bidirectional replication at the DBF4 origin.227 Using 1-way PCR-based primer extension, the DBF4 origin was found to contain 2 initiation zones, one on the sense strand and one on the antisense strand, separated by approximately 400 bp that include the transcription start site (Fig. 2C). DBF4 replication starts from initiation zone I, which has more open chromatin, and then proceeds in the sense direction, that is, the direction of DBF4 transcription, toward initiation zone II. Replication of the antisense strand from initiation zone II began after the replication on the sense strand had reached or passed through this initiation zone. ORC binds both initiation zones, both have DNase I–hypersensitive regions, and replication of both strands proceeds as though it is a leading strand. Combining the asymmetric replication model with the fact that origins are often at the beginning of genes implies that the replication could be specifically oriented to follow the direction of transcription, with a subsequent origin firing to replicate the opposite strand.
It remains to be determined whether this asymmetric bidirectional replication model is more widely used. In a follow-up study, the same authors used 1-way PCR-based mapping to monitor the lamin B origin and concluded that it likely also contains 2 short initiation zones with a 40-bp noninitiation zone in between.228 Further, when Cayrou and colleagues148 used nascent strand purification to identify origins in Drosophila and mouse cells, a bimodal distribution was observed. There were 2 initiation sites that flanked transcription start sites with CpG islands. The data would be consistent with bimodal origins in which there is initiation from 2 start sites that move in opposite directions followed by a fusion of the 2 replication bubbles. More research will need to be done to clarify this issue. For instance, the first report of the lamin B promoter identified Okazaki fragments consistent with lagging strand replication.229 In addition, the observation that there is nucleotide skewing would argue that there are leading and lagging strands during DNA replication, although some skewing of nucleotide composition may result from transcription as well as replication.230,231
By using the same transcription factors that facilitate both transcription and origin firing, it would be possible for the cell to flexibly coordinate these processes. If the cell takes on a new fate or differentiates, it can both induce the expression of a particular gene, for instance, a hemoglobin or immunoglobulin component, and at the same time ensure that the portion of the genome encoding this newly critical gene will be replicated early. The reprogramming of cells when they take on a different fate, both in terms of replication and 3-dimensional chromatin structure, may help to explain why there is so little correlation between, for example, the origins found in an ovarian cancer cell line and a lymphoblastoid cell line.80 This would be consistent with findings that cells from different tissue take on different 3-dimensional chromatin structures.232 It would also be consistent with findings of significant overlap in origin firing between 2 similar types of cells, which would be expected to have much more similar higher order chromatin structures.232
What causes origin interference?
In addition to the selection of a specific origin, there is likely also a method to deselect origins. Indeed, if there is a selection of one and only one origin within a replicon, there is likely a method of origin interference, that is, a mechanism whereby the selection of one origin for firing inhibits the selection of nearby origins.233 In budding and fission yeast, when 2 origins are located close together, it is rare that both origins are active in the same cell during S phase.57,60,233 Origin interference is unlikely to be related to ORC binding or prereplication complex assembly as these factors bind efficiently to all origins. Certainly, as the replication fork from the selected origin passes, it will inactivate nearby forks.234 Thus, potential origins that are not as efficient will be inactivated as a consequence of the selection of a more efficient origin nearby.
In addition to this passive mechanism, the ATM and ATR checkpoint kinases also play an active role in the unfolding of S phase, even in unstressed cells.235-239 ATR–/– mice are embryonic lethal, and cells from the mouse are not viable,240 supporting an important role for ATR without checkpoint activation. The ATM and ATR kinases downregulate the Cdk2 and Cdc7 kinases and thereby slow down the rate of DNA replication by blocking origin firing.241 Inhibiting ATM and ATR kinases, for instance, by adding caffeine or neutralizing antibodies, increases the replication rate.241 The likely activator of ATM and ATR is RPA-bound, single-stranded DNA, which would be expected to be high near an actively firing origin. With this mechanism, the cell can inhibit replication forks near a replication zone. The role of ATR as an inhibitor of S-phase progression could also explain its role in halting DNA replication when there are replication problems. The role of ATM and ATR in limiting origin firing in the context of DNA damage can be viewed from this perspective as an extension of its role in origin interference in unstressed cells. DNA damage or stalled forks would result in more extensive single-stranded DNA and thus a stronger checkpoint response.236
Why have late origins?
The role of the ATM and ATR proteins in limiting origin firing raises the question: why have early and late origins? Does the existence of both early and late replication origins allow the cell to use the early origins as sensors for replication conditions and then adjust the rate of replication for late origins based on the results of the early firings using the checkpoint response? Or alternatively, would firing all available origins at the same time lead to a depletion of replication factors, fork-stabilizing proteins, or dNTPs? Would it result in too many replication fork convergences for the cell to accommodate? Consistent with these theories, there have been suggestions of a checkpoint that limits the total number of replication forks at any time during S phase.226 However, any hypotheses about the need for both early and late origins would have to incorporate the fact that embryonic frog and fly cells do manage to faithfully replicate their genomes with a pattern that involves firing many origins simultaneously, at least for a few cell cycles.
One current model is there are proteins or protein complexes that are rate limiting for origin firing.242 Indeed, substrates of the kinases that activate S phase have been reported to be rate limiting for origin firing in budding yeast.242 The formation of replicon foci could reflect the need to bring early origins in close contact with each other to benefit from the availability of these factors. The distance between these origins is consistent with replication initiation sites being defined by an optimal loop size that correlates with the intrinsic stiffness of DNA.182 An origin exclusion zone would be created as those origins that are physically too far from the limiting replication reagents. In this case, the physical position of the origin, in addition to ATR/ATM activation, would serve to limit the activity of origins near those that were selected. As S phase progresses, the limiting factors may be released or increase in abundance, allowing more origin firing.185 Origins fired later in S phase could reuse rate-limiting proteins released by the early firing origins. In addition, the organization of DNA replication in higher order substructures could allow the cell to compartmentalize important proteins.45 Certain types of topoisomerases, chromatin remodelers, or histone chaperones might be concentrated in specific nuclear subcompartments.45 This might be consistent with theories that replication organization facilitates the propagation of chromatin states during DNA synthesis.110,243
Does the Organization of the Genome for Replication Contribute to Carcinogenesis?
There is substantial evidence that inappropriate organization of DNA replication may be central to the process of tumorigenesis. Some of the data to support this perspective are based on the demonstration that tumors exhibit a replication-induced DNA damage response. Early stages of tumorigenesis are consistently associated with the engagement of the DNA damage checkpoint response in multiple tissues.244,245 Analysis of precancerous and cancerous lesions from human patients revealed foci of DNA repair proteins, suggesting the presence of DNA double-stranded breaks not found in normal tissues.244-246
According to this model, early in tumor progression, cell proliferation and transformation are inhibited by senescence. Analysis of colon and bladder precancerous lesions shows that senescence markers coincide with DNA damage response markers.247 Di Micco and colleagues,248 for instance, showed that senescence triggered by the expression of an activated oncogene H-RasV12 in normal human cells is a consequence of a robust DNA damage response. When Ras was expressed in DNA damage response–deficient cells, the cells continued to proliferate rather than senesce. With DNA combing, they found that oncogene activation led to an increased number of active replicons and an increased asymmetry in the progression between right and left forks emanating from the same origin in Ras-expressing cells. Such discontinuous fork advancement might reflect increased fork instability or extensive fork pausing, possibly resulting from excessive origin firing. An outstanding question for the field is whether the activation of oncogenic factors mediates increased origin firing and a DNA damage response through an increase in the levels of replication proteins like CDC6 or whether oncogene activation stimulates origin firing more directly through epigenetic changes.
Several other lines of evidence are consistent with oncogenes affecting DNA replication. The oncogenic transcription factors c-Myc,128 E2F,115 c-Jun, and c-Fos105 have been associated not only with transcription but also with origins of replication and with promoting DNA replication. Indeed, the transcription factors most closely associated with oncogenic transformation are also those that are enriched near origins of replication. In both precancerous lesions and cancers, oncogene activation induces the stalling and collapse of DNA replication forks, which in turn leads to the formation of DNA double-stranded breaks.249 Activation of oncogenes and more generally of growth signaling pathways induces a loss of heterozygosity and genomic instability in mammalian cells cultured in vitro, human xenografts, mouse models, and yeast.244,245,247,248,250-254 In yeast, deregulation of CDK activity compromises DNA replication and leads to the formation of double-stranded breaks and genomic instability.255 The association between oncogenes and replication-induced DNA damage could reflect higher levels of proteins involved in prereplication complex assembly, higher levels of rate-limiting S-phase kinase activity, higher levels of transcription and gate gating, effects on the accessibility of chromatin surrounding origins of replication or changes in higher order chromatin structure.5
The types of mutations observed in human cancers are consistent with the types of mutations that occur as a result of DNA replication errors. Double-stranded breaks resulting from excessive origin firing could contribute to the genomic instability that characterizes most human cancers. This has been tested by monitoring fragile sites at which replication forks preferentially collapse.256 These loci are prone to the formation of microdeletions and gross chromosomal rearrangements and thus represent an indicator of the presence of replication stress. In both human precancerous lesions and in a human skin xenograft hyperplasia model, loss of heterozygosity was associated with common fragile sites, indicating that the lesions likely experience DNA replication stress.244,245 In oral precancerous lesions, loss of heterozygosity at the common fragile site Fra3B was a better predictor of progression to cancer than the other markers investigated.257
As described above for genetic disorders, the types of mutations observed in cancers are also indicative of DNA damage response. Recently, data from deep sequencing of tumors have resulted in a new term “chromothripsis” to define instability in 1% to 3% of cancers, resulting in a highly complex pattern of genomic rearrangements with multiple copy number variants.258 This type of mutagenic event is consistent with the expected errors from fork failure and template switching.28 Multiple copy number and structural changes consistent with microhomology-mediated, break-induced replication are thus common to both cancers and genetic disorders.40,258,259
The relationship between the organization of DNA for replication and somatic copy number alterations, a hallmark of cancer, has been investigated directly.198 Analysis of thousands of cancer samples including 26 cancer types revealed that the 2 boundaries of copy number alterations tend to be close to each other in the nucleus and replicate at a similar time. In fact, long-range interaction and replication timing data were sufficient to identify copy number variations. The authors concluded that the spatial proximity of regions replicating at the same time is an important contributor to the mutations observed in cancer.198,260
It is possible that a similar model will prove to be explanatory for the recurrent chromosomal translocations that are hallmarks of many cancers. Translocations, like copy number variants, tend to join genetic loci that are in close spatial proximity.232,261 As an example, BCR and ABL are not only in close nuclear proximity but are also replicated at a similar time in S phase, which might help to explain their frequent fusion to form the BCR-ABL oncogene that drives leukemogenesis.198 A recent high-throughput, genome-wide translocation sequencing study262 revealed that double-stranded, break-induced translocations to the IgH or c-Myc loci are much more likely to occur at sites that are either close to those loci or about 300 to 600 bp on the sense side of active transcription start sites. Microhomologies characterized the translocations. The correlation with sites that are origins of replication is striking and suggests that a 3-dimensional clustering of early origins into replicons might provide an opportunity for the formation of translocations when double-stranded breaks formed by the DNA polymerase at these genomic loci are clustered together in S phase. Thus, the association between cancer and fragile site mutations, the examples of “shattered” chromosomes that might reflect DNA replication failure, the association between DNA replication and the formation of copy number alterations, and the possibility that common translocations are a reflection of the organization of chromatin around DNA replication all suggest that DNA replication is a central driver of the genetic component of tumorigenesis.
Finally, the recent discovery that the TET enzymes are important for preparing CpG sequences in E-boxes for c-Myc binding and origin firing132 could have repercussions for our interpretation of the role of mutations in isocitrate dehydrogenases in cancer. Gain-of-function mutations in isocitrate dehydrogenase enzymes IDH1 and IDH2 are frequently observed in tumors including acute myelogenous leukemia (AML) and glioblastoma.263 The mutant versions of these enzymes have a neomorphic ability to generate 2-hydroxyglutarate (2-HG), and high levels of this metabolite have been identified in the serum of patients with IDH1 or IDH2 mutations.264 2-HG can inhibit the activity of TET enzymes, which use α-ketoglutarate as an oxygen donor. By inhibiting TET enzymes, the IDH1/2 mutations limit the removal of cytosine methyl groups. Indeed, AML patients with IDH1/2 mutations have hypermethylated DNA, and the hypermethylation preferentially targets promoter regions and CpG islands neighboring transcription start sites.265 2-HG can also inhibit the activity of other enzymes that rely on α-ketoglutarate as an oxygen donor, including histone demethylases, and consistent with this hypothesis, tumors with IDH1/2 mutations contain hypermethylated histones.266 Further, mutations in the TET enzymes themselves have also been identified in AML, and these are found in a distinct set of tumors from those with IDH1/2 mutations, indicating that inhibition of TET enzymes is a common pathway to tumorigenesis.267 The prevailing model is that altered CpG methylation and histone methylation in these patients are causative for tumor growth, and certainly, this may be the most predominant effect. The new findings relating TET enzymes to the preparation of origins for firing132 raise the possibility that IDH mutations affect replication as well as transcription. IDH mutations would be expected to result in an accumulation of methylated CpG dinucleotides, thus inhibiting c-Myc from binding to its E-box at specific replication origins. It is interesting to note that IDH mutations are associated with a good prognosis in gliomas,268 glioblastomas,269 and acute myeloid leukemias.270 There are a number of reasons that these tumors could have a favorable prognosis, including that they tend to have normal karyotypes,271 they have altered methylation patterns, and they are associated with metabolic changes. It will be interesting to determine whether origin firing is impaired in patients with tumors with IDH mutations and whether this limits the tumor’s ability to grow.
Anticipated Future Directions
Recent studies have highlighted the importance of DNA replication for genetic diseases and cancer. We anticipate several emerging areas for the field of DNA replication. Application of methods for systematically identifying large numbers of origins of replication combined with methods to assess higher order chromatin structure will permit us to better understand the relationship between chromatin structure, origin selection, and replication kinetics. Our hope is that it will be possible to perform such studies in samples isolated directly from tissues. Such studies will likely help us to define the most important principles of DNA replication in higher eukaryotes: for example, what controls origin selection, origin interference, and the orderly timing of S phase. We anticipate that these technologies will allow us to better understand and classify the types of S phases in different cells, in different differentiation states, in response to different stimuli and in pathological states.
We also anticipate that more careful studies of the biophysical properties of replication forks, transcription loops, and nuclear gating will be forthcoming. Such studies might illuminate the biomechanical importance of the mechanisms that relieve the torsional stress associated with DNA replication such as topoisomerases, helicases, and checkpoint proteins. They might also provide more information on the topological state of DNA when it is replicated, transcribed, and subjected to both at the same time.
Further detailed analyses of the replication patterns at individual origins and of the factors required for DNA replication will also advance the field by providing more mechanistic insights and by potentially implicating other important pathways in the control of replication. It will also be important to test further some of the hypotheses put forward here such as whether the presence of a transcription factor binding site is an important regulator of origin firing, whether most promoters fire from a single origin or use bivalent origins, whether CpGs at myc-binding origins are demethylated with each cell division,272-274 and whether IDH mutations affect DNA replication as well as transcription.
Another question that we anticipate will be addressed is about the organization of genetic information into genomes. Issues surrounding DNA replication can explain many of the properties of bacterial genomes including the positioning of genes, the orientation of genes, nucleotide composition, and the locus-specific mutation rate. We wonder whether there are important rules of replication in higher eukaryotes that will have explanatory power for the locations of genes, their orientation and spacing, as well as the nucleotide composition and evolution rate that will be discovered.
Our expectation is that the field will also provide information on origin selection, origin firing, and origin dynamics in tumors. Correlating information about replication dynamics, 3-dimensional chromatin structure and the types of genetic events that drive tumorigenesis will allow us to better understand the molecular basis for this disease. For example, it will also be important to assess whether transcription factors or chromatin remodelers promote tumorigenesis through their roles in DNA replication in addition to their roles in the activation of cyclin-dependent kinases and transcription. From this perspective, it is especially interesting that mutations in the Rif1 protein that organizes S-phase progression in mice have been found in breast cancer patients.275,276 Many anticancer treatments involve drugs that interfere with DNA replication. A better understanding of the role of checkpoints and replication forks might help us to design better anticancer treatments.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received the following financial support for the research, authorship, and/or publication of this article: National Institute of General Medical Sciences (NIGMS) Center of Excellence grant P50 GM071508 (David Botstein, PI), National Institutes of Health (NIH) Oncology: Molecular Basis of Cancer grant 2T32 CA009538 (James Broach, PI), NIH/NIGMS grant 1R01 GM081686, and NIH/NIGMS grant 1R01 GM086465.
References
- 1. Jacob F, Brenner S. [On the regulation of DNA synthesis in bacteria: the hypothesis of the replicon]. C R Hebd Seances Acad Sci. 1963;256:298-300 [PubMed] [Google Scholar]
- 2. Mechali M. Eukaryotic DNA replication origins: many choices for appropriate answers. Nat Rev Mol Cell Biol. 2010;11:728-38 [DOI] [PubMed] [Google Scholar]
- 3. Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem. 2002;71:333-74 [DOI] [PubMed] [Google Scholar]
- 4. Todorovic V, Falaschi A, Giacca M. Replication origins of mammalian chromosomes: the happy few. Front Biosci. 1999;4:D859-68 [DOI] [PubMed] [Google Scholar]
- 5. Kohzaki H, Murakami Y. Transcription factors and DNA replication origin selection. Bioessays. 2005;27:1107-16 [DOI] [PubMed] [Google Scholar]
- 6. DePamphilis ML. Replication origins in metazoan chromosomes: fact or fiction? Bioessays. 1999;21:5-16 [DOI] [PubMed] [Google Scholar]
- 7. Zou L, Mitchell J, Stillman B. CDC45, a novel yeast gene that functions with the origin recognition complex and Mcm proteins in initiation of DNA replication. Mol Cell Biol. 1997;17:553-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Walter JC. Evidence for sequential action of cdc7 and cdk2 protein kinases during initiation of DNA replication in Xenopus egg extracts. J Biol Chem. 2000;275:39773-8 [DOI] [PubMed] [Google Scholar]
- 9. Jiang W, McDonald D, Hope TJ, Hunter T. Mammalian Cdc7-Dbf4 protein kinase complex is essential for initiation of DNA replication. EMBO J. 1999;18:5703-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Porter AC. Preventing DNA over-replication: a Cdk perspective. Cell Div. 2008;3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arias EE, Walter JC. Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes Dev. 2007;21:497-518 [DOI] [PubMed] [Google Scholar]
- 12. Rampakakis E, Arvanitis DN, Di Paola D, Zannis-Hadjopoulos M. Metazoan origins of DNA replication: regulation through dynamic chromatin structure. J Cell Biochem. 2009;106:512-20 [DOI] [PubMed] [Google Scholar]
- 13. Sclafani RA, Holzen TM. Cell cycle regulation of DNA replication. Annu Rev Genet. 2007;41:237-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Machida YJ, Hamlin JL, Dutta A. Right place, right time, and only once: replication initiation in metazoans. Cell. 2005;123:13-24 [DOI] [PubMed] [Google Scholar]
- 15. Takeda DY, Dutta A. DNA replication and progression through S phase. Oncogene. 2005;24:2827-43 [DOI] [PubMed] [Google Scholar]
- 16. Nguyen VQ, Co C, Li JJ. Cyclin-dependent kinases prevent DNA re-replication through multiple mechanisms. Nature. 2001;411:1068-73 [DOI] [PubMed] [Google Scholar]
- 17. Diffley JF. Once and only once upon a time: specifying and regulating origins of DNA replication in eukaryotic cells. Genes Dev. 1996;10:2819-30 [DOI] [PubMed] [Google Scholar]
- 18. Narayanan V, Mieczkowski PA, Kim HM, Petes TD, Lobachev KS. The pattern of gene amplification is determined by the chromosomal location of hairpin-capped breaks. Cell. 2006;125:1283-96 [DOI] [PubMed] [Google Scholar]
- 19. Lopes M, Cotta-Ramusino C, Pellicioli A, et al. The DNA replication checkpoint response stabilizes stalled replication forks. Nature. 2001;412:557-61 [DOI] [PubMed] [Google Scholar]
- 20. Ivessa AS, Lenzmeier BA, Bessler JB, Goudsouzian LK, Schnakenberg SL, Zakian VA. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol Cell. 2003;12:1525-36 [DOI] [PubMed] [Google Scholar]
- 21. Pomerantz RT, O’Donnell M. Direct restart of a replication fork stalled by a head-on RNA polymerase. Science. 2010;327:590-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arlt MF, Mulle JG, Schaibley VM, et al. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am J Hum Genet. 2009;84:339-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arlt MF, Ozdemir AC, Birkeland SR, Wilson TE, Glover TW. Hydroxyurea induces de novo copy number variants in human cells. Proc Natl Acad Sci U S A. 2011;108:17360-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542-8 [DOI] [PubMed] [Google Scholar]
- 26. Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11:208-19 [DOI] [PubMed] [Google Scholar]
- 27. Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599-602 [DOI] [PubMed] [Google Scholar]
- 28. Carr AM, Paek AL, Weinert T. DNA replication: failures and inverted fusions. Semin Cell Dev Biol. 2011;22:866-74 [DOI] [PubMed] [Google Scholar]
- 29. Lange J, Skaletsky H, van Daalen SK, et al. Isodicentric Y chromosomes and sex disorders as byproducts of homologous recombination that maintains palindromes. Cell. 2009;138:855- 69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aguilera A, Gomez-Gonzalez B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. 2008;9:204-17 [DOI] [PubMed] [Google Scholar]
- 31. Weinert T, Kaochar S, Jones H, Paek A, Clark AJ. The replication fork’s five degrees of freedom, their failure and genome rearrangements. Curr Opin Cell Biol. 2009;21:778-84 [DOI] [PubMed] [Google Scholar]
- 32. Kolodner RD, Putnam CD, Myung K. Maintenance of genome stability in Saccharomyces cerevisiae. Science. 2002;297:552-7 [DOI] [PubMed] [Google Scholar]
- 33. Sharp AJ, Hansen S, Selzer RR, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38:1038-42 [DOI] [PubMed] [Google Scholar]
- 34. Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235-47 [DOI] [PubMed] [Google Scholar]
- 35. Mefford HC, Eichler EE. Duplication hotspots, rare genomic disorders, and common disease. Curr Opin Genet Dev. 2009;19:196-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tercero JA, Longhese MP, Diffley JF. A central role for DNA replication forks in checkpoint activation and response. Mol Cell. 2003;11:1323-36 [DOI] [PubMed] [Google Scholar]
- 37. Pevzner P, Tesler G. Genome rearrangements in mammalian evolution: lessons from human and mouse genomes. Genome Res. 2003;13:37-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carvalho CM, Zhang F, Liu P, et al. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum Mol Genet. 2009;18:2188-203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Slack A, Thornton PC, Magner DB, Rosenberg SM, Hastings PJ. On the mechanism of gene amplification induced under stress in Escherichia coli. PLoS Genet. 2006;2:e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu P, Carvalho CM, Hastings PJ, Lupski JR. Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev. 2012;22:211-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ankala A, Kohn JN, Hegde A, et al. Aberrant firing of replication origins potentially explains intragenic nonrecurrent rearrangements within genes, including the human DMD gene. Genome Res. 2012;22:25-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet. 2009;41:849-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen JM, Cooper DN, Ferec C, Kehrer-Sawatzki H, Patrinos GP. Genomic rearrangements in inherited disease and cancer. Semin Cancer Biol. 2010;20:222-33 [DOI] [PubMed] [Google Scholar]
- 44. Hwang DS, Kornberg A. Opening of the replication origin of Escherichia coli by DnaA protein with protein HU or IHF. J Biol Chem. 1992;267:23083-6 [PubMed] [Google Scholar]
- 45. Gilbert DM. Making sense of eukaryotic DNA replication origins. Science. 2001;294:96-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Erzberger JP, Mott ML, Berger JM. Structural basis for ATP-dependent DnaA assembly and replication-origin remodeling. Nat Struct Mol Biol. 2006;13:676-83 [DOI] [PubMed] [Google Scholar]
- 47. Ardell DH, Kirsebom LA. The genomic pattern of tDNA operon expression in E. coli. PLoS Comput Biol. 2005;1:e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Couturier E, Rocha EP. Replication-associated gene dosage effects shape the genomes of fast-growing bacteria but only for transcription and translation genes. Mol Microbiol. 2006;59:1506-18 [DOI] [PubMed] [Google Scholar]
- 49. Rocha EP, Danchin A. Essentiality, not expressiveness, drives gene-strand bias in bacteria. Nat Genet. 2003;34:377-8 [DOI] [PubMed] [Google Scholar]
- 50. Louarn JM, Bouche JP, Legendre F, Louarn J, Patte J. Characterization and properties of very large inversions of the E. coli chromosome along the origin-to-terminus axis. Mol Gen Genet. 1985;201:467-76 [DOI] [PubMed] [Google Scholar]
- 51. Theis JF, Newlon CS. The ARS309 chromosomal replicator of Saccharomyces cerevisiae depends on an exceptional ARS consensus sequence. Proc Natl Acad Sci U S A. 1997;94:10786-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stinchcomb DT, Thomas M, Kelly J, Selker E, Davis RW. Eukaryotic DNA segments capable of autonomous replication in yeast. Proc Natl Acad Sci U S A. 1980;77:4559-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chan CS, Tye BK. Autonomously replicating sequences in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1980;77:6329-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nieduszynski CA, Knox Y, Donaldson AD. Genome-wide identification of replication origins in yeast by comparative genomics. Genes Dev. 2006;20:1874-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Berbenetz NM, Nislow C, Brown GW. Diversity of eukaryotic DNA replication origins revealed by genome-wide analysis of chromatin structure. PLoS Genet. 2010;6:e1001092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Eaton ML, Galani K, Kang S, Bell SP, MacAlpine DM. Conserved nucleosome positioning defines replication origins. Genes Dev. 2010;24:748-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Brewer BJ, Fangman WL. Initiation preference at a yeast origin of replication. Proc Natl Acad Sci U S A. 1994;91:3418-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hu YF, Hao ZL, Li R. Chromatin remodeling and activation of chromosomal DNA replication by an acidic transcriptional activation domain from BRCA1. Genes Dev. 1999;13:637-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Danis E, Brodolin K, Menut S, Maiorano D, Girard-Reydet C, Mechali M. Specification of a DNA replication origin by a transcription complex. Nat Cell Biol. 2004;6:721-30 [DOI] [PubMed] [Google Scholar]
- 60. Marahrens Y, Stillman B. A yeast chromosomal origin of DNA replication defined by multiple functional elements. Science. 1992;255:817-23 [DOI] [PubMed] [Google Scholar]
- 61. Stevenson JB, Gottschling DE. Telomeric chromatin modulates replication timing near chromosome ends. Genes Dev. 1999;13:146-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Raghuraman MK, Winzeler EA, Collingwood D, et al. Replication dynamics of the yeast genome. Science. 2001;294:115-21 [DOI] [PubMed] [Google Scholar]
- 63. Segurado M, de Luis A, Antequera F. Genome-wide distribution of DNA replication origins at A+T-rich islands in Schizosaccharomyces pombe. EMBO Rep. 2003;4:1048-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dai J, Chuang RY, Kelly TJ. DNA replication origins in the Schizosaccharomyces pombe genome. Proc Natl Acad Sci U S A. 2005;102:337-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chuang RY, Kelly TJ. The fission yeast homologue of Orc4p binds to replication origin DNA via multiple AT-hooks. Proc Natl Acad Sci U S A. 1999;96:2656-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Iyer V, Struhl K. Poly(dA:dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure. EMBO J. 1995;14:2570-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Drew HR, Travers AA. DNA bending and its relation to nucleosome positioning. J Mol Biol. 1985;186:773-90 [DOI] [PubMed] [Google Scholar]
- 68. Xu J, Yanagisawa Y, Tsankov AM, et al. Genome-wide identification and characterization of replication origins by deep sequencing. Genome Biol. 2012;13:R27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tsankov A, Yanagisawa Y, Rhind N, Regev A, Rando OJ. Evolutionary divergence of intrinsic and trans-regulated nucleosome positioning sequences reveals plastic rules for chromatin organization. Genome Res. 2011;21:1851-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Harland RM, Laskey RA. Regulated replication of DNA microinjected into eggs of Xenopus laevis. Cell. 1980;21:761-71 [DOI] [PubMed] [Google Scholar]
- 71. Blumenthal AB, Kriegstein HJ, Hogness DS. The units of DNA replication in Drosophila melanogaster chromosomes. Cold Spring Harb Symp Quant Biol. 1974;38:205-23 [DOI] [PubMed] [Google Scholar]
- 72. Laskey RA, Harland RM. Replication origins in the eucaryotic chromosome. Cell. 1981;24:283-4 [DOI] [PubMed] [Google Scholar]
- 73. Stanojcic S, Lemaitre JM, Brodolin K, Danis E, Mechali M. In Xenopus egg extracts, DNA replication initiates preferentially at or near asymmetric AT sequences. Mol Cell Biol. 2008;28:5265-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mechali M, Kearsey S. Lack of specific sequence requirement for DNA replication in Xenopus eggs compared with high sequence specificity in yeast. Cell. 1984;38:55-64 [DOI] [PubMed] [Google Scholar]
- 75. Dolfini S, Courgeon AM, Tiepolo L. The cell cycle of an established line of Drosophila melanogaster cells in vitro. Experientia. 1970;26:1020-1 [DOI] [PubMed] [Google Scholar]
- 76. Blumenthal AB, Kriegstein HJ, Hogness DS. The units of DNA replication in Drosophila melanogaster chromosomes. Cold Spring Harb Symp Quant Biol. 1973;38:205-23 [DOI] [PubMed] [Google Scholar]
- 77. Callan HG. Replication of DNA in eukaryotic chromosomes. Br Med Bull. 1973;29:192-5 [DOI] [PubMed] [Google Scholar]
- 78. Sasaki T, Sawado T, Yamaguchi M, Shinomiya T. Specification of regions of DNA replication initiation during embryogenesis in the 65-kilobase DNApolalpha-dE2F locus of Drosophila melanogaster. Mol Cell Biol. 1999;19:547-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hyrien O, Maric C, Mechali M. Transition in specification of embryonic metazoan DNA replication origins. Science. 1995;270:994-7 [DOI] [PubMed] [Google Scholar]
- 80. Mesner LD, Valsakumar V, Karnani N, Dutta A, Hamlin JL, Bekiranov S. Bubble-chip analysis of human origin distributions demonstrates on a genomic scale significant clustering into zones and significant association with transcription. Genome Res. 2011;21:377-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Remus D, Beall EL, Botchan MR. DNA topology, not DNA sequence, is a critical determinant for Drosophila ORC-DNA binding. EMBO J. 2004;23:897-907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. MacAlpine DM, Rodriguez HK, Bell SP. Coordination of replication and transcription along a Drosophila chromosome. Genes Dev. 2004;18:3094-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Heichinger C, Penkett CJ, Bahler J, Nurse P. Genome-wide characterization of fission yeast DNA replication origins. EMBO J. 2006;25:5171-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Friedman KL, Brewer BJ, Fangman WL. Replication profile of Saccharomyces cerevisiae chromosome VI. Genes Cells. 1997;2:667-78 [DOI] [PubMed] [Google Scholar]
- 85. Lebofsky R, Heilig R, Sonnleitner M, Weissenbach J, Bensimon A. DNA replication origin interference increases the spacing between initiation events in human cells. Mol Biol Cell. 2006;17:5337-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hamlin JL, Mesner LD, Lar O, Torres R, Chodaparambil SV, Wang L. A revisionist replicon model for higher eukaryotic genomes. J Cell Biochem. 2008;105:321-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Huberman JA, Riggs AD. Autoradiography of chromosomal DNA fibers from Chinese hamster cells. Proc Natl Acad Sci U S A. 1966;55:599-606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hyrien O, Marheineke K, Goldar A. Paradoxes of eukaryotic DNA replication: MCM proteins and the random completion problem. Bioessays. 2003;25:116-25 [DOI] [PubMed] [Google Scholar]
- 89. Czajkowsky DM, Liu J, Hamlin JL, Shao Z. DNA combing reveals intrinsic temporal disorder in the replication of yeast chromosome VI. J Mol Biol. 2008;375:12-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Patel PK, Arcangioli B, Baker SP, Bensimon A, Rhind N. DNA replication origins fire stochastically in fission yeast. Mol Biol Cell. 2006;17:308-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. DePamphilis ML. Eukaryotic DNA replication: anatomy of an origin. Annu Rev Biochem. 1993;62:29-63 [DOI] [PubMed] [Google Scholar]
- 92. Biamonti G, Giacca M, Perini G, et al. The gene for a novel human lamin maps at a highly transcribed locus of chromosome 19 which replicates at the onset of S-phase. Mol Cell Biol. 1992;12:3499-506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Delgado S, Gomez M, Bird A, Antequera F. Initiation of DNA replication at CpG islands in mammalian chromosomes. EMBO J. 1998;17:2426-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tao L, Dong Z, Leffak M, Zannis-Hadjopoulos M, Price G. Major DNA replication initiation sites in the c-myc locus in human cells. J Cell Biochem. 2000;78:442-57 [DOI] [PubMed] [Google Scholar]
- 95. Gilbert DM. Replication timing and transcriptional control: beyond cause and effect. Curr Opin Cell Biol. 2002;14:277-83 [DOI] [PubMed] [Google Scholar]
- 96. Tabancay AP, Jr, Forsburg SL. Curr Top Dev Biol. 2006;76:129-84 [DOI] [PubMed] [Google Scholar]
- 97. Rampakakis E, Di Paola D, Chan MK, Zannis-Hadjopoulos M. Dynamic changes in chromatin structure through post-translational modifications of histone H3 during replication origin activation. J Cell Biochem. 2009;108:400-7 [DOI] [PubMed] [Google Scholar]
- 98. Birney E, Stamatoyannopoulos JA, Dutta A, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799-816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Karnani N, Taylor CM, Malhotra A, Dutta A. Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol Biol Cell. 2010;21:393-404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Karnani N, Taylor C, Malhotra A, Dutta A. Pan-S replication patterns and chromosomal domains defined by genome-tiling arrays of ENCODE genomic areas. Genome Res. 2007;17:865-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Vogelauer M, Rubbi L, Lucas I, Brewer BJ, Grunstein M. Histone acetylation regulates the time of replication origin firing. Mol Cell. 2002;10:1223-33 [DOI] [PubMed] [Google Scholar]
- 102. Aparicio JG, Viggiani CJ, Gibson DG, Aparicio OM. The Rpd3-Sin3 histone deacetylase regulates replication timing and enables intra-S origin control in Saccharomyces cerevisiae. Mol Cell Biol. 2004;24:4769-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Crampton A, Chang F, Pappas DL, Jr., Frisch RL, Weinreich M. An ARS element inhibits DNA replication through a SIR2-dependent mechanism. Mol Cell. 2008;30:156-66 [DOI] [PubMed] [Google Scholar]
- 104. Knott SR, Viggiani CJ, Tavare S, Aparicio OM. Genome-wide replication profiles indicate an expansive role for Rpd3L in regulating replication initiation timing or efficiency, and reveal genomic loci of Rpd3 function in Saccharomyces cerevisiae. Genes Dev. 2009;23:1077-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Cadoret JC, Meisch F, Hassan-Zadeh V, et al. Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc Natl Acad Sci U S A. 2008;105:15837-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Sequeira-Mendes J, Diaz-Uriarte R, Apedaile A, Huntley D, Brockdorff N, Gomez M. Transcription initiation activity sets replication origin efficiency in mammalian cells. PLoS Genet. 2009;5:e1000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Gomez M, Brockdorff N. Heterochromatin on the inactive X chromosome delays replication timing without affecting origin usage. Proc Natl Acad Sci U S A. 2004;101:6923-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Schwaiger M, Schubeler D. A question of timing: emerging links between transcription and replication. Curr Opin Genet Dev. 2006;16:177-83 [DOI] [PubMed] [Google Scholar]
- 109. Dazy S, Gandrillon O, Hyrien O, Prioleau MN. Broadening of DNA replication origin usage during metazoan cell differentiation. EMBO Rep. 2006;7:806-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Schwaiger M, Stadler MB, Bell O, Kohler H, Oakeley EJ, Schubeler D. Chromatin state marks cell-type- and gender-specific replication of the Drosophila genome. Genes Dev. 2009;23:589-601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Cohen SM, Brylawski BP, Cordeiro-Stone M, Kaufman DG. Same origins of DNA replication function on the active and inactive human X chromosomes. J Cell Biochem. 2003;88:923-31 [DOI] [PubMed] [Google Scholar]
- 112. Aladjem MI, Groudine M, Brody LL, et al. Participation of the human beta-globin locus control region in initiation of DNA replication. Science. 1995;270:815-9 [DOI] [PubMed] [Google Scholar]
- 113. Kitsberg D, Selig S, Keshet I, Cedar H. Replication structure of the human beta-globin gene domain. Nature. 1993;366:588-90 [DOI] [PubMed] [Google Scholar]
- 114. Forrester WC, Epner E, Driscoll MC, et al. A deletion of the human beta-globin locus activation region causes a major alteration in chromatin structure and replication across the entire beta-globin locus. Genes Dev. 1990;4:1637-49 [DOI] [PubMed] [Google Scholar]
- 115. Bosco G, Du W, Orr-Weaver TL. DNA replication control through interaction of E2F-RB and the origin recognition complex. Nat Cell Biol. 2001;3:289-95 [DOI] [PubMed] [Google Scholar]
- 116. Beall EL, Manak JR, Zhou S, Bell M, Lipsick JS, Botchan MR. Role for a Drosophila Myb-containing protein complex in site-specific DNA replication. Nature. 2002;420:833-7 [DOI] [PubMed] [Google Scholar]
- 117. Dimitrova DS, Giacca M, Demarchi F, Biamonti G, Riva S, Falaschi A. In vivo protein-DNA interactions at human DNA replication origin. Proc Natl Acad Sci U S A. 1996;93:1498-503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lipford JR, Bell SP. Nucleosomes positioned by ORC facilitate the initiation of DNA replication. Mol Cell. 2001;7:21-30 [DOI] [PubMed] [Google Scholar]
- 119. Ghosh M, Kemp M, Liu G, Ritzi M, Schepers A, Leffak M. Differential binding of replication proteins across the human c-myc replicator. Mol Cell Biol. 2006;26:5270-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ito K, Asano M, Hughes P, et al. c-Jun stimulates origin-dependent DNA unwinding by polyomavirus large T antigen. EMBO J. 1996;15:5636-46 [PMC free article] [PubMed] [Google Scholar]
- 121. Baru M, Shlissel M, Manor H. The yeast GAL4 protein transactivates the polyomavirus origin of DNA replication in mouse cells. J Virol. 1991;65:3496-503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Nilsson M, Forsberg M, You ZY, Westin G, Magnusson G. Enhancer effect of bovine papillomavirus E2 protein in replication of polyomavirus DNA. Nucleic Acids Res. 1991;19:7061-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Ishikawa H, Asano M, Kanda T, Kumar S, Gelinas C, Ito Y. Two novel functions associated with the Rel oncoproteins: DNA replication and cell-specific transcriptional activation. Oncogene. 1993;8:2889-96 [PubMed] [Google Scholar]
- 124. Kanda T, Segawa K, Ohuchi N, Mori S, Ito Y. Stimulation of polyomavirus DNA replication by wild-type p53 through the DNA-binding site. Mol Cell Biol. 1994;14:2651-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Yang L, Li R, Mohr IJ, Clark R, Botchan MR. Activation of BPV-1 replication in vitro by the transcription factor E2. Nature. 1991;353:628-32 [DOI] [PubMed] [Google Scholar]
- 126. Seo YS, Muller F, Lusky M, et al. Bovine papilloma virus (BPV)-encoded E2 protein enhances binding of E1 protein to the BPV replication origin. Proc Natl Acad Sci U S A. 1993;90:2865-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sedman J, Stenlund A. Co-operative interaction between the initiator E1 and the transcriptional activator E2 is required for replicator specific DNA replication of bovine papillomavirus in vivo and in vitro. EMBO J. 1995;14:6218-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Dominguez-Sola D, Ying CY, Grandori C, et al. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007;448:445-51 [DOI] [PubMed] [Google Scholar]
- 129. Cole MD, Cowling VH. Transcription-independent functions of MYC: regulation of translation and DNA replication. Nat Rev Mol Cell Biol. 2008;9:810-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Levens DL. Reconstructing MYC. Genes Dev. 2003;17:1071-7 [DOI] [PubMed] [Google Scholar]
- 131. Robinson K, Asawachaicharn N, Galloway DA, Grandori C. c-Myc accelerates S-phase and requires WRN to avoid replication stress. PloS One. 2009;4:e5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Swarnalatha M, Singh AK, Kumar V. The epigenetic control of E-box and Myc-dependent chromatin modifications regulate the licensing of lamin B2 origin during cell cycle. Nucleic Acids Res. 2012;40:9021-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Falaschi A, Abdurashidova G, Sandoval O, Radulescu S, Biamonti G, Riva S. Molecular and structural transactions at human DNA replication origins. Cell Cycle. 2007;6:1705-12 [DOI] [PubMed] [Google Scholar]
- 134. Jorgensen HF, Azuara V, Amoils S, et al. The impact of chromatin modifiers on the timing of locus replication in mouse embryonic stem cells. Genome Biol. 2007;8:R169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Aladjem MI, Fanning E. The replicon revisited: an old model learns new tricks in metazoan chromosomes. EMBO Rep. 2004;5:686-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Francino MP, Ochman H. Strand asymmetries in DNA evolution. Trends Genet. 1997;13:240-5 [DOI] [PubMed] [Google Scholar]
- 137. Fijalkowska IJ, Jonczyk P, Tkaczyk MM, Bialoskorska M, Schaaper RM. Unequal fidelity of leading strand and lagging strand DNA replication on the Escherichia coli chromosome. Proc Natl Acad Sci U S A. 1998;95:10020-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Chen CL, Duquenne L, Audit B, et al. Replication-associated mutational asymmetry in the human genome. Mol Biol Evol. 2011;28:2327-37 [DOI] [PubMed] [Google Scholar]
- 139. Lobry JR. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol Biol Evol. 1996;13:660-5 [DOI] [PubMed] [Google Scholar]
- 140. Frank AC, Lobry JR. Asymmetric substitution patterns: a review of possible underlying mutational or selective mechanisms. Gene. 1999;238:65-77 [DOI] [PubMed] [Google Scholar]
- 141. Lutsenko E, Bhagwat AS. Principal causes of hot spots for cytosine to thymine mutations at sites of cytosine methylation in growing cells: a model, its experimental support and implications. Mutat Res. 1999;437:11-20 [DOI] [PubMed] [Google Scholar]
- 142. Rocha EP. The replication-related organization of bacterial genomes. Microbiology. 2004;150:1609-27 [DOI] [PubMed] [Google Scholar]
- 143. Grigoriev A. Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res. 1998;26:2286-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Lobry JR. Origin of replication of Mycoplasma genitalium. Science. 1996;272:745-6 [DOI] [PubMed] [Google Scholar]
- 145. Gierlik A, Kowalczuk M, Mackiewicz P, Dudek MR, Cebrat S. Is there replication-associated mutational pressure in the Saccharomyces cerevisiae genome? J Theor Biol. 2000;202:305-14 [DOI] [PubMed] [Google Scholar]
- 146. Touchon M, Nicolay S, Audit B, et al. Replication-associated strand asymmetries in mammalian genomes: toward detection of replication origins. Proc Natl Acad Sci U S A. 2005;102:9836-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Huvet M, Nicolay S, Touchon M, et al. Human gene organization driven by the coordination of replication and transcription. Genome Res. 2007;17:1278-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Cayrou C, Coulombe P, Vigneron A, et al. Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res. 2011;21:1438-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Mesner LD, Crawford EL, Hamlin JL. Isolating apparently pure libraries of replication origins from complex genomes. Mol Cell. 2006;21:719-26 [DOI] [PubMed] [Google Scholar]
- 150. Necsulea A, Guillet C, Cadoret JC, Prioleau MN, Duret L. The relationship between DNA replication and human genome organization. Mol Biol Evol. 2009;26:729-41 [DOI] [PubMed] [Google Scholar]
- 151. Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430-40 [DOI] [PubMed] [Google Scholar]
- 152. Bermejo R, Lai MS, Foiani M. Preventing replication stress to maintain genome stability: resolving conflicts between replication and transcription. Mol Cell. 2012;45:710-8 [DOI] [PubMed] [Google Scholar]
- 153. Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem. 2001;70:369-413 [DOI] [PubMed] [Google Scholar]
- 154. Bermejo R, Doksani Y, Capra T, et al. Top1- and Top2-mediated topological transitions at replication forks ensure fork progression and stability and prevent DNA damage checkpoint activation. Genes Dev. 2007;21:1921-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Ansari A, Hampsey M. A role for the CPF 3’-end processing machinery in RNAP II-dependent gene looping. Genes Dev. 2005;19:2969-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Cabal GG, Genovesio A, Rodriguez-Navarro S, et al. SAGA interacting factors confine sub-diffusion of transcribed genes to the nuclear envelope. Nature. 2006;441:770-3 [DOI] [PubMed] [Google Scholar]
- 157. Rougemaille M, Dieppois G, Kisseleva-Romanova E, et al. THO/Sub2p functions to coordinate 3′-end processing with gene-nuclear pore association. Cell. 2008;135:308-21 [DOI] [PubMed] [Google Scholar]
- 158. Tan-Wong SM, Wijayatilake HD, Proudfoot NJ. Gene loops function to maintain transcriptional memory through interaction with the nuclear pore complex. Genes Dev. 2009;23:2610-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Chavez S, Beilharz T, Rondon AG, et al. A protein complex containing Tho2, Hpr1, Mft1 and a novel protein, Thp2, connects transcription elongation with mitotic recombination in Saccharomyces cerevisiae. EMBO J. 2000;19:5824-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Bentley DL. Rules of engagement: co-transcriptional recruitment of pre-mRNA processing factors. Curr Opin Cell Biol. 2005;17:251-6 [DOI] [PubMed] [Google Scholar]
- 161. Luna R, Gaillard H, Gonzalez-Aguilera C, Aguilera A. Biogenesis of mRNPs: integrating different processes in the eukaryotic nucleus. Chromosoma. 2008;117:319-31 [DOI] [PubMed] [Google Scholar]
- 162. Azvolinsky A, Giresi PG, Lieb JD, Zakian VA. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol Cell. 2009;34:722-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Bermejo R, Capra T, Gonzalez-Huici V, et al. Genome-organizing factors Top2 and Hmo1 prevent chromosome fragility at sites of S phase transcription. Cell. 2009;138:870-84 [DOI] [PubMed] [Google Scholar]
- 164. Aguilera A, Klein HL. HPR1, a novel yeast gene that prevents intrachromosomal excision recombination, shows carboxy-terminal homology to the Saccharomyces cerevisiae TOP1 gene. Mol Cell Biol. 1990;10:1439-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Rondon AG, Jimeno S, Aguilera A. The interface between transcription and mRNP export: from THO to THSC/TREX-2. Biochim Biophys Acta. 2010;1799:533-8 [DOI] [PubMed] [Google Scholar]
- 166. Dominguez-Sanchez MS, Barroso S, Gomez-Gonzalez B, Luna R, Aguilera A. Genome instability and transcription elongation impairment in human cells depleted of THO/TREX. PLoS Genet. 2011;7:e1002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Gilbert DM, Takebayashi SI, Ryba T, et al. Space and time in the nucleus: developmental control of replication timing and chromosome architecture. Cold Spring Harb Symp Quant Biol. 2010;75:143-53 [DOI] [PubMed] [Google Scholar]
- 168. Ryba T, Battaglia D, Chang BH, et al. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 2012;22:1833-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Yaffe E, Farkash-Amar S, Polten A, Yakhini Z, Tanay A, Simon I. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 2010;6:e1001011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol. 1998;140:1285-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Ma H, Samarabandu J, Devdhar RS, et al. Spatial and temporal dynamics of DNA replication sites in mammalian cells. J Cell Biol. 1998;143:1415-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Dimitrova DS, Gilbert DM. The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol Cell. 1999;4:983-93 [DOI] [PubMed] [Google Scholar]
- 173. Sadoni N, Cardoso MC, Stelzer EH, Leonhardt H, Zink D. Stable chromosomal units determine the spatial and temporal organization of DNA replication. J Cell Sci. 2004;117:5353-65 [DOI] [PubMed] [Google Scholar]
- 174. Woodfine K, Beare DM, Ichimura K, et al. Replication timing of human chromosome 6. Cell Cycle. 2005;4:172-6 [DOI] [PubMed] [Google Scholar]
- 175. Woodfine K, Fiegler H, Beare DM, et al. Replication timing of the human genome. Hum Mol Genet. 2004;13:191-202 [DOI] [PubMed] [Google Scholar]
- 176. Hiratani I, Ryba T, Itoh M, et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008;6:e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Lieberman-Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Meister P, Taddei A, Gasser SM. In and out of the replication factory. Cell. 2006;125:1233-5 [DOI] [PubMed] [Google Scholar]
- 179. Ryba T, Hiratani I, Lu J, et al. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010;20:761-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Cornacchia D, Dileep V, Quivy JP, et al. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J. 2012;31:3678-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Knott SR, Peace JM, Ostrow AZ, et al. Forkhead transcription factors establish origin timing and long-range clustering in S. cerevisiae. Cell. 2012;148:99-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Jun S, Herrick J, Bensimon A, Bechhoefer J. Persistence length of chromatin determines origin spacing in Xenopus early-embryo DNA replication: quantitative comparisons between theory and experiment. Cell Cycle. 2004;3:223-9 [PubMed] [Google Scholar]
- 183. Guilbaud G, Rappailles A, Baker A, et al. Evidence for sequential and increasing activation of replication origins along replication timing gradients in the human genome. PLoS Comput Biol. 2011;7:e1002322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Lucas I, Chevrier-Miller M, Sogo JM, Hyrien O. Mechanisms ensuring rapid and complete DNA replication despite random initiation in Xenopus early embryos. J Mol Biol. 2000;296:769-86 [DOI] [PubMed] [Google Scholar]
- 185. Rhind N. DNA replication timing: random thoughts about origin firing. Nat Cell Biol. 2006;8:1313-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Hyrien O, Goldar A. Mathematical modelling of eukaryotic DNA replication. Chromosome Res. 2010;18:147-61 [DOI] [PubMed] [Google Scholar]
- 187. Ma E, Hyrien O, Goldar A. Do replication forks control late origin firing in Saccharomyces cerevisiae? Nucleic Acids Res. 2012;40:2010-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Baker A, Audit B, Chen CL, et al. Replication fork polarity gradients revealed by megabase-sized U-shaped replication timing domains in human cell lines. PLoS Comput Biol. 2012;8:e1002443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Herrick J, Stanislawski P, Hyrien O, Bensimon A. Replication fork density increases during DNA synthesis in X. laevisegg extracts. J Mol Biol. 2000;300:1133-42 [DOI] [PubMed] [Google Scholar]
- 190. Blow JJ, Gillespie PJ, Francis D, Jackson DA. Replication origins in Xenopus egg extract are 5-15 kilobases apart and are activated in clusters that fire at different times. J Cell Biol. 2001;152:15-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Holmquist GP. Role of replication time in the control of tissue-specific gene expression. Am J Hum Genet. 1987;40:151-73 [PMC free article] [PubMed] [Google Scholar]
- 192. Hiratani I, Takebayashi S, Lu J, Gilbert DM. Replication timing and transcriptional control: beyond cause and effect, part II. Curr Opin Genet Dev. 2009;19:142-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Breier AM, Chatterji S, Cozzarelli NR. Prediction of Saccharomyces cerevisiae replication origins. Genome Biol. 2004;5:R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Rocha EP, Danchin A. Base composition bias might result from competition for metabolic resources. Trends Genet. 2002;18:291-4 [DOI] [PubMed] [Google Scholar]
- 195. Sharp PM, Shields DC, Wolfe KH, Li WH. Chromosomal location and evolutionary rate variation in enterobacterial genes. Science. 1989;246:808-10 [DOI] [PubMed] [Google Scholar]
- 196. Danchin A, Dondon L, Daniel J. Metabolic alterations mediated by 2-ketobutyrate in Escherichia coli K12. Mol Gen Genet. 1984;193:473-8 [DOI] [PubMed] [Google Scholar]
- 197. Mira A, Ochman H. Gene location and bacterial sequence divergence. Mol Biol Evol. 2002;19:1350-8 [DOI] [PubMed] [Google Scholar]
- 198. De S, Michor F. DNA replication timing and long-range DNA interactions predict mutational landscapes of cancer genomes. Nat Biotechnol. 2011;29:1103-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Liu B, Alberts BM. Head-on collision between a DNA replication apparatus and RNA polymerase transcription complex. Science. 1995; 267:1131-7 [DOI] [PubMed] [Google Scholar]
- 200. Pomerantz RT, O’Donnell M. What happens when replication and transcription complexes collide? Cell Cycle. 2010;9:2537-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011;25:1320-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. French S. Consequences of replication fork movement through transcription units in vivo. Science. 1992;258:1362-5 [DOI] [PubMed] [Google Scholar]
- 203. Mirkin EV, Mirkin SM. Mechanisms of transcription-replication collisions in bacteria. Mol Cell Biol. 2005;25:888-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Srivatsan A, Tehranchi A, MacAlpine DM, Wang JD. Co-orientation of replication and transcription preserves genome integrity. PLoS Genet. 2010;6:e1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Wang JD, Berkmen MB, Grossman AD. Genome-wide coorientation of replication and transcription reduces adverse effects on replication in Bacillus subtilis. Proc Natl Acad Sci U S A. 2007;104:5608-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Pomerantz RT, O’Donnell M. The replisome uses mRNA as a primer after colliding with RNA polymerase. Nature. 2008;456:762-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Elias-Arnanz M, Salas M. Bacteriophage phi29 DNA replication arrest caused by codirectional collisions with the transcription machinery. EMBO J. 1997;16:5775-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Liu B, Wong ML, Tinker RL, Geiduschek EP, Alberts BM. The DNA replication fork can pass RNA polymerase without displacing the nascent transcript. Nature. 1993;366:33-9 [DOI] [PubMed] [Google Scholar]
- 209. Brewer BJ. When polymerases collide: replication and the transcriptional organization of the E. coli chromosome. Cell. 1988;53:679-86 [DOI] [PubMed] [Google Scholar]
- 210. McLean MJ, Wolfe KH, Devine KM. Base composition skews, replication orientation, and gene orientation in 12 prokaryote genomes. J Mol Evol. 1998;47:691-6 [DOI] [PubMed] [Google Scholar]
- 211. Mrazek J, Karlin S. Strand compositional asymmetry in bacterial and large viral genomes. Proc Natl Acad Sci U S A. 1998;95:3720-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Rocha EP, Danchin A. Gene essentiality determines chromosome organisation in bacteria. Nucleic Acids Res. 2003;31:6570-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Ellwood M, Nomura M. Chromosomal locations of the genes for rRNA in Escherichia coli K-12. J Bacteriol. 1982;149:458-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Nomura M, Morgan EA. Genetics of bacterial ribosomes. Annu Rev Genet. 1977;11:297-347 [DOI] [PubMed] [Google Scholar]
- 215. Epshtein V, Toulme F, Rahmouni AR, Borukhov S, Nudler E. Transcription through the roadblocks: the role of RNA polymerase cooperation. EMBO J. 2003;22:4719-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Kunst F, Ogasawara N, Moszer I, et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249-56 [DOI] [PubMed] [Google Scholar]
- 217. Blattner FR, Plunkett G, 3rd, Bloch CA, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453-62 [DOI] [PubMed] [Google Scholar]
- 218. Rocha E. Is there a role for replication fork asymmetry in the distribution of genes in bacterial genomes? Trends Microbiol. 2002;10: 393-5 [DOI] [PubMed] [Google Scholar]
- 219. Sanchez JA, Kim SM, Huberman JA. Ribosomal DNA replication in the fission yeast, Schizosaccharomyces pombe. Exp Cell Res. 1998;238:220-30 [DOI] [PubMed] [Google Scholar]
- 220. Brewer BJ, Fangman WL. A replication fork barrier at the 3′ end of yeast ribosomal RNA genes. Cell. 1988;55:637-43 [DOI] [PubMed] [Google Scholar]
- 221. Linskens MH, Huberman JA. Organization of replication of ribosomal DNA in Saccharomyces cerevisiae. Mol Cell Biol. 1988;8:4927-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Dalgaard JZ, Klar AJ. A DNA replication-arrest site RTS1 regulates imprinting by determining the direction of replication at mat1 in S. pombe. Genes Dev. 2001;15:2060-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Krings G, Bastia D. swi1- and swi3-dependent and independent replication fork arrest at the ribosomal DNA of Schizosaccharomyces pombe. Proc Natl Acad Sci U S A. 2004;101:14085-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Little RD, Platt TH, Schildkraut CL. Initiation and termination of DNA replication in human rRNA genes. Mol Cell Biol. 1993;13:6600-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Wiesendanger B, Lucchini R, Koller T, Sogo JM. Replication fork barriers in the Xenopus rDNA. Nucleic Acids Res. 1994;22:5038-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Bermejo R, Capra T, Jossen R, et al. The replication checkpoint protects fork stability by releasing transcribed genes from nuclear pores. Cell. 2011;146:233-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Romero J, Lee H. Asymmetric bidirectional replication at the human DBF4 origin. Nat Struct Mol Biol. 2008;15:722-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Lee H, Romero J. Origin of DNA replication at the human lamin B2 locus: OBR or ABR? Cell Cycle. 2012;11:4281-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Abdurashidova G, Deganuto M, Klima R, et al. Start sites of bidirectional DNA synthesis at the human lamin B2 origin. Science. 2000;287:2023-6 [DOI] [PubMed] [Google Scholar]
- 230. Beletskii A, Bhagwat AS. Transcription-induced mutations: increase in C to T mutations in the nontranscribed strand during transcription in Escherichia coli. Proc Natl Acad Sci U S A. 1996;93:13919-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Francino MP, Chao L, Riley MA, Ochman H. Asymmetries generated by transcription-coupled repair in enterobacterial genes. Science. 1996;272:107-9 [DOI] [PubMed] [Google Scholar]
- 232. Engreitz JM, Agarwala V, Mirny LA. Three-dimensional genome architecture influences partner selection for chromosomal translocations in human disease. PloS One. 2012;7:e44196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Brewer BJ, Fangman WL. Initiation at closely spaced replication origins in a yeast chromosome. Science. 1993;262:1728-31 [DOI] [PubMed] [Google Scholar]
- 234. Santocanale C, Sharma K, Diffley JF. Activation of dormant origins of DNA replication in budding yeast. Genes Dev. 1999;13:2360- 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Shechter D, Costanzo V, Gautier J. Regulation of DNA replication by ATR: signaling in response to DNA intermediates. DNA Repair. 2004;3:901-8 [DOI] [PubMed] [Google Scholar]
- 236. Shechter D, Gautier J. ATM and ATR check in on origins: a dynamic model for origin selection and activation. Cell Cycle. 2005;4:235-8 [PubMed] [Google Scholar]
- 237. Marheineke K, Hyrien O. Control of replication origin density and firing time in Xenopus egg extracts: role of a caffeine-sensitive, ATR-dependent checkpoint. J Biol Chem. 2004;279:28071-81 [DOI] [PubMed] [Google Scholar]
- 238. Sorensen CS, Syljuasen RG, Lukas J, Bartek J. ATR, claspin and the Rad9-Rad1-Hus1 complex regulate Chk1 and Cdc25A in the absence of DNA damage. Cell Cycle. 2004;3:941-5 [PubMed] [Google Scholar]
- 239. Miao H, Seiler JA, Burhans WC. Regulation of cellular and SV40 virus origins of replication by Chk1-dependent intrinsic and UVC radiation-induced checkpoints. J Biol Chem. 2003;278:4295-304 [DOI] [PubMed] [Google Scholar]
- 240. de Klein A, Muijtjens M, van Os R, et al. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol. 2000;10:479-82 [DOI] [PubMed] [Google Scholar]
- 241. Shechter D, Costanzo V, Gautier J. ATR and ATM regulate the timing of DNA replication origin firing. Nat Cell Biol. 2004;6:648-55 [DOI] [PubMed] [Google Scholar]
- 242. Mantiero D, Mackenzie A, Donaldson A, Zegerman P. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J. 2011;30:4805-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Goren A, Cedar H. Replicating by the clock. Nat Rev Mol Cell Biol. 2003;4:25-32 [DOI] [PubMed] [Google Scholar]
- 244. Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907-13 [DOI] [PubMed] [Google Scholar]
- 245. Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864-70 [DOI] [PubMed] [Google Scholar]
- 246. DiTullio RA, Jr., Mochan TA, Venere M, et al. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat Cell Biol. 2002;4:998-1002 [DOI] [PubMed] [Google Scholar]
- 247. Bartkova J, Rezaei N, Liontos M, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633-7 [DOI] [PubMed] [Google Scholar]
- 248. Di Micco R, Fumagalli M, Cicalese A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper- replication. Nature. 2006;444:638-42 [DOI] [PubMed] [Google Scholar]
- 249. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352-5 [DOI] [PubMed] [Google Scholar]
- 250. Denko NC, Giaccia AJ, Stringer JR, Stambrook PJ. The human Ha-ras oncogene induces genomic instability in murine fibroblasts within one cell cycle. Proc Natl Acad Sci U S A. 1994;91:5124-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Mai S, Fluri M, Siwarski D, Huppi K. Genomic instability in MycER-activated Rat1A-MycER cells. Chromosome Res. 1996;4:365-71 [DOI] [PubMed] [Google Scholar]
- 252. Felsher DW, Bishop JM. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc Natl Acad Sci U S A. 1999;96:3940-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature. 1999;401:297-300 [DOI] [PubMed] [Google Scholar]
- 254. Minella AC, Grim JE, Welcker M, Clurman BE. p53 and SCFFbw7 cooperatively restrain cyclin E-associated genome instability. Oncogene. 2007;26:6948-53 [DOI] [PubMed] [Google Scholar]
- 255. Lengronne A, Schwob E. The yeast CDK inhibitor Sic1 prevents genomic instability by promoting replication origin licensing in late G(1). Mol Cell. 2002;9:1067-78 [DOI] [PubMed] [Google Scholar]
- 256. Arlt MF, Durkin SG, Ragland RL, Glover TW. Common fragile sites as targets for chromosome rearrangements. DNA Repair. 2006;5:1126-35 [DOI] [PubMed] [Google Scholar]
- 257. Mao L, Lee JS, Fan YH, et al. Frequent microsatellite alterations at chromosomes 9p21 and 3p14 in oral premalignant lesions and their value in cancer risk assessment. Nat Med. 1996;2:682-5 [DOI] [PubMed] [Google Scholar]
- 258. Stephens PJ, Greenman CD, Fu B, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Liu P, Erez A, Nagamani SC, et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell. 2011;146:889-903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Fudenberg G, Getz G, Meyerson M, Mirny LA. High order chromatin architecture shapes the landscape of chromosomal alterations in cancer. Nat Biotechnol. 2011;29:1109-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Zhang Y, McCord RP, Ho YJ, et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148:908-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Chiarle R, Zhang Y, Frock RL, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147:107-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Leonardi R, Subramanian C, Jackowski S, Rock CO. Cancer-associated isocitrate dehydrogenase mutations inactivate NADPH-dependent reductive carboxylation. J Biol Chem. 2012;287:14615-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Gaidzik VI, Paschka P, Spath D, et al. TET2 mutations in acute myeloid leukemia (AML): results from a comprehensive genetic and clinical analysis of the AML study group. J Clin Oncol. 2012;30:1350-7 [DOI] [PubMed] [Google Scholar]
- 268. Okita Y, Narita Y, Miyakita Y, et al. IDH1/2 mutation is a prognostic marker for survival and predicts response to chemotherapy for grade II gliomas concomitantly treated with radiation therapy. Int J Oncol. Epub 2012. July 20 [DOI] [PubMed] [Google Scholar]
- 269. SongTao Q, Lei Y, Si G, et al. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012;103:269-73 [DOI] [PubMed] [Google Scholar]
- 270. Zhou KG, Jiang LJ, Shang Z, Wang J, Huang L, Zhou JF. Potential application of IDH1 and IDH2 mutations as prognostic indicators in non-promyelocytic acute myeloid leukemia: a meta-analysis. Leuk Lymphoma. 2012;53:2423-9 [DOI] [PubMed] [Google Scholar]
- 271. Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst. 2010;102:932-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Bruniquel D, Schwartz RH. Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nat Immunol. 2003;4:235-40 [DOI] [PubMed] [Google Scholar]
- 273. Metivier R, Gallais R, Tiffoche C, et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45-50 [DOI] [PubMed] [Google Scholar]
- 274. Kangaspeska S, Stride B, Metivier R, et al. Transient cyclical methylation of promoter DNA. Nature. 2008;452:112-5 [DOI] [PubMed] [Google Scholar]
- 275. Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268-74 [DOI] [PubMed] [Google Scholar]
- 276. Howarth KD, Blood KA, Ng BL, et al. Array painting reveals a high frequency of balanced translocations in breast cancer cell lines that break in cancer-relevant genes. Oncogene. 2008;27:3345-59 [DOI] [PMC free article] [PubMed] [Google Scholar]