

Abstract
Several genes were duplicated during human evolution. It seems that one such duplication gave rise to a gene that may have helped to make human brains bigger and more adaptable than those of our ancestors.
The decoding of the human and chimpanzee genomes was heralded as an opportunity to truly understand how changes in DNA resulted in the evolution of our cognitive features. However, more than a decade and much detective work later, the functional consequences of such changes have proved elusive, with a few exceptions1,2. Now, writing in Cell, Dennis et al.3 and Charrier et al.4 describe the evolutionary history and function of the human gene SRGAP2 and provide evidence for molecular and cellular mechanisms that may link the gene’s evolution with that of our brain*.
It was already known that SRGAP2 is involved in brain development5 and that humans have at least three similar copies of the gene, whereas non-human primates carry only one6. However, the study of duplicated, or very similar, segments of DNA is hampered by the fact that most human cells carry two sets of chromosomes (one inherited from each parent), which makes it difficult to distinguish duplicated copies from the different parental forms of the gene. To circumvent this problem, Dennis et al.3 searched for copies of SRGAP2 in the genome of a hydatidiform mole — an abnormal, non-viable human embryo that results from the fusion of a sperm with an egg that has lost its genetic material; it therefore has chromosomes derived from a single parent.
The authors showed that humans carry four non-identical copies (named A-D) of SRGAP2 at different locations on chromosome 1. By comparing the genes’ sequences with that of the SRGAP2 gene from the orangutan and chimpanzee, the authors estimated that SRGAP2 was duplicated in the human lineage about 3.4 million years ago, resulting in SRGAP2A (the ancestral version that we share with other primates) and SRGAP2B. Further duplications of SRGAP2B gave rise to SRGAP2C about 2.4 million years ago and to SRGAP2D about 1 million years ago (Fig. 1a).
Figure 1. Evolution and function of a human gene.
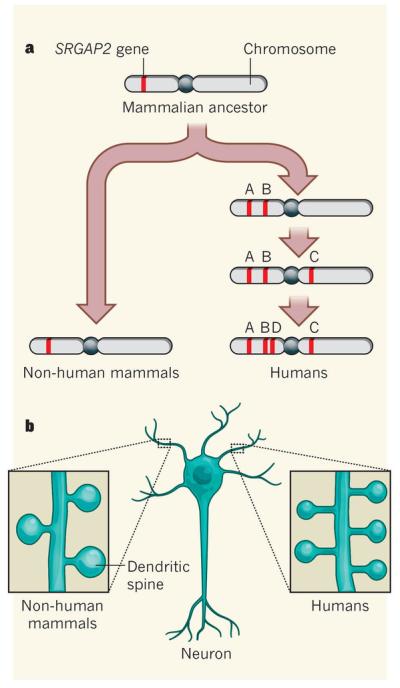
a, Dennis et al.3 and Charrier et al.4 detail how the SRGAP2 gene, which is found as a single copy in the genomes of most mammals, was duplicated three times during the evolution of human ancestors to give rise to four similar versions of the gene, named A–D. b, Charrier et al. demonstrate that the ‘ancestral’ version, SRGAP2A, stimulates the maturation of dendritic spines (protuberances) on the surfaces of neurons, whereas SRGAP2C promotes an increased number of immature spines in humans. This development might have contributed to the evolution of human cognitive abilities.
Dennis et al. found that SRGAP2B and SRGAP2D are expressed at much lower levels, and are more prone to sequence variations among humans, than are the A and C copies. They therefore suggest that SRGAP2C might have played a major part in the emergence of the Homo lineage 2 million to 3 million years ago, when human brain specializations (such as those leading to the development of language, social cognition and problem solving) were probably evolving.
In a complementary study, Charrier and colleagues4 investigated the role of the SRGAP2 genes in the development of spines — not those that permit us to walk upright, but dendritic spines. These are small protrusions from the surface of neurons that have an important role in the transmission of nerve impulses. Previous studies had documented the fact that humans have greater numbers and densities of dendritic spines than other primates and rodents7,8, but the molecular mechanisms driving this feature were unknown. By studying cells from genetically modified mice and human brain tissue, the authors show that SRGAP2A promotes the maturation of spines and slows down the migration of neurons within the developing cerebral cortex, whereas the human-specific SRGAP2C has opposite effects and so favours the formation of further spines (Fig. 1b). These results suggest that the emergence of SRGAP2C could have contributed to expansion of the cortex and increase in spine numbers in human ancestors, and therefore to the evolution of brain function and plasticity (the ability to alter neural connections in response to new experiences).
Interestingly, Dennis et al. studied a group of young patients who had developmental disorders, and identified a few individuals — out of several thousand — who had DNA duplications or deletions that affected SRGAP2A or SRGAP2C. Although such numbers are too small to be statistically significant, they indicate that mutations in SRGAP2A and SRGAP2C could be linked to disease. This hypothesis is consistent with previous suggestions that the evolution of a bigger and more complex brain in the human lineage was accompanied by an increased susceptibility to neurological disorders9.
Taken together, the findings reported by Dennis et al. and Charrier et al. significantly add to the current working version of the human genome, and provide an example of how human-specific gene duplications can modulate brain function. They also give functional context to the long-known prolonged immaturity of the developing human brain (a process known as neoteny)10,11. By slowing spine maturation and promoting neuronal migration, SRGAP2C might allow the environment to have a more protracted influence on brain development than is possible during the shorter brain maturation times seen in other mammals, providing additional malleability.
Furthermore, Charrier et al. demonstrate how evolutionary hypotheses derived from the comparative genomics of humans and other primates can be tested in cell culture and animal models. This elegant work provides a launching point for unravelling a more detailed mechanistic understanding of the role of human-specific duplicated genes in brain development, and of their potential contribution to brain evolution and neurodevelopmental disorders.
Footnotes
This News & Views article was published online on 20 June 2012.
Contributor Information
Daniel H. Geschwind, Department of Neurology and the Semel Institute, David Geffen School of Medicine at UCLA, University of California, Los Angeles, Los Angeles, California 90095-1761, USA
Genevieve Konopka, Department of Neuroscience, University of Texas Southwestern Medical Center, Dallas, Texas 75390-9111, USA.
References
- 1.Konopka G, et al. Nature. 2009;462:213–217. doi: 10.1038/nature08549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Enard W, et al. Cell. 2009;137:961–971. doi: 10.1016/j.cell.2009.03.041. [DOI] [PubMed] [Google Scholar]
- 3.Dennis MY, et al. Cell. 2012;149:912–922. doi: 10.1016/j.cell.2012.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Charrier C, et al. Cell. 2012;149:923–935. doi: 10.1016/j.cell.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guerrier S, et al. Cell. 2009;138:990–1004. doi: 10.1016/j.cell.2009.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sudmant PH, et al. Science. 2010;330:641–646. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elston GN, Benavides-Piccione R, DeFelipe JJ. Neurosci. 2001;21:RC163. doi: 10.1523/JNEUROSCI.21-17-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benavides-Piccione R, Ballesteros-Yañez I, DeFelipe J, Yuste RJ. Neurocytol. 2002;31:337–346. doi: 10.1023/a:1024134312173. [DOI] [PubMed] [Google Scholar]
- 9.Preuss TM, Cáceres M, Oldham MC, Geschwind DH. Nature Rev. Genet. 2004;5:850–860. doi: 10.1038/nrg1469. [DOI] [PubMed] [Google Scholar]
- 10.Petanjek Z, et al. Proc. Natl Acad. Sci. USA. 2011;108:13281–13286. doi: 10.1073/pnas.1105108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Somel M, et al. Proc. Natl Acad. Sci. USA. 2009;106:5743–5748. doi: 10.1073/pnas.0900544106. [DOI] [PMC free article] [PubMed] [Google Scholar]