

Abstract
In the principal cells of the renal collecting duct, arginine vasopressin (AVP) stimulates the synthesis of cAMP, leading to signaling events that culminate in the phosphorylation of aquaporin-2 water channels and their redistribution from intracellular domains to the plasma membrane via vesicular trafficking. The molecular mechanisms that control aquaporin-2 trafficking and the consequent water reabsorption, however, are not completely understood. Here, we used a cell-based assay and automated immunofluorescence microscopy to screen 17,700 small molecules for inhibitors of the cAMP-dependent redistribution of aquaporin-2. This approach identified 17 inhibitors, including 4-acetyldiphyllin, a selective blocker of vacuolar H+-ATPase that increases the pH of intracellular vesicles and causes accumulation of aquaporin-2 in the Golgi compartment. Although 4-acetyldiphyllin did not inhibit forskolin-induced increases in cAMP formation and downstream activation of protein kinase A (PKA), it did prevent cAMP/PKA-dependent phosphorylation at serine 256 of aquaporin-2, which triggers the redistribution to the plasma membrane. It did not, however, prevent cAMP-induced changes to the phosphorylation status at serines 261 or 269. Last, we identified the fungicide fluconazole as an inhibitor of cAMP-mediated redistribution of aquaporin-2, but its target in this pathway remains unknown. In conclusion, our screening approach provides a method to begin dissecting molecular mechanisms underlying AVP-mediated water reabsorption, evidenced by our identification of 4-acetyldiphyllin as a modulator of aquaporin-2 trafficking.
Arginine-vasopressin (AVP) stimulates vasopressin V2 receptors on the surface of renal collecting duct principal cells and thereby production of cAMP, which activates protein kinase A (PKA). Initiation of this signaling results in the redistribution of the water channel aquaporin-2 (AQP2) from intracellular vesicles into the plasma membrane by an exocytosis-like process. The membrane insertion of AQP2 facilitates water reabsorption from primary urine and fine-tunes blood osmolality.1,2 Loss of AVP secretion causes central diabetes insipidus, loss of function mutations in vasopressin V2 receptors, or AQP2 lead to nephrogenic diabetes insipidus,3–5 whereas pathologically elevated levels of AVP with excessive water retention are associated with chronic heart failure or the syndrome of inappropriate antidiuretic hormone secretion.6
AVP induces the PKA-catalyzed phosphorylation of AQP2 at serine 256 (S256). This phosphorylation is the key trigger for the redistribution of AQP2 from intracellular vesicles into the plasma membrane.7–11 AVP also induces phosphorylations of S264 and S269, which are associated with a predominant plasma membrane localization of AQP2.12–17 Under resting conditions, AQP2 is phosphorylated at S261.12 AVP mediates dephosphorylation of S261.15,18 This is associated with decreased polyubiquitination and proteasomal degradation and an enhanced AQP2 abundance, which contributes to the increase in water reabsorption of the collecting duct in response to AVP.19
Although several proteins controlling AQP2 trafficking were identified and the paths of AQP2 to and from the plasma membrane are defined in general terms,1,17 the molecular details underlying AQP2 trafficking are unclear. We present a novel, unbiased, high-throughput cell-based assay that identifies small-molecule inhibitors of the cAMP-dependent redistribution of AQP2. Identification of the targets of candidate molecules reveals new proteins and mechanisms controlling AQP2.
Results
High-Throughput Screening Identifies Small-Molecule Inhibitors of the cAMP-Dependent AQP2 Redistribution
Mouse collecting-duct cells stably expressing human AQP2 (MCD4 cells20) were used to establish a high-throughput assay to identify small-molecule inhibitors of the cAMP-dependent redistribution of AQP2 from intracellular vesicles into the plasma membrane (Supplemental Figure 1). MCD4 cells were incubated with each of the 17,700 small molecules from the ChemBioNet library (40 µM).21 Forskolin, a direct activator of adenylyl cyclases, was added to induce the redistribution of AQP2. The localization of AQP2 and cortical F-actin as plasma membrane marker were evaluated by automated immunofluorescence microscopy (Figure 1A). The localization of AQP2 was expressed as the ratio of fluorescence signal intensity at the plasma membrane to intracellular fluorescence signal intensity (Figure 1B).11,22 Forskolin induced the redistribution of AQP2 from a perinuclear localization to the plasma membrane (ratio, 1.40±0.1). As expected, blocking PKA with the kinase inhibitor H8923,24 prevented the AQP2 redistribution7,8,11,19 (ratio, 0.91±0.1) (Figure 1, A and B). In previous studies cells were incubated with H89 for 30 minutes,11,22 whereas we incubated cells for 2 hours, as long as with the library compounds. This most likely leads to the dramatic change in the localization of AQP2 compared with earlier studies. On the basis of the ratios determined in the presence of forskolin (1.4) and the combination of forskolin and H89 (0.9), ratios ≤1.2 were considered to indicate low plasma membrane abundance of AQP2 (Supplemental Figure 2). Treatment with forskolin in the presence of 83 of the library compounds resulted in ratios ≤ 1.2 (Supplemental Figure 2 and Supplemental Table 1), defining them as inhibitors of the forskolin-induced AQP2 redistribution; 17 of these compounds (Table 1) inhibited in a concentration dependent manner (40 µM, 4 µM, and 0.4 µM). Fourteen of the 17 were commercially available and could thus be tested in secondary screens using rat primary inner medullary collecting duct (IMCD) cells expressing AQP2 endogenously.19,22 In this model, five of the compounds inhibited the forskolin-induced redistribution of AQP2 (Table 1), two of which were further characterized: 4-acetyldiphyllin (4AD; ratio, 0.88; Table 1) and triazolpropenon (ratio, 1.06; Table 1).
Figure 1.

Identification of 4AD as an inhibitor of the forskolin (FSK)-induced AQP2 redistribution in MCD4 cells by cell-based high-throughput screening. (A) MCD4 cells were seeded into 384-well plates, left untreated, treated with forskolin (10 µM, 20 minutes), or incubated with each of the 17,700 small molecules from the ChemBioNet small-molecule library (40 µM) for 100 minutes before forskolin (10 µM) was added for an additional 20 minutes. As a control, the forskolin-induced redistribution of AQP2 was inhibited with H89 (30 µM, 2 hours), a known blocker of the AQP2 translocation. The cells were fixed with paraformaldehyde, and AQP2 was detected by immunofluorescence microscopy using specific antibodies (H27) and Cy5-coupled antirabbit secondary antibodies (red).51 F-actin was visualized using TRITC-conjugated phalloidin (green). Fluorescence signals were detected using an automated screening microscope (10× magnification). The magnified views are from the indicated squares in the merge. Shown are representative images from one of three independent experiments. (B) The chemical structure represents 4AD. The intensities of intracellular and plasma membrane immunofluorescence signals arising from AQP2 were determined and related to nuclear signal intensities (mean ± SEM; three independent experiments). The ratios of plasma membrane to intracellular fluorescence signal intensities were calculated. According to the ratios determined for cells treated with forskolin alone (1.4) and forskolin in combination with H89 (0.9), ratios <1.2 indicate a predominant intracellular localization. Values significantly different from forskolin-treated cells are indicated (***P<0.001).
Table 1.
Small molecule inhibitors of the cAMP-induced redistribution of AQP2.
| No. | ID | Structure | Formula | Ratio | Plate | Well | Molecular Weight | Partition Coefficient | H-Acc | H-Don | IUPAC Nomenclature |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 4-acetydiphillin (4AD) 216407 | 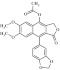 |
C23H18O8 | 0.88 | 115 | N9 | 422.4 | 3.08 | 6 | 0 | 9-benzo[d][1,3] dioxol- 5-yl)-6,7-dimethoxy-1-oxo-1,3-dihydronaphtho[2,3-c]furan-4-yl acetate |
| 2 | 212678 | 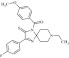 |
C23H24FN3O2S | 0.84 | 105 | K1 | 425.5 | 4.99 | 4 | 0 | (8-ethyl-3-(4-fluorophenyl)-2-thioxo-1,4,8-triazaspiro[4.5]dec-3-en-1-yl)(4-methoxyphenyl)methanone |
| 3 | 212634 | 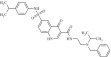 |
C31H36N4O4S | 0.88 | 104 | D14 | 560.7 | 5.2 | 6 | 3 | N-{2[benzyl (isopropyl)amino] ethyl}-6-[(4isopropylphenyl)sulfamoyl]-4-oxo-1H-quinoline-3-carboxamide |
| 4 | 202336 | 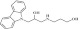 |
C18H22N2O2 | 1 | 75 | P11 | 298.4 | 1.69 | 3 | 3 | 3-((3-(9H-carbazol-9-yl) 2 hydroxypropyl)amino)propan-1-ol |
| 5 | 209614 triazolpropenon | 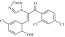 |
C17H10Cl3N3O2 | 1.06 | 96 | K6 | 394.6 | 4.58 | 4 | 1 | (Z)-3-(3-chloro-6-hydroxycyclohexa-2,4-dienyl)-1-(2,4-dichlorophenyl)-2-(11H,2,4-triazol-1-yl)prop-2-en-1-one |
| 6 | 214590 | 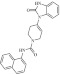 |
C23H20N4O2 | 0.69 | 110 | K18 | 384.4 | 3.24 | 2 | 2 | N-(naphthalen-1-yl)-4-(2-oxo-3H-1,3-benzodiazol-1-yl)-3,6-dihydro-2H-pyridine-1-carboxamide |
| 7 | 205653 | 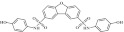 |
C24H18N2O7S2 | 0.78 | 85 | I5 | 510.5 | 3.52 | 6 | 4 | N,N′-bis(4-hydroxyphenyl)dibenzo[b,d]furan-2,8-disulfonamide |
| 8 | 216086 | 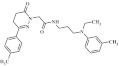 |
C25H32N4O2 | 0.79 | 114 | L17 | 420.5 | 3.47 | 4 | 1 | N-{3-[ethyl(3-methylphenyl)amino]propyl}-2-[3-(4-methylphenyl)-6-oxo-4,5-dihydropyridazin-1-yl]acetamide |
| 9 | 214645 | 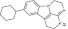 |
C19H23N3 | 0.8 | 110 | J9 | 293.4 | 3.62 | 2 | 1 | 9-cyclohexyl-2,3,5,6-tetrahydro-1H-3,4,6a-triazafluoranthene |
| 10 | 203240 | 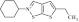 |
C13H18NS2 | 0.89 | 78 | O17 | 252.4 | 1.6 | 0 | 0 | 2-cyclohexyl-5-ethyl-2,3-dihydrothieno[3,2-d]isothiazole |
| 11 | 215223 | 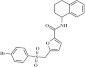 |
C22H20BrNO4S | 0.9 | 112 | M21 | 474.4 | 4.23 | 3 | 1 | 5-[(4-bromobenzenesulfonyl)methyl]-N-(1,2,3,4-tetrahydronaphthalen-1-yl)furan-2-carboxamide |
| 12 | 203313 | 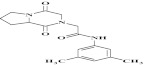 |
C17H21N3O3 | 1 | 78 | A16 | 315.4 | 0.79 | 3 | 1 | N-(3,5-dimethylphenyl)-2-(1,4 dioxohexahydropyrrolo[1,2-a] pyrazin-2(1H)-yl) acetamide |
| 13 | 201213 | 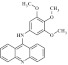 |
C22H20N2O3 | 1.02 | 72 | I18 | 360.4 | 4.47 | 5 | 1 | N-(3,4,5-trimethoxyphenyl)acridin-9-amine |
| 14 | 208677 | 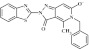 |
C21H15N4O2S | 1.07 | 93 | J13 | 387.4 | 1.13 | 5 | 0 | 2-(1,3-benzothiazol-2-yl)-5-benzyl-4-methyl-3-oxo-3,5-dihydro-2H-pyrazolo[4,3-c]pyridin-6-olate |
| 15 | 202329 | 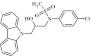 |
C22H21ClN2O3S | 1.12 | 75 | B11 | 428.9 | 3.69 | 3 | 1 | N-(3-(9H-carbazol-9-yl)-2-hydroxypropyl)-N-(4-chlorophe nyl)methane-sulfonamide |
| 16 | 204332 | 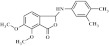 |
C18H19NO4 | 1.16 | 81 | G6 | 313.3 | 3.84 | 4 | 1 | 3-[(3,4dimethyl-phenyl)amino]-6,7-dimethoxy-3H-2-benzofuran-1-one |
| 17 | 209044 | 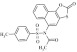 |
C20H15NO5S2 | 1.18 | 94 | H17 | 413.5 | 4.34 | 5 | 0 | N-(2-oxonaphtho[2,1-d][1,3]oxathiol-5-yl)-N-tosylacetamide |
Primary screening of 17,700 small molecules identified 83 inhibitors of the cAMP-induced redistribution of AQP2 in MCD4 cells. Secondary screening revealed that 17 chemically highly diverse hits inhibited the AQP2 redistribution in a concentration-dependent manner in MCD4 cells (compounds 1–17). Compounds 1–5 also inhibited the redistribution in IMCD cells. Compounds are ranked according to increasing ratios of plasma membrane/intracellular AQP2 fluorescence signal intensities. Compounds with ratios ≤ 1.2 were defined as inhibitory (a predominant intracellular localization of AQP2). Compound ID numbers (ID), compound library positions (plate, well), molecular weight, numbers of proton donor (H-Don.) and acceptor (H-Acc.) moieties are given for each substance. Compound names are according to the International Union of Pure and Applied Chemistry nomenclature. IUPAC, International Union of Pure and Applied Chemistry.
4AD Prevents the cAMP-Induced Redistribution of AQP2
In resting IMCD cells, AQP2 is located intracellularly; in the presence of forskolin, it predominantly resides in the basolateral plasma membrane, while a minor fraction of AQP2 redistributes to the apical plasma membrane (Figure 2A).11,19,22 This resembles the situation in the collecting duct where in response to AVP the AQP2 amount in the basolateral plasma membrane can be equal to or greater than that in the apical plasma membrane.2 4AD concentration-dependently inhibited the forskolin-induced AQP2 redistribution (Figure 2A). Cell-surface biotinylation assays were used to quantify the effect. IMCD cells were treated as indicated above; cell-surface proteins were biotinylated and precipitated, and AQP2 and cadherin (loading control) were detected by Western blotting. Densitometric analysis confirmed that 4AD significantly reduced the forskolin-induced increase in plasma membrane abundance of AQP2 (Figure 2B); 4AD alone did not alter the membrane abundance of AQP2 compared with untreated controls. In line with these observations, 4AD prevented the forskolin-induced increase of AQP2 phosphorylation at S256 (AQP2-pS256) and S269 (AQP2-pS269) at the plasma membrane (Figure 3). The reason for the nuclear staining with the anti-pS269 antibody in the absence of forskolin is unclear; it also occurred with a custom-made antibody25 (data not shown), suggesting that the antibodies cross-react with a nuclear protein containing a similar epitope. The effective concentrations of 4AD did not induce any visible signs of toxicity in MCD4 or IMCD cells (24-hour toxicity assay) (Supplemental Figure 3B).
Figure 2.

4AD concentration-dependently inhibits the forskolin (FSK)-induced redistribution of AQP2 in primary IMCD cells. (A) Primary IMCD cells were left untreated and were stimulated with forskolin (10 µM, 20 minutes) or with a combination of 4AD (40, 4, or 0.4 µM, 30 minutes) and forskolin. AQP2 was detected by immunofluorescence microscopy using specific primary antibodies (H27) and Cy3-coupled antirabbit secondary antibodies (red).51 Nuclei were stained with DAPI (blue). Shown are representative images from one of three independent experiments. (B) Upper panel. Cell-surface biotinylation assays were carried out to quantify the effect of 4AD on the localization of AQP2. To detect AQP2 on the surface of IMCD cells, surface proteins were biotinylated with EZ-Link Sulfo-NHS-SS-Biotin, precipitated with streptavidin-agarose, separated by SDS-PAGE, and AQP2, and as a loading control cadherin were detected by Western blotting (shown is a representative blot of three independent experiments). Lower panel. The amounts of complex (compl) glycosylated AQP2 and the high mannose (hm) form of AQP2 and nonglycosylated (ng) AQP2 on the blots were quantitatively analyzed by densitometry; each bar of the histogram represents the total amounts of the three AQP2 forms (mean ± SEM). *P<0.05, **P<0.01, and ***P<0.001 versus forskolin-treated cells.
Figure 3.

4AD directs the forskolin (FSK) induced phosphorylation of S269 (pS269) to intracellular domains and prevents S256 phosphorylation of AQP2 in primary IMCD cells. (A) The cells were left untreated, incubated with forskolin (10 µM, 20 minutes), 4AD (4 µM, 30 minutes), or a combination of 4AD and FSK. AQP2 was detected using specific primary antibodies (C-17, Santa Cruz) and Cy2-coupled antigoat secondary antibodies (green).19 AQP2-pS256 was detected using specific antiphospho primary and Cy3-coupled secondary antibodies.25 AQP2-pS269 was detected using specific antiphospho primary (p112–269, Phosphosolutions) and Cy3-coupled secondary antibodies. Nuclei were stained with DAPI (blue). Shown are representative images from one of three independent experiments. (B) The intensities of intracellular and plasma membrane immunofluorescence signals arising from AQP2, AQP2-pS256, and AQP2-S269 were determined and related to nuclear signal intensities (n≥30 cells per condition; mean ±SEM; three independent experiments). The ratios of plasma membrane to intracellular fluorescence signal intensities were calculated. Ratios >1 indicate a predominant localization at the plasma membrane. Values significantly different from control cells and cells treated with 4AD or forskolin and 4AD are indicated (***P<0.001).
4AD Increases Intravesicular pH and Causes Accumulation of AQP2 in the Golgi Compartment
4AD is a selective blocker of vacuolar H+-ATPases(V-ATPases);26 to compare its effects with another inhibitor of V-ATPases, we used bafilomycin A1 (baf A1).27,28 Baf A1 also blocked the forskolin-induced AQP2 translocation in IMCD cells (Figure 4A). To investigate whether baf A1 and 4AD inhibited intravesicular acidification, MCD4 cells were incubated with LysoTracker RED DND-99, which stains acidic intracellular compartments (red vesicular structures in Figure 4B). Both 4AD and baf A1 shifted intravesicular acidity to a higher pH, as indicated by the strong decrease of the mean fluorescence signal intensity per µm2 of individual cells (Figure 4B).
Figure 4.

Inhibition of V-ATPase with 4AD or baf A1 increases acidity of intracellular vesicles in IMCD cells and is associated with inhibition of the FSK-induced redistribution of AQP2 from intracellular vesicles to the plasma membrane. (A) IMCD cells were left untreated or treated with baf A1 in the indicated concentrations (30 minutes) in the absence or presence of forskolin (10 µM, 20 minutes). AQP2 was detected by immunofluorescence microscopy using specific primary antibody H27 and Cy3-coupled antirabbit secondary antibodies (red).51 Nuclei were stained with DAPI (blue). Representative images from one of three independent experiments are shown. (B) Upper panel. To visualize acidic compartments in MCD4 cells in the absence or presence of the V-ATPase inhibitors 4AD or baf A1, the cells were incubated with the acidotropic dye LysoTracker Red DND-99 (75 nM, 30 minutes), which is retained in acidified compartments (red vesicular structures). Nuclei were stained with Hoechst 33258 (blue). Shown are representative images from one of three independent experiments. Lower panel. The intensities of fluorescence signals arising from Lysotracker Red DND-99 were determined in individual cells (>50 cells per condition; three independent experiments; mean ± SEM). Statistically significant differences versus untreated cells are indicated (***P<0.001).
Baf A1 has previously been shown to arrest AQP2 in the trans-Golgi network (TGN) in LLC-PK1 cells, a porcine cell line stably expressing AQP2.29,30 In MCD4 cells, 4AD caused an accumulation of AQP2 in perinuclear patches (Figure 1, A and B) resembling those in LLC-PK1 cells that represent the TGN.17,30 In IMCD cells, 4AD did not induce the formation of patches clearly classifiable as TGN (Figure 2A). To confirm, however, that AQP2 is located in the Golgi in the presence of 4AD, we performed co-localization analyses of AQP2 and two Golgi proteins: (1) Golgi 58K (formiminotransferase cyclodeaminase), distributed throughout the Golgi, and (2) a marker for the endoplasmic reticulum Golgi intermediate compartment, ERGIC-53/p58. Figure 5 shows co-localization of AQP2 with both proteins in resting IMCD cells and a decrease in the co-localization upon treatment with forskolin when AQP2 exits intracellular compartments, including the Golgi, en route to the plasma membrane. Combined with forskolin 4AD decreased the co-localization with ERGIC-53/p58 and increased it with Golgi 58K, indicating that 4AD caused AQP2 accumulation in the Golgi. Whether this effect involves inhibition of Golgi V-ATPase is unclear; it is likely because inhibition of V-ATPase with baf A1 inhibits acidification of Golgi vesicles.31
Figure 5.

4AD causes an accumulation of AQP2 in the Golgi compartment. (A) IMCD cells were seeded on 12-mm cover slides, left untreated or treated with 4AD (4 µM or 0.4 µM, 30 minutes). AQP2 was detected with specific primary (C17, Santa Cruz) and secondary Cy2-coupled antibodies (red). ERGIC-53/p58 and Golgi apparatus 58K proteins were detected with specific primary and Cy5-conjugated secondary antibodies (both green). Fluorescence signals were visualized by laser-scanning microscopy. Shown are representative images from one of three independent experiments. (B) The co-localization of AQP2 with ERGIC-53 and Golgi apparatus 58K was determined from the images in part A using ZEN2010 software (Carl Zeiss; >25 individual cells per condition; three independent experiments; mean ±SEM). Statistically significant differences are indicated (*P<0.05; ***P<0.001). FSK, forskolin.
4AD Inhibits Forskolin-Induced Phosphorylation of AQP2 at S256 without Affecting cAMP Levels, Global PKA Activity, or Phosphorylations at S261 and S269
To further elucidate the molecular mechanisms underlying the inhibitory effect of 4AD, we investigated whether 4AD influences the cAMP/PKA signaling pathway. 4AD did not alter AVP- or forskolin-induced cAMP formation in IMCD cells (Figure 6A). cAMP is hydrolyzed by phosphodiesterases (PDEs). We had previously shown that nonselective inhibition of PDEs with 3-isobutyl-1-methylxanthine (IBMX) or specific inhibition of the PDE4 family with rolipram enhances AVP- and forskolin-induced increases in cAMP and the redistribution of AQP2 to the plasma membrane in IMCD cells.22 4AD did not visibly modify the effect of IBMX on the localization of AQP2 (Figure 6B), suggesting that it does not influence cAMP hydrolysis by PDEs.
Figure 6.

4AD does not affect formation of cAMP or global PKA activity. (A) Primary IMCD cells were left untreated, treated with 4AD (30 minutes), forskolin (FSK; 20 minutes), or AVP (20 minutes) in the indicated concentrations. Where indicated, cells were treated with 4AD for 30 minutes before addition of AVP or forskolin for an additional 20 minutes. Cell lysates were prepared and cAMP concentrations were determined by radioimmunoassay (three independent experiments, triplicates per condition; mean ± SEM; ***P< 0.001). (B) IMCD cells were seeded on 12-mm cover slides, left untreated, or treated with forskolin (10 µM, 20 minutes), IBMX (250 µM, 30 minutes), or the indicated combinations of forskolin and 4AD. AQP2 was detected by immunofluorescence microscopy using specific primary antibody H27 and Cy3-coupled antirabbit secondary antibodies (red).51 Nuclei were stained with DAPI (blue). Shown are representative images from one of three independent experiments. (C) MCD4 cells were left untreated, treated with forskolin (10 µM, 20 minutes), or treated with 4AD in the absence or presence of forskolin as indicated. Upper panels. Cell lysates were prepared and PKA activity was determined by measuring its ability to phosphorylate the substrate peptide PepTag A1. Where indicated, cAMP (1 µM) was added to induce maximal PKA activity. Agarose gels from one of three representative experiments show PKA-phosphorylated and nonphosphorylated PepTag A1 peptide. Lower panels. The amounts of phosphorylated and nonphosphorylated PepTag A1 peptides were densitometrically evaluated. PKA activity is expressed as the ratio of phosphorylated to nonphosphorylated PepTag A1 peptides. Values are mean ± SEM; ***P<0.001.
A potential influence of 4AD on total cellular PKA activity was assessed by measuring the kinase’s ability to phosphorylate a PKA-specific substrate peptide added to lysates derived from MCD4 cells (Figure 6C). In the absence or presence of forskolin, 4AD did not influence the phosphorylation of the peptide, indicating that 4AD does not globally affect PKA activity.
In IMCD cells, elevation of cAMP in response to forskolin or AVP increases the abundance of endogenous AQP2 approximately two-fold within 30 minutes.19 We had previously shown that this increase is detectable with antibodies directed against both the N and the C terminus of AQP2 and is thus independent from phosphorylations in the C terminus.19 The forskolin-induced increase in AQP2 abundance occurred similarly in the presence of 4AD (Figure 7A).
Figure 7.

4AD inhibits the forskolin (FSK)-induced phosphorylation of AQP2 at S256 but does not affect cAMP-regulated phosphorylations at S261 and S269. IMCD cells were left untreated or were treated with forskolin (10 µM, 20 minutes), 4AD (0.4 or 4 µM, 30 minutes), or the indicated combinations of forskolin and 4AD. Lysates were prepared, proteins were separated by SDS-PAGE, and AQP2 was detected using specific antibodies (C17, Santa Cruz) (A). AQP2 phosphorylated at S256 (pS256-AQP2) (B) was detected with specific custom-made antibodies.25 pS261-AQP2 (C) and pS269 (D), and, as a loading control, α-tubulin were detected using commercially available antibodies. Representative blots from one of five or more independent experiments are shown. Upper panels. The signals emerging from the complex glycosylated form of AQP2 were quantified densitometrically. Statistically significant differences are indicated (mean ± SEM; *P<0.05; **P<0.01, ***P<0.001). compl, complex glycosylated APQ2; hm, high mannose form of AQP2; ng, nonglycosylated AQP2.
Surprisingly, although 4AD did not inhibit forskolin-induced increases in cAMP formation and cellular PKA activity, 4AD prevented the forskolin-induced cAMP/PKA-dependent AQP2 phosphorylation at S256, the key trigger for its redistribution to the plasma membrane (Figures 3 and 7B). In contrast, 4AD did not affect the forskolin-induced dephosphorylation at S261 or the forskolin-induced phosphorylation of S269 (Figure 7, C and D). Thus, 4AD uncouples the cAMP-induced S256 phosphorylation from cAMP-induced phosphorylation of S269 and dephosphorylation of S261 of AQP2.
Fluconazole Inhibits the cAMP-Induced Redistribution of AQP2
Another promising inhibitory compound from our primary screen is triazolpropenon (Table 1). However, it inhibited the AQP2 redistribution in concentrations close to its 50% lethal concentration (34 µM), and the less toxic derivatives (50% lethal concentration, 235 µM) that we synthesized lost their inhibitory potency (data not shown). Triazolpropenon possesses structural similarity to the fungistatic agent fluconazole. Fluconazole blocks fungal lanosterol 14α-demethylase32 and thereby inhibits ergosterol biosynthesis, causing membrane defects. In IMCD cells, fluconazole inhibits the forskolin-induced redistribution of AQP2 (Figure 8). This inhibitory effect is apparently not due to obvious membrane defects similar to those fluconazole causes in fungi. In humans, fluconazole is a substrate for elimination and an inhibitor of several cytochrome P450 enzymes (CYP2C9, CYP2C19, CYP3A4).33 Thus, such P450 systems may be involved in the control of AQP2 or fluconazole modulates another yet unknown target controlling AQP2.
Figure 8.

Fluconazole inhibits the forskolin (FSK)-induced redistribution of AQP2 in primary IMCD cells. (A) Primary IMCD cells were left untreated, stimulated with forskolin (10 µM, 20 minutes), or treated with fluconazole (10 µM, 30 minutes) alone or in combination with forskolin. AQP2 was detected by immunofluorescence microscopy using specific primary antibodies (H27) and Cy3-coupled antirabbit secondary antibodies (red).51 Nuclei were stained with DAPI (blue). Shown are representative images from one of three independent experiments. (B) The intensities of intracellular and plasma membrane immunofluorescence signals arising from AQP2 were determined and related to nuclear signal intensities (n≥30 cells per condition; mean ±SEM; three independent experiments). The ratios of plasma membrane to intracellular fluorescence signal intensities were calculated. Ratios >1 indicate a predominant localization at the plasma membrane. ***P<0.001.
Discussion
We report here a novel, cell-based, high-throughput small-molecule screening approach as a feasible starting point for dissecting molecular mechanisms underlying AVP-mediated water reabsorption. The system permits monitoring effects of small molecules on AQP2 localization, and cell-damaging hits can easily be recognized and excluded from follow-up studies.
One of the small-molecule inhibitors of the AQP2 redistribution, which we identified, is 4AD, a blocker of V-ATPase.26 In renal principal cells, 4AD interferes with acidification of intracellular vesicles (Figure 4) and leads to inhibition of the PKA-dependent phosphorylation of AQP2 at S256, the key trigger for its redistribution to the plasma membrane (Figure 7B). This effect cannot be explained by global inhibition of PKA because 4AD does not affect total cellular PKA activity (Figure 6C). However, 4AD may inhibit PKA locally. PKA is tethered to AQP2-bearing vesicles by A-kinase anchoring proteins (AKAPs), such as AKAP220 and AKAP18δ, and AKAP-PKA interactions are crucial for the cAMP-induced phosphorylation of S256 and the consequent redistribution of AQP2.11,34–36 AKAP18δ is recruited to AQP2-bearing vesicles through ionic interaction with negatively charged membrane lipids and, in addition, through protein-protein interactions.37 Ablation of the proton gradient across vesicular membranes by 4AD could interfere with the electrostatic interaction of AKAP18δ with AQP2-bearing vesicles and thereby with the local AKAP18δ-dependent PKA activation. Because AKAPs generally interact with membranes through protein-protein or electrostatic interactions or lipid modifications,38 an altered proton gradient may also interfere with other AKAPs on AQP2-bearing vesicles. Notably, V-ATPase binds PKA.39 It acts as a pH sensor and binds the small GTPase ARNO at low vesicular pH. Baf A1 prevents this interaction.40 Similarly, the tethering of PKA by V-ATPase to AQP2-bearing vesicles may be pH-dependent and could be disrupted by 4AD. An altered proton gradient could also directly inhibit local PKA by affecting PKA’s “catalytically competent protonation state”41 between a pH of 6.5 and 8.42 Whether changes in protonation affect AQP2, in particular its accessibility for phosphorylation by PKA, is unknown.
AQP2-pS256 is slightly elevated (1.5-fold; Figure 7B) in the presence of forskolin and 4AD. 4AD (Figure 4) and baf A130 arrest AQP2 in the Golgi compartment. However, we have not detected PKA and Golgi markers in the same preparations of immunoisolated AQP2-bearing vesicles.22,43 Thus, if PKA plays a role in phosphorylating S256 in the presence of 4AD and forskolin, it occurs outside the Golgi. In line, in IMCD cells AVP initiates activation of a perinuclear pool of PKA co-localizing with AQP2, apparently outside the Golgi, prior to mediating activation of PKA in other compartments.22 Alternatively, the S256 phosphorylation may be catalyzed by kinases other than PKA. One candidate is Golgi casein kinase. It is located within the Golgi and has been suggested to phosphorylate AQP2 in this compartment (Figure 5).44 Other candidates are casein kinase II and protein kinase Cα, which phosphorylates S256 in vitro.15,45
In IMCD cells, 4AD does not affect the phosphorylations at S261 and S269 (Figures 3 and 7), suggesting that the responsible kinases (e.g., p38 mitogen-activated protein kinases or Jun kinase19) are insensitive to 4AD or that they catalyze the phosphorylations independently from V-ATPase activity. Because 4AD inhibits only the S256 phosphorylation, our data corroborate the many earlier studies in providing further evidence that the S256 phosphorylation is the crucial trigger for the membrane insertion of AQP2.7–9,15,25 Our data also show that the cAMP-dependent dephosphorylation at S261 and the phosphorylation at S269 are independent from the S256 phosphorylation by PKA. Tamma et al. found that the cAMP/PKA-dependent S261 phosphorylation also occurs in the AQP2-S256A mutant, which cannot be phosphorylated at S256, and that forskolin induces dephosphorylation of S261 in this mutant.18 However, our data seem in contrast to studies by Hoffert and coworkers, who had observed that cAMP-stimulated and PKA-dependent phosphorylation of S256 precedes and primes for phosphorylation of S269.15 The reason for the discrepancy is unclear. The residual phosphorylation of S256 in the presence of 4AD and forskolin (Figure 7B) may be sufficient to facilitate phosphorylation of S269. Alternatively, S269 phosphorylation may occur independently from S256 phosphorylation, and location may play a role: Because the combination of 4AD and forskolin maintains AQP2 mainly inside IMCD cells (Figure 5), S269 may be accessible for phosphorylation at a different location compared with the normal situation, in which forskolin stimulates its phosphorylation at the plasma membrane. This is underlined by immunofluorescence microscopic analysis (Figure 3). Under resting conditions, AQP2-pS269 is hardly detectable, whereas in the presence of forskolin alone, AQP2-pS269 is almost exclusively located at the plasma membrane, supporting the proposed inhibitory role of pS269 for endocytosis of AQP2.15 In the presence of both 4AD and forskolin, AQP2-pS269 is detectable mainly intracellularly; the observed arrest of AQP2 in Golgi compartments in the presence of 4AD (Figure 5) suggests that S269 phosphorylation could be catalyzed there.
So far, baf A1 is widely used to block V-ATPases in studies of protein and vesicle trafficking. However, baf A1 not only inhibits V-ATPase but also functions as an ionophore that causes influx of potassium into mitochondria. This can be lethal for various cells, including cancer and osteoclast-like cells,46–48 but most importantly this function blurs interpretation of experimental data because intracellular ion homeostasis is disturbed. We are not aware of such effects of 4AD, suggesting that 4AD and other novel V-ATPase inhibitors, such as new derivatives of baf A1 that selectively inhibit V-ATPases,49 represent alternatives to baf A1 as molecular tools for studying cellular trafficking processes.
4AD and structurally related compounds were suggested for treatment of bone resorption disorders (e.g., osteopetrosis) and various cancers.26,49,50 However, because of the vital role of V-ATPases in normal physiology, adverse effects on blocking of V-ATPase may be expected. Hence, it is unclear whether such candidates reach the clinic. With regard to the work presented here, our identification of 4AD confirms the feasibility of our approach to discover new candidate molecules for further development into drugs for the treatment of diseases associated with dysregulated AVP-mediated water reabsorption (e.g., heart failure or syndrome of inappropriate antidiuretic hormone secretion).
With fluconazole, we discovered a small molecule targeting apparently a yet unknown player in the control of AQP2 (Figure 8). In addition, we identified three further inhibitors of the AQP2 redistribution, which we have not yet characterized but which will potentially identify additional novel proteins controlling AQP2 (compound IDs 212678, 212634 and 202336) (Table 1). Moreover, through screening further small-molecule libraries covering the chemical space more extensively, our approach has the potential for discovering further molecules and their targets controlling AQP2.
In summary, our screening lays the foundation for uncovering molecular mechanisms directing AQP2. In addition, identified molecules may constitute novel tools to study cellular trafficking processes and may pave the way to innovative treatments of diseases associated with disturbances of AVP-mediated water reabsorption.
Concise Methods
Materials and Reagents
AVP was synthesized by M. Beyermann, Leibniz-Institut für Molekulare Pharmakologie (Berlin, Germany), and forskolin was from BIOLOG Life Sciences Institute (Bremen, Germany). All other chemicals, if not stated otherwise, were purchased from Sigma-Aldrich (Munich, Germany).
Cell Culture
Primary rat IMCD cells were cultured as described previously.19,22 Mouse collecting-duct cells stably expressing human AQP2 (MCD4 cell line) were grown in Dulbecco’s modified Eagle medium: Nutrient Mixture F-12 (DMEM/F-12) 1:1 with GlutaMAX (catalog number 31331–028; Gibco) supplemented with 5% FBS, 5 µM dexamethasone, 100 IU/ml penicillin, and 100 µg/ml streptomycin.20
High-Throughput Screening
The ChemBioNet compound collection at the Leibniz-Institut für Molekulare Pharmakologie was purchased from ChemDiv (San Diego, CA). All library compounds are accessible at www.fmp-berlin.de, and further information on every compound is publically available at http://www.ncbi.nlm.nih.gov/pccompound. All dispensing and washing steps were performed using the EL406 washer/dispenser combination (BioTek Instruments, Bad Friedrichshall, Germany). The screen was performed in a 384-well plate format, with each plate containing 32 control wells (16 with forskolin alone and 16 with a mixture of forskolin and the PKA inhibitor, H89) (Supplemental Figure 2). MCD4 cells were seeded and after 24 hours incubated with each of the 17,700 library compounds (40 µM, 2 hours) or H89 (30 µM, 2 hours) and stimulated with forskolin (10 µM within the last 20 minutes). The cells were fixed with paraformaldehyde (2%), and AQP2 was detected using rabbit anti-AQP2 antibody H2751 and Cy5-coupled secondary antirabbit antibody (Dianova, Hamburg, Germany). Nuclei were stained with DAPI, and F-actin was labeled with TRITC-phalloidin.52 Fluorescence signals were visualized with an automated ArrayScan VTI HCS reader microscope (Cellomics, ThermoFisher, Pittsburgh, PA) fitted with a 10× objective, in which the microscope automatically focused on the DAPI-stained nuclei. The localization of AQP2 was assessed by CytoCellMemTrans V3 application software (Thermo Fisher Scientific Inc., Waltham, MA). Automated object recognition of nucleus and plasma membrane was optimized on the basis of F-actin and DAPI staining. From these objects a cytosolic object mask with a variable distance from the nucleus was defined. AQP2 localization was semi-quantitatively expressed as the ratio of plasma membrane to intracellular fluorescence signal intensities, as described previously.11,34,52 On the basis of the controls (forskolin alone or in combination with 4AD; see also Supplemental Figure 2) ratios <1.2 were defined as a predominant intracellular localization of AQP2. Acquired fluorescence signal data were processed with Pipeline Pilot software (Accelrys, Inc.; San Diego, CA), integrating the obtained information from this screen with compound details from the library, such as false-positive activity in previous screening assays.
Immunofluorescence Microscopy and Visualization of Acidic Intracellular Vesicles
MCD4 and IMCD cells were seeded on 12-mm cover slides. Immunofluorescence microscopy was carried out as described (LSM 710; Carl Zeiss MicroImaging, Jena, Germany).19,22 In brief, AQP2 was detected employing primary anti-AQP2 H27 and Cy3-coupled secondary donkey antirabbit antibodies or with goat polyclonal anti-AQP2 (1:100; C-17, Santa Cruz Biotechnology, Inc.) and Cy2-coupled secondary donkey antigoat antibodies.19,22 ERGIC-53/p58 and Golgi 58K protein were detected with polyclonal antirabbit (1:500; Sigma-Aldrich, Munich, Germany) and monoclonal mouse (1:100; Abcam; Cambridge, United Kingdom) antibodies, respectively, and Cy3-coupled polyclonal donkey antirabbit antibodies. AQP2-pS256 was detected with phospho-specific custom-made polyclonal rabbit antibody (Eurogentec Deutschland GmbH, Germany; 1:200 and 1:25, respectively) and antirabbit Cy3-coupled secondary antibodies.19,25 AQP2-pS269 was detected with phospho-specific primary antibody (p112–269, Phosphosolutions, Aurora, CO) and antirabbit Cy3-coupled secondary antibodies. All secondary antibodies were purchased from Dianova (Hamburg, Germany) and used in a dilution of 1:600.
For co-localization studies of AQP2 with ERGIC-53/p58 and Golgi 58K, ZEN2010 software (Carl Zeiss, Jena, Germany) was used. Co-localizations were analyzed in defined regions of interest (i.e., the intracellular region between the nucleus and the plasma membrane).
LysoTracker RED DND-99 (Invitrogen) selectively accumulates in acidic intracellular vesicles. When pH levels shift to >6.5–7, LysoTracker RED DND-99 protonation and retention are impaired, indicated by decreased fluorescence signal intensity (Figure 4B). MCD4 cells were seeded on 30-mm cover slides and incubated with LysoTracker RED DND-99 (75 nM, 30 minutes, 37°C) and washed twice with Dulbecco PBS. Incubation with 4AD or baf A1 with Hoechst 33258 (5 µM, nuclei staining) was carried out simultaneously. Cover slides were transferred to a custom-made cuvette, and fluorescence signals were evaluated by confocal laser-scanning microscopy (LSM 510 META microscope, Carl Zeiss, Jena, Germany). Mean fluorescence signal intensities of LysoTracker RED DND-99 were measured in 10 fields per condition using ImageJ software.
Western Blotting, cAMP Measurements by Radioimmunoassay, Biotinylation of Cell Surface Proteins, and PKA Activity Assay
Western blotting was carried out as described elsewhere.19,22 AQP2 was detected with goat polyclonal anti-AQP2 antibody C-17 (see above). AQP2-pS261 was detected with phospho-specific polyclonal phospho-S261 rabbit antibody (Abcam; Cambridge, UK). AQP2-pS256 was detected with phospho-specific custom-made polyclonal rabbit antibody (see above),19,25 and AQP2-pS269 with a commercially available antibody (Phosphosolutions, see above). We detected α-tubulin with a CalBiochem antibody (CP06). Secondary antibodies, all coupled to horseradish peroxidase, were purchased from Dianova. Signals were visualized by utilizing chemiluminescence, i.e., incubation of the Western blot membranes with Lumi-Light Western blotting substrate solution (Roche Applied Science) and imaging at the Lumi Imager F1 (Roche Applied Science). Protein amounts were semi-quantified by densitometry (LumiAnalyst 3.0 software; Roche Diagnostics GmbH, Mannheim, Germany). Determination of cAMP levels by RIA, cell-surface biotinylation assays19,22,53 and analysis of PKA activity using Promega’s PepTag A1 peptide assay kit54 were carried out as described.
Statistical Analyses
Statistical analyses of data were carried out with GraphPad Prism 5.0 software using a one-way ANOVA test combined with a Bonferroni post hoc comparison test; P<0.05 was considered to represent statistically significant differences. All values are expressed as mean ± SEM.
Disclosures
None.
Acknowledgments
We thank Andrea Geelhaar, Beate Eisermann, Aline Kirschner, Christoph Erdmann, and Carola Seyffarth for excellent technical assistance. This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG KL1415/3-2 and KL1415/4-2) and the GoBio program of the Bundesministerium für Bildung und Forschung (grant 0315516). P.M.T.D. is the recipient of the VICI grant 865.07.002 of the Netherlands Organization for Scientific research (NWO.). This study was supported by this NWO grant to P.M.T.D. and a Marie Curie Intra European Fellowship grant (272375) to C.T.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “Aquaporin-2 Inhibitors: Fishing in the Chemical Pool,” on pages 685–687.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2012030295/-/DCSupplemental.
References
- 1.Nedvetsky PI, Tamma G, Beulshausen S, Valenti G, Rosenthal W, Klussmann E: Regulation of aquaporin-2 trafficking. Handb Exp Pharmacol: 133–157, 2009 [DOI] [PubMed]
- 2.Brown D, Bouley R, Păunescu TG, Breton S, Lu HA: New insights into the dynamic regulation of water and acid-base balance by renal epithelial cells. Am J Physiol Cell Physiol 302: C1421–C1433, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deen PM, Knoers NV: Vasopressin type-2 receptor and aquaporin-2 water channel mutants in nephrogenic diabetes insipidus. Am J Med Sci 316: 300–309, 1998 [DOI] [PubMed] [Google Scholar]
- 4.Bichet DG, Oksche A, Rosenthal W: Congenital nephrogenic diabetes insipidus. J Am Soc Nephrol 8: 1951–1958, 1997 [DOI] [PubMed] [Google Scholar]
- 5.Wesche D, Deen PM, Knoers NV: Congenital nephrogenic diabetes insipidus: The current state of affairs. Pediatr Nephrol 27: 2183–2204, 2012 [DOI] [PubMed] [Google Scholar]
- 6.Schrier RW: Use of diuretics in heart failure and cirrhosis. Semin Nephrol 31: 503–512, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Katsura T, Gustafson CE, Ausiello DA, Brown D: Protein kinase A phosphorylation is involved in regulated exocytosis of aquaporin-2 in transfected LLC-PK1 cells. Am J Physiol 272: F817–F822, 1997 [PubMed] [Google Scholar]
- 8.Fushimi K, Sasaki S, Marumo F: Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J Biol Chem 272: 14800–14804, 1997 [DOI] [PubMed] [Google Scholar]
- 9.van Balkom BW, Savelkoul PJ, Markovich D, Hofman E, Nielsen S, van der Sluijs P, Deen PM: The role of putative phosphorylation sites in the targeting and shuttling of the aquaporin-2 water channel. J Biol Chem 277: 41473–41479, 2002 [DOI] [PubMed] [Google Scholar]
- 10.Kuwahara M, Asai T, Terada Y, Sasaki S: The C-terminal tail of aquaporin-2 determines apical trafficking. Kidney Int 68: 1999–2009, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Klussmann E, Maric K, Wiesner B, Beyermann M, Rosenthal W: Protein kinase A anchoring proteins are required for vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J Biol Chem 274: 4934–4938, 1999 [DOI] [PubMed] [Google Scholar]
- 12.Hoffert JD, Pisitkun T, Wang G, Shen RF, Knepper MA: Quantitative phosphoproteomics of vasopressin-sensitive renal cells: regulation of aquaporin-2 phosphorylation at two sites. Proc Natl Acad Sci U S A 103: 7159–7164, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fenton RA, Moeller HB, Hoffert JD, Yu MJ, Nielsen S, Knepper MA: Acute regulation of aquaporin-2 phosphorylation at Ser-264 by vasopressin. Proc Natl Acad Sci U S A 105: 3134–3139, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moeller HB, Praetorius J, Rützler MR, Fenton RA: Phosphorylation of aquaporin-2 regulates its endocytosis and protein-protein interactions. Proc Natl Acad Sci U S A 107: 424–429, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffert JD, Fenton RA, Moeller HB, Simons B, Tchapyjnikov D, McDill BW, Yu MJ, Pisitkun T, Chen F, Knepper MA: Vasopressin-stimulated increase in phosphorylation at Ser269 potentiates plasma membrane retention of aquaporin-2. J Biol Chem 283: 24617–24627, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moeller HB, Knepper MA, Fenton RA: Serine 269 phosphorylated aquaporin-2 is targeted to the apical membrane of collecting duct principal cells. Kidney Int 75: 295–303, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rice WL, Zhang Y, Chen Y, Matsuzaki T, Brown D, Lu HA: Differential, phosphorylation dependent trafficking of AQP2 in LLC-PK1 cells. PLoS ONE 7: e32843, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamma G, Robben JH, Trimpert C, Boone M, Deen PM: Regulation of AQP2 localization by S256 and S261 phosphorylation and ubiquitination. Am J Physiol Cell Physiol 300: C636–C646, 2011 [DOI] [PubMed] [Google Scholar]
- 19.Nedvetsky PI, Tabor V, Tamma G, Beulshausen S, Skroblin P, Kirschner A, Mutig K, Boltzen M, Petrucci O, Vossenkämper A, Wiesner B, Bachmann S, Rosenthal W, Klussmann E: Reciprocal regulation of aquaporin-2 abundance and degradation by protein kinase A and p38-MAP kinase. J Am Soc Nephrol 21: 1645–1656, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iolascon A, Aglio V, Tamma G, D'Apolito M, Addabbo F, Procino G, Simonetti MC, Montini G, Gesualdo L, Debler EW, Svelto M, Valenti G: Characterization of two novel missense mutations in the AQP2 gene causing nephrogenic diabetes insipidus. Nephron Physiol 105: p33–41, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Choi M, Eulenberg C, Rolle S, von Kries JP, Luft FC, Kettritz R: The use of small molecule high-throughput screening to identify inhibitors of the proteinase 3-NB1 interaction. Clin Exp Immunol 161: 389–396, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stefan E, Wiesner B, Baillie GS, Mollajew R, Henn V, Lorenz D, Furkert J, Santamaria K, Nedvetsky P, Hundsrucker C, Beyermann M, Krause E, Pohl P, Gall I, MacIntyre AN, Bachmann S, Houslay MD, Rosenthal W, Klussmann E: Compartmentalization of cAMP-dependent signaling by phosphodiesterase-4D is involved in the regulation of vasopressin-mediated water reabsorption in renal principal cells. J Am Soc Nephrol 18: 199–212, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Murray AJ: Pharmacological PKA inhibition: All may not be what it seems. Sci Signal 1: re4, 2008 [DOI] [PubMed] [Google Scholar]
- 24.Davies SP, Reddy H, Caivano M, Cohen P: Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351: 95–105, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trimpert C, van den Berg DT, Fenton RA, Klussmann E, Deen PM: Vasopressin increases S261 phosphorylation in AQP2-P262L, a mutant in recessive nephrogenic diabetes insipidus. Nephrol Dial Transplant 27: 4389–4397, 2012 [DOI] [PubMed] [Google Scholar]
- 26.Sørensen MG, Henriksen K, Neutzsky-Wulff AV, Dziegiel MH, Karsdal MA: Diphyllin, a novel and naturally potent V-ATPase inhibitor, abrogates acidification of the osteoclastic resorption lacunae and bone resorption. J Bone Miner Res 22: 1640–1648, 2007 [DOI] [PubMed] [Google Scholar]
- 27.Clague MJ, Urbé S, Aniento F, Gruenberg J: Vacuolar ATPase activity is required for endosomal carrier vesicle formation. J Biol Chem 269: 21–24, 1994 [PubMed] [Google Scholar]
- 28.Bowman EJ, Siebers A, Altendorf K: Bafilomycins: A class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci U S A 85: 7972–7976, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown D, Breton S, Ausiello DA, Marshansky V: Sensing, signaling and sorting events in kidney epithelial cell physiology. Traffic 10: 275–284, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gustafson CE, Katsura T, McKee M, Bouley R, Casanova JE, Brown D: Recycling of AQP2 occurs through a temperature- and bafilomycin-sensitive trans-Golgi-associated compartment. Am J Physiol Renal Physiol 278: F317–F326, 2000 [DOI] [PubMed] [Google Scholar]
- 31.Moriyama Y, Nelson N: H+-translocating ATPase in Golgi apparatus. Characterization as vacuolar H+-ATPase and its subunit structures. J Biol Chem 264: 18445–18450, 1989 [PubMed] [Google Scholar]
- 32.Lepesheva GI, Waterman MR: Sterol 14alpha-demethylase (CYP51) as a therapeutic target for human trypanosomiasis and leishmaniasis. Curr Top Med Chem 11: 2060–2071, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niwa T, Shiraga T, Takagi A: Effect of antifungal drugs on cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 activities in human liver microsomes. Biol Pharm Bull 28: 1805–1808, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Henn V, Edemir B, Stefan E, Wiesner B, Lorenz D, Theilig F, Schmitt R, Vossebein L, Tamma G, Beyermann M, Krause E, Herberg FW, Valenti G, Bachmann S, Rosenthal W, Klussmann E: Identification of a novel A-kinase anchoring protein 18 isoform and evidence for its role in the vasopressin-induced aquaporin-2 shuttle in renal principal cells. J Biol Chem 279: 26654–26665, 2004 [DOI] [PubMed] [Google Scholar]
- 35.Okutsu R, Rai T, Kikuchi A, Ohno M, Uchida K, Sasaki S, Uchida S: AKAP220 colocalizes with AQP2 in the inner medullary collecting ducts. Kidney Int 74: 1429–1433, 2008 [DOI] [PubMed] [Google Scholar]
- 36.Skroblin P, Grossmann S, Schäfer G, Rosenthal W, Klussmann E: Mechanisms of protein kinase A anchoring. Int Rev Cell Mol Biol 283: 235–330, 2010 [DOI] [PubMed] [Google Scholar]
- 37.Horner A, Goetz F, Tampé R, Klussmann E, Pohl P: Mechanism for targeting the A-kinase anchoring protein AKAP18δ to the membrane. J Biol Chem 287: 42495–42501, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Troger J, Moutty MC, Skroblin P, Klussmann E: A-kinase anchoring proteins as potential drug targets. Br J Pharmacol 166: 420–433, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hundsrucker C, Skroblin P, Christian F, Zenn HM, Popara V, Joshi M, Eichhorst J, Wiesner B, Herberg FW, Reif B, Rosenthal W, Klussmann E: Glycogen synthase kinase 3beta interaction protein functions as an A-kinase anchoring protein. J Biol Chem 285: 5507–5521, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hurtado-Lorenzo A, Skinner M, El Annan J, Futai M, Sun-Wada GH, Bourgoin S, Casanova J, Wildeman A, Bechoua S, Ausiello DA, Brown D, Marshansky V: V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nat Cell Biol 8: 124–136, 2006 [DOI] [PubMed] [Google Scholar]
- 41.Bjarnadottir U, Nielsen JE: Calculating pKa values in the cAMP-dependent protein kinase: The effect of conformational change and ligand binding. Protein Sci 19: 2485–2497, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carrey EA: A protonated histidine residue in a phosphorylation site for cyclic AMP-dependent protein kinase. Comparison of a synthetic peptide with the exposed linking region in the multienzyme polypeptide CAD. Biochem J 287: 791–795, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nedvetsky PI, Stefan E, Frische S, Santamaria K, Wiesner B, Valenti G, Hammer JA, 3rd, Nielsen S, Goldenring JR, Rosenthal W, Klussmann E: A Role of myosin Vb and Rab11-FIP2 in the aquaporin-2 shuttle. Traffic 8: 110–123, 2007 [DOI] [PubMed] [Google Scholar]
- 44.Procino G, Carmosino M, Marin O, Brunati AM, Contri A, Pinna LA, Mannucci R, Nielsen S, Kwon TH, Svelto M, Valenti G: Ser-256 phosphorylation dynamics of Aquaporin 2 during maturation from the ER to the vesicular compartment in renal cells. FASEB J 17: 1886–1888, 2003 [DOI] [PubMed] [Google Scholar]
- 45.Brown D, Hasler U, Nunes P, Bouley R, Lu HA: Phosphorylation events and the modulation of aquaporin 2 cell surface expression. Curr Opin Nephrol Hypertens 17: 491–498, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teplova VV, Tonshin AA, Grigoriev PA, Saris NE, Salkinoja-Salonen MS: Bafilomycin A1 is a potassium ionophore that impairs mitochondrial functions. J Bioenerg Biomembr 39: 321–329, 2007 [DOI] [PubMed] [Google Scholar]
- 47.Ohta T, Arakawa H, Futagami F, Fushida S, Kitagawa H, Kayahara M, Nagakawa T, Miwa K, Kurashima K, Numata M, Kitamura Y, Terada T, Ohkuma S: Bafilomycin A1 induces apoptosis in the human pancreatic cancer cell line Capan-1. J Pathol 185: 324–330, 1998 [DOI] [PubMed] [Google Scholar]
- 48.Okahashi N, Nakamura I, Jimi E, Koide M, Suda T, Nishihara T: Specific inhibitors of vacuolar H(+)-ATPase trigger apoptotic cell death of osteoclasts. J Bone Miner Res 12: 1116–1123, 1997 [DOI] [PubMed] [Google Scholar]
- 49.Pérez-Sayáns M, Somoza-Martín JM, Barros-Angueira F, Rey JM, García-García A: V-ATPase inhibitors and implication in cancer treatment. Cancer Treat Rev 35: 707–713, 2009 [DOI] [PubMed] [Google Scholar]
- 50.Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson JP, Keeling DJ, Andersson AK, Wallbrandt P, Zecca L, Notarangelo LD, Vezzoni P, Villa A: Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet 25: 343–346, 2000 [DOI] [PubMed] [Google Scholar]
- 51.Maric K, Oksche A, Rosenthal W: Aquaporin-2 expression in primary cultured rat inner medullary collecting duct cells. Am J Physiol 275: F796–F801, 1998 [DOI] [PubMed] [Google Scholar]
- 52.Klussmann E, Tamma G, Lorenz D, Wiesner B, Maric K, Hofmann F, Aktories K, Valenti G, Rosenthal W: An inhibitory role of Rho in the vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J Biol Chem 276: 20451–20457, 2001 [DOI] [PubMed] [Google Scholar]
- 53.Haas AK, Kleinau G, Hoyer I, Neumann S, Furkert J, Rutz C, Schülein R, Gershengorn MC, Krause G: Mutations that silence constitutive signaling activity in the allosteric ligand-binding site of the thyrotropin receptor. Cell Mol Life Sci 68: 159–167, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Christian F, Szaszák M, Friedl S, Drewianka S, Lorenz D, Goncalves A, Furkert J, Vargas C, Schmieder P, Götz F, Zühlke K, Moutty M, Göttert H, Joshi M, Reif B, Haase H, Morano I, Grossmann S, Klukovits A, Verli J, Gáspár R, Noack C, Bergmann M, Kass R, Hampel K, Kashin D, Genieser HG, Herberg FW, Willoughby D, Cooper DM, Baillie GS, Houslay MD, von Kries JP, Zimmermann B, Rosenthal W, Klussmann E: Small molecule AKAP-protein kinase A (PKA) interaction disruptors that activate PKA interfere with compartmentalized cAMP signaling in cardiac myocytes. J Biol Chem 286: 9079–9096, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]