

Abstract
Serum phosphate independently predicts cardiovascular mortality in the general population and CKD, even when levels are in the normal range. Associations between serum phosphate, arterial stiffness, and left ventricular (LV) mass suggest a possible pathophysiological mechanism, potentially mediated by the phosphaturic hormone fibroblast growth factor-23 (FGF-23). To what extent the phosphate binder sevelamer modulates these effects is not well understood. In this single-center, randomized, double-blind, placebo-controlled trial, we enrolled 120 patients with stage 3 nondiabetic CKD. After a 4-week open-label run-in period, during which time all patients received sevelamer carbonate, we randomly assigned 109 patients to sevelamer (n=55) or placebo (n=54) for an additional 36 weeks. We assessed LV mass and systolic and diastolic function with cardiovascular magnetic resonance imaging and echocardiography, and we assessed arterial stiffness by carotid–femoral pulse wave velocity. The mean age was 55 years, and the mean eGFR was 50 ml/min per 1.73 m2. After 40 weeks, we found no statistically significant differences between sevelamer and placebo with regard to LV mass, systolic and diastolic function, or pulse wave velocity. Only 56% of subjects took ≥80% of prescribed therapy; in this compliant subgroup, treatment with sevelamer associated with lower urinary phosphate excretion and serum FGF-23 but not serum phosphate, klotho, vitamin D, or cardiovascular-related outcomes of interest. In conclusion, this study does not provide evidence that sevelamer carbonate improves LV mass, LV function, or arterial stiffness in stage 3 nondiabetic CKD.
Over the last decade, phosphate has emerged as an important cardiovascular risk factor.1 Serum phosphate within the normal range independently predicted mortality in the Framingham Offspring Study.2 CKD is the most common cause of abnormal phosphate handling, and the relationship between phosphate and adverse cardiovascular outcome was first identified in hemodialysis patients.3,4 Additional studies have shown this relationship in early stage CKD,5,6 type 2 diabetes mellitus,7 and patients with coronary artery disease and normal renal function.8 The strong association between serum phosphate and increased arterial stiffness9 as well as increased left ventricular (LV) mass10–12 suggests a potential mechanism, although the cellular processes remain unclear. The ability of the kidneys to excrete phosphate is reduced at a GFR below 60 ml/min per 1.73 m2. Serum phosphate, however, remains within the normal range until the GFR falls below 30 ml/min per 1.73 m2 because of increased production of the phosphatonins parathyroid hormone (PTH) and fibroblast growth factor-23 (FGF-23), which promote urinary phosphate excretion.13,14 Recent data have linked FGF-23 to increases in LV mass and development of LV hypertrophy (LVH).15
The increased cardiovascular risk observed in CKD cannot be fully attributed to conventional atherosclerotic risk factors; it is likely that nonatheromatous pathogenic processes, such as increased arterial stiffness and structural heart disease, are the major contributors to this adverse outcome.16 Heart failure and arrhythmias are the most common cause of cardiovascular death, and the current paradigm is that these events are a consequence of LV disease, predominantly LVH and fibrosis.17 Although the higher cardiovascular risk conferred by the presence of LVH is well recognized, it is becoming increasingly accepted that no biologic dichotomy exists; LV mass as a continuous variable has a graded relationship with cardiovascular risk.18 Furthermore, given that over 70% of incident dialysis patients have established LVH,19 an understanding of the early pathophysiology of increased LV mass and LVH is essential for designing novel therapeutic strategies to attenuate cardiovascular disease in CKD.
We hypothesized that lowering gastrointestinal phosphate absorption through use of the noncalcium-based phosphate binder sevelamer carbonate would reduce serum levels of phosphatonins such as FGF-23, thereby reducing LV mass and arterial stiffness as well as improving LV systolic and diastolic function in patients with early stage CKD.
Results
Baseline
A total of 120 subjects was recruited (Figure 1); 11 subjects were withdrawn during run-in, leaving 109 subjects for randomization to sevelamer (n=55) or placebo (n=54). Renal disease was diagnosed by biopsy in 52% and ultrasound in 34%. Patients were well matched at baseline (Table 1). Mean serum phosphate, mean serum calcium, and median PTH were within normal limits along with serum klotho and 1,25-hydroxyvitamin D. Serum FGF-23 and urinary fractional excretion of phosphate were both elevated. BP was well controlled in both groups. Baseline aortic calcification was present in 45% (35% placebo group versus 55% treatment group, P=0.06). Mean LV mass, LV systolic and diastolic function, and volumes were within normal limits. Only one patient had LVH.
Figure 1.

Study consort diagram. Of 1297 patients screened, 120 were recruited into the study. During the 4-week run-in phase 11 patients were withdrawn, leaving 109 to be randomized to treatment (n=55) or placebo (n=54). During the blinded treatment phase an additional five patients were withdrawn from the treatment group and seven from the placebo group. In total, 97 patients completed follow-up to 40 weeks. GI, gastrointestinal.
Table 1.
Baseline demographics
| Placebo (n=54) | Sevelamer (n=55) | |
|---|---|---|
| Age (yr) | 55±14 | 55±13 |
| Men (%) | 28 (52) | 32 (58) |
| Renal diagnosis (%) | ||
| Hypertensive nephropathy | 10 (19) | 3 (6)a |
| IgA nephropathy | 3 (6) | 7 (13) |
| Reflux nephropathy | 4 (7) | 7 (13) |
| Adult polycystic kidney disease | 8 (15) | 4 (7) |
| Systemic vasculitis | 5 (9) | 4 (7) |
| Lupus nephritis | 0 (0) | 3 (6) |
| FSGS | 4 (7) | 3 (6) |
| Nephrectomy for neoplasm | 2 (4) | 3 (6) |
| Other GN | 2 (4) | 3 (6) |
| Other | 15 (28) | 16 (29) |
| Unknown | 1 (2) | 2 (4) |
| Previous cardiovascular disease (%) | 5 (9) | 6 (11) |
| Smoking status (%) | ||
| Current smoker | 8 (15) | 8 (15) |
| Previous smoker | 25 (46) | 32 (58) |
| Medication (%) | ||
| Angiotensin-converting enzyme inhibitor | 32 (59) | 25 (46) |
| Angiotensin receptor blocker | 16 (30) | 13 (24) |
| Aspirin | 9 (17) | 10 (18) |
| Aldosterone antagonist | 1 (2) | 1 (2) |
| β-blocker | 12 (22) | 6 (11) |
| Calcium channel blocker | 15 (28) | 14 (26) |
| Diuretic | 19 (35) | 10 (18) |
| Statin | 26 (48) | 24 (44) |
| Other lipid lowering therapy | 3 (6) | 3 (6) |
| Prednisolone | 8 (15) | 9 (16) |
| Immunosuppressant | 11 (20) | 11 (20) |
| Body mass index (kg/m2) | 29.7±6.7 | 28.6±4.3 |
| Hip to waist ratio | 1.08±0.10 | 1.08±0.09 |
| Aortic calcification present (%) | 19 (35) | 30 (55) |
Data are mean ± SD, median (interquartile range), or frequency (%) analyzed using independent samples t or chi-squared test.
P<0.05.
Week 4 Data
After 4 weeks of open-label treatment with sevelamer, significant reductions were seen in serum cholesterol, LDL, serum urate, urinary albumin, creatinine ratio, and brachial systolic BP (Table 2). There was a small increase in serum phosphate (3.16±0.53 versus 3.25±0.50 mg/dl, P<0.05). Levels of 1,25-dihydroxyvitamin D and 25-hydroxyvitamin D decreased after 4 weeks, but this result was not accompanied by any change in FGF-23, klotho, or urinary phosphate excretion. There were no changes in central BP or indices of arterial stiffness. In subgroup analyses, the tertile of patients with higher FGF-23 at baseline (n=38) showed a modest but significant decrease in serum FGF-23 after 4 weeks of sevelamer treatment (from 94 [83–117] to 86 [77–98] pg/ml, P=0.001). Similar results were shown in the tertile of patients with higher serum phosphate at baseline (n=36, mean serum phosphate=3.7±0.3 mg/dl, decrease in FGF-23 from 77 [68–110] to 73 [58–86] pg/ml, P=0.003). In the middle and lower tertiles of serum FGF-23 and phosphate, no changes in serum FGF-23 levels were detected over the 4-week run-in period.
Table 2.
Open-label data
| Week 0 | Week 4 | |
|---|---|---|
| Biochemical | ||
| eGFR (ml/min per 1.73 m2) | 50±13 | 49±13 |
| Phosphate (mg/dl) | 3.16±0.53 | 3.25±0.50a |
| PTH (pg/ml)b | 53.3 (38.5–71.2) | 55.0 (43.9–70.8) |
| FGF-23 (pg/ml)b | 68.8 (51.2–85.2) | 68.1 (52.5–85.7) |
| Klotho (pg/ml) | 936±403 | 945±359 |
| 1,25-dihydroxyvitamin D (pg/ml) | 29.1±12.1 | 26.2±9.7c |
| 25-hydroxyvitamin D (ng/ml) | 22.0±11.7 | 20.7±11.3c |
| Bicarbonate (mEq/L) | 26.7±2.9 | 27.0±2.8 |
| Total cholesterol (mg/dl) | 190±46 | 172±44d |
| LDL cholesterol (mg/dl) | 106±32 | 87±33d |
| Urate (mg/dl) | 6.98±1.87 | 6.66±1.63c |
| NT-proBNP (pg/ml)b | 720 (317–1637) | 677 (326–1491) |
| Urine | ||
| Albumin to creatinine ratio (mg/g)b | 50 (7–377) | 37 (17–413)c |
| Fractional excretion of phosphateb | 21.3 (12.5–32.3) | 19.7 (12.4–32.9) |
| Hemodynamics | ||
| Brachial systolic BP (mmHg) | 128±16 | 125±16a |
| Brachial diastolic BP (mmHg) | 71±11 | 71±10 |
| Brachial mean arterial pressure (mmHg) | 90±11 | 90±11 |
| Central systolic BP (mmHg) | 117±16 | 115±16 |
| Arterial measures | ||
| PWV (m/s)b | 9.0 (7.5–11.2) | 8.8 (7.5–10.6) |
| PWVadj (m/s)b | 9.1 (7.7–10.6) | 8.7 (7.6–10.3) |
| AIx75 | 19.7±11.9 | 19.8±10.8 |
Data are mean ± SD or median (interquartile range). Data were analyzed with paired t tests (n=112). NT-proBNP, N-terminal pro-brain natriuretic peptide; PWVadj, PWV adjusted for mean arterial pressure; AIx75, augmentation index adjusted to heart rate of 75 beats/min.
P<0.05.
Log-transformed before analysis.
P<0.005.
P<0.001.
Treatment Effects
Twelve patients (11%) did not complete follow-up to 40 weeks after randomization (seven patients in the placebo group and five patients in the sevelamer group, P=0.56) (Figure 1). Six patients declined continued participation because of difficulties with tablet frequency and a wish to discontinue study visits, four patients experienced persistent hypophosphatemia, and two patients were intolerant of medication. No patients died.
After 40 weeks of treatment with sevelamer carbonate, there were no significant changes in any biochemical parameters compared with placebo (Table 3). There were no differences in the primary end point of LV mass (Figure 2) or LV mass indexed to body surface area. There were no changes in any indices of LV systolic function, LV diastolic function, strain or twist, arterial stiffness or peripheral resting, or central and ambulatory BP. Bone density remained unaltered in both groups. When patients with aortic calcification were excluded from analyses, no differences were observed in LV mass (P=0.18), augmentation index (AIx; P=0.62), or pulse wave velocity (PWV; P=0.54) at 40 weeks.
Table 3.
Treatment effects
| Placebo (n=50) | Sevelamer (n=54) | Mean Difference in Change between Groups | 95% CI | |||
|---|---|---|---|---|---|---|
| Week 0 | Week 40 | Week 0 | Week 40 | |||
| Biochemical | ||||||
| eGFR (ml/min per 1.73 m2) | 49±13 | 50±14 | 49±13 | 48±14 | 1.2 | −1.3–3.6 |
| Bicarbonate (mEq/L) | 26.4±2.8 | 27.2±3.4 | 27.0±2.7 | 27.2±6.2 | 0.6 | −1.3–2.6 |
| Total cholesterol (mg/dl) | 181±42 | 170±46 | 193±50 | 166±54 | 18.7 | −1.9–39.3 |
| LDL cholesterol (mg/dl) | 105±36 | 100±42 | 106±35 | 92±39 | 9.2 | −2.6–21.1 |
| Phosphate (mg/dl) | 3.25±0.53 | 3.31±0.53 | 3.16±0.50 | 3.16±0.71 | 0.06 | −0.18–0.30 |
| Corrected calcium (mg/dl) | 8.80±0.40 | 8.76±0.32 | 8.88±0.36 | 8.84±0.32 | 0.00 | −0.14–0.15 |
| PTH (pg/ml)a | 54 (37–73) | 51 (39–72) | 52 (39–70) | 52 (35–75) | −3.1 | −10.4–4.3 |
| FGF-23 (pg/ml)a | 67.6 (51.1–87.7) | 63.6 (52.0–83.6) | 70.8 (52.5–83.0) | 65.9 (49.2–90.7) | 0.8 | −7.7–9.3 |
| Klotho (pg/ml) | 869±279 | 873±320 | 1001±500 | 980±533 | 40 | −58–139 |
| 1,25-dihydroxyvitamin D (pg/ml) | 28.8±12.7 | 26.1±10.8 | 28.5±10.3 | 27.3±10.4 | −3.6 | −13.8–6.7 |
| 25-hydroxyvitamin D (ng/ml) | 22.2±12.5 | 21.9±12.3 | 23.0±11.2 | 23.4±13.3 | −2.2 | −11.1–6.8 |
| NT-proBNP (pg/ml)a | 746 (343–1697) | 789 (394–1748) | 514 (309–1586) | 694 (283–1294) | −247 | −593–100 |
| Urate (mg/dl) | 6.89±1.77 | 6.96±1.71 | 6.96±1.78 | 6.91±1.73 | 0.12 | −0.29–0.53 |
| Urinary fractional excretion of phosphatea | 21.2 (12.6–28.3) | 19.8 (10.2–23.7) | 21.7 (12.6–33.7) | 17.7 (12.9–29.9) | 0.6 | −4.1–5.3 |
| Hemodynamics | ||||||
| Brachial systolic BP (mmHg) | 127±17 | 124±21 | 128±15 | 124±15 | 1.5 | −3.6–6.7 |
| Brachial mean arterial pressure (mmHg) | 89±12 | 87±13 | 92±10 | 89±11 | 0.1 | −3.8–4.0 |
| Central systolic BP (mmHg) | 115±18 | 113±21 | 119±15 | 114±16 | 2.8 | −2.7–7.8 |
| Ambulatory systolic BP (mmHg) | 125±15 | 123±16 | 124±10 | 122±13 | −0.5 | −4.4–3.5 |
| PWV (m/s)a | 9.3 (7.6–11.2) | 8.8 (7.9–10.6) | 8.5 (7.3–10.7) | 8.6 (7.5–10.7) | −0.1 | −0.5–0.3 |
| AIx75 | 18.9±12.0 | 20.1±13.2 | 21.4±10.9 | 20.8±12.2 | 1.8 | −1.1–4.6 |
| CMR datab | ||||||
| LV mass (g) | 99±31 | 99±30 | 102±29 | 101±27 | 0.8 | −2.1–3.7 |
| LV mass index (g/m2) | 51±13 | 51±12 | 52±13 | 52±13 | −0.07 | −1.71–1.58 |
| LV end systolic volume index (ml/m2) | 15±6 | 14±6 | 17±7 | 16±7 | 0.3 | −1.8–2.4 |
| LV ejection fraction (%) | 76±7 | 76±6 | 73±6 | 72±13 | 1.2 | −2.5–4.9 |
| Ascending aortic distensibility (×10−3 mmHg−1) | 2.34±1.79 | 2.40±1.49 | 2.54±1.64 | 2.70±1.88 | −0.27 | −0.67–0.12 |
| Diastolic function | ||||||
| Transmitral E/Aa | 0.96 (0.81–1.13) | 0.99 (0.81–1.17) | 0.93 (0.77–1.06) | 0.97 (0.73–1.16) | −0.04 | −0.14–0.06 |
| Mean e′ (m/s) | 9.3±3.4 | 9.1±2.8 | 8.6±2.6 | 8.8±2.6 | −0.4 | −1.1–0.3 |
| E/mean e′a | 7.3 (6.6–9.3) | 7.4 (6.5–9.3) | 7.8 (6.5–9.4) | 7.2 (6.2–9.4) | 0.2 | −0.6–1.0 |
| Left atrial volume index (ml/m2) | 28.9±8.2 | 26.4±7.3 | 27.3±8.1 | 25.7±8.1 | −1.0 | −3.9–1.9 |
| Echocardiographic strain | ||||||
| Global peak longitudinal systolic strain (%) | −19.9±2.6 | −20.8±2.0 | −19.2±2.6 | −19.5±2.8 | −0.5 | −1.6–0.5 |
| Global early diastolic strain rate (s−1) | 1.58±0.30 | 1.60±0.32 | 1.57±0.32 | 1.53±0.31 | 0.05 | −0.04–0.15 |
| Peak systolic torsion (°/cm) | 2.72±0.61 | 2.57±0.84 | 2.75±0.76 | 2.47±0.65 | −0.30 | −0.97–0.38 |
| Early diastolic untwist rate (°/s) | −129±28 | −115±43 | −129±46 | −135±37 | 20.8 | −12.4–54.0 |
| Mean bone mineral density (g/cm2) | ||||||
| Lumbar spine | 1.14±0.20 | 1.14±0.20 | 1.06±0.16 | 1.06±0.16 | 0.00 | −0.01–0.01 |
| Hips | 1.02±0.13 | 1.03±0.18 | 1.00±0.14 | 1.00±0.15 | 0.01 | −0.04–0.05 |
Data are mean ± SD or median (interquartile range). Repeated-measures ANOVA with time point (week 0 or 40) as the within-subjects factor and group (placebo or sevelamer) as the between-subjects factor tested the difference in change over time between groups. NT-proBNP, N-terminal pro-brain natriuretic peptide; AIx75, augmentation index adjusted to heart rate of 75 beats/min.
Log-transformed before analysis.
CMR data are available in 45 from placebo group and 45 from treatment group.
Figure 2.

After 40 weeks of treatment there was no significant difference in the change in LV mass between sevelamer and placebo groups. Data were analyzed with repeated-measures ANOVA (P=0.58).
A mean of 79±21% of tablets was taken during the run-in phase. During the blinded phase, there was no difference in compliance between sevelamer and placebo groups (80±20% versus 81±30%, P=0.82). Overall, 61 patients (56%) returned <20% of medication prescribed (Figure 3). A subgroup analysis of only these highly compliant subjects revealed significant decreases in FGF-23 (−8.9±19.8 versus 1.0±17.7 pg/ml, P<0.05) and urinary phosphate concentration (−7.4±13.3 versus 1.9±11.1 mg/dl, P<0.01) over 40 weeks in those patients receiving sevelamer compared with placebo but no differences in serum phosphate, klotho, or vitamin D. There were also no differences in BP, arterial stiffness, LV mass, or measures of LV systolic or diastolic function.
Figure 3.

Compliance with study medication was assessed through pill counts of returned medication. Compliance was low with only 56% of patients taking ≥80% of prescribed medication despite regular monitoring under controlled conditions.
Adverse Effects
During run-in, nine subjects (8%) developed hypophosphatemia (phosphate<2.5 mg/dl), necessitating a reduction in sevelamer dose to 800 mg with meals; one subject was withdrawn because of persistent hypophosphatemia despite dose reduction. Nine patients (8%) experienced dyspepsia, six patients (5%) developed constipation, and two patients (2%) complained of nausea and vomiting. Two patients were withdrawn because of persistence of adverse effects despite dose reduction (Figure 1). After randomization, another seven subjects developed hypophosphatemia (four patients in the treatment group and three patients in the placebo group, P=0.70) requiring dose reduction. Three subjects from the treatment group and one patient from the placebo group were withdrawn because of persistent hypophosphatemia. Twelve patients were hospitalized during their time in the study for unrelated noncardiovascular medical conditions (two patients during run-in, six patients from the treatment group, and four patients from the placebo group).
Reproducibility
Intraobserver variability for LV mass was low, with an intraclass correlation coefficient of 0.99 (95% confidence interval [CI]=0.98–1.00, P<0.01). Interstudy reproducibility for LV mass was also good (0.92, 95% CI=0.74–0.98, P<0.01).
Discussion
In patients with stage 3 nondiabetic CKD, 40 weeks of treatment with sevelamer carbonate resulted in no change in LV mass compared with placebo. Furthermore, there were no significant effects on arterial stiffness, wave reflection, or LV systolic and diastolic function. Although a small reduction in brachial systolic BP was observed after 4 weeks of open-label sevelamer, this finding did not persist in the double-blind, placebo-controlled phase. Despite repeated reminders and suggested methods to maximize compliance, adherence to medication was low, with only 56% of subjects taking ≥80% of study medication. Furthermore, several patients withdrew from the study because of difficulty with the frequency and tolerability of the study agents.
CKD is the most common condition associated with deranged phosphate homeostasis. Abnormal phosphate handling, coupled with phosphate-rich Western diets, may contribute to the elevated cardiovascular risk observed in this population.20 Observational studies have also linked phosphate to an increased risk of cardiovascular events8 and development of symptomatic heart failure in the general population2,11 together with a higher prevalence of vascular calcification.21–23 Cellular mechanisms of phosphate cardiovascular toxicity are unclear, but postprandial increases in serum phosphate seen after acute phosphate loading are associated with transient endothelial dysfunction.24 Studies in vitro have shown that vascular smooth muscle cells undergo osteogenic transformation in the presence of hyperphosphatemia, with upregulation of genes that promote matrix mineralization and vascular calcium deposition.25 The presence of vascular medial calcification is associated with increased arterial stiffness,26–28 which seems to be a principle driver of abnormalities in cardiac structure and function.16 Relationships have been shown between serum phosphate and increased arterial stiffness9 as well as increased LV mass10,11 and LVH,12 which are all independent predictors of adverse cardiovascular outcome and may be responsible for the excess cardiovascular morbidity and mortality observed in CKD.16 Furthermore, evidence from hemodialysis patients suggests that lowering of serum phosphate may reduce LV mass.29 These observations led to our hypothesis that, in early stage CKD, when phosphate levels are normal, reducing phosphate exposure using a noncalcium-based phosphate binder would have beneficial effects on both arterial stiffness and LV mass.
The absence of improvement in cardiac and vascular parameters after 40 weeks of treatment with sevelamer would suggest that our primary hypothesis has been refuted. Before this concept is discarded, however, a number of issues must be considered. We chose subjects with stage 3 CKD, because at this level of kidney function, cardiovascular risk is increased despite normal serum phosphate levels, and FGF-23 and urinary phosphate excretion are elevated.13 This result was, indeed, the case, although mean levels of klotho, 1,25-dihydroxyvitamin D, and PTH were normal, indicating that most subjects had only mildly impaired renal function and therefore, only a minimally impaired capacity to excrete a phosphate load. Furthermore, at this level of kidney function, use of phosphate binders reduces urinary phosphate excretion because of a poorly understood gut–renal axis, such that overall phosphate flux remains unchanged.30,31 Given the inaccuracy of the four-variable Modified Diet in Renal Disease equation in near-normal renal function, it is possible that a significant number of participants had true GFR values >60 ml/min per 1.73 m2.32 Despite this result, 86% of patients enrolled had histologic or radiologic evidence of kidney disease.
We selected patients with well controlled BP, and almost all had baseline LV mass within normal limits. We did this selection to examine whether our intervention would have beneficial cardiovascular effects over and above optimal conventional treatment, a concept validated in our research with spironolactone.33 This method could have potentially masked any benefits of reducing phosphate absorption. However, there is increasing evidence highlighting the prognostic value of LV mass as a continuous variable, even when values are within normal limits.18 Furthermore, over 70% of incident dialysis patients have established LVH, indicating that structural heart disease develops in the earlier stages of CKD.19 We did examine multiple indices of cardiovascular structure and function, which remained unchanged during the controlled phase of the study. We also used cardiovascular magnetic resonance imaging (CMR), a highly sensitive modality for detecting changes in LV mass that permits a smaller sample size compared with echocardiography.34 We cannot exclude the possibility that the cardiovascular effects of sevelamer were too small to detect with a sample size of 60 in each group. Our power calculations were based on changes in LV mass observed in a previous trial of the same size, duration, and design using spironolactone as the treatment agent.33 Although LV mass data were only available for 45 subjects in each arm of the study, detection of a change in LV mass of 8 g with an SD of the change in LV mass of 12 g was still possible with 88% power. Similarly, we chose 40 weeks for the study duration based on this previous trial, which showed notable changes in LV mass, arterial stiffness, and LV function over this time period.33 It is possible that 40 weeks is too short of a time period for changes in LV remodeling to manifest; a longer treatment duration with sevelamer may be required to exert detectable effects on cardiovascular structure and function, particularly with normal BP and LV mass at baseline. Our findings parallel those of the recently published PRIMO study (Paricalcitol Capsules Benefits in Renal Failure Induced Cardiac Morbidity in Subjects with Chronic Kidney Disease Stage 3/4), in which 227 patients with stages 3 and 4 CKD (estimated GFR [eGFR]=15–59 ml/min per 1.73 m2) failed to show any reductions in LV mass after 48 weeks of treatment with paricalcitol, a one-time daily vitamin D analog, compared with placebo.35 This result was despite a lower mean eGFR at baseline compared with our cohort and inclusion of patients with mild to moderate LVH at study entry.
Adherence to both active treatment and placebo was low, with 56% of subjects taking ≥80% of prescribed study medication. This result is likely to be an underestimate of the actual tablets taken given the known limitations of pill counts.36 This low level of compliance occurred despite regular encouragement and monitoring throughout the study period, and it may account for the modest effects on urinary phosphate excretion, cholesterol, and urate levels. Several participants were unable to maintain a regimen of two tablets three times a day, and this lack of maintenance contributed significantly to reasons given by patients when withdrawing from the study. A subgroup analysis of 61 subjects with ≥80% compliance indicated some biochemical changes in those patients taking sevelamer compared with placebo, with significant reductions in serum FGF-23 and urinary phosphate excretion; this result is consistent with findings from a recently published study by Block et al.31 examining biochemical effects of phosphate binder therapy in early stage CKD. There were, however, no changes in LV mass or arterial stiffness between these groups. Such subgroup analyses are likely underpowered to detect any differences in these parameters. Effective testing of our original hypothesis may, thus, require the introduction of a well tolerated one-time daily agent that significantly lowers gastrointestinal phosphate absorption and is associated with improved compliance. An analogous situation may have occurred with cholesterol lowering; early studies with cholesterol-lowering agents failed to show significant benefits,37 and the concept that cholesterol lowering was beneficial was not supported until statins were introduced.
Despite excluding patients with diabetes mellitus and including a study population with early stage CKD and no history of phosphate binder or vitamin D therapy, 48% of subjects had aortic calcification. This percentage is higher than the reported prevalence in nondiabetic stage 3 CKD.38 The use of more sensitive computed tomography would only have increased the proportion of patients with detectable calcification. It is possible that this high prevalence would have made it very difficult, if not impossible, to lower arterial stiffness and therefore, LV mass in these patients over 40 weeks. Treatment with sevelamer over 12 months has been shown to reduce progression of arterial calcification in hemodialysis patients but not cause regression.39,40 In a subgroup analysis that only included subjects without aortic calcification, there were no differences in arterial stiffness or LV mass between groups at 40 weeks, although only 25 patients were included in each group.
It remains possible that the association between phosphate and adverse cardiovascular outcome is mediated by other pathophysiological processes unrelated to arterial stiffness and structural heart disease. Phosphate correlates with angiographic severity of coronary artery disease41 and may contribute to changes in plaque structure.1 Such changes would not have been detected by any of the techniques used in our study. Finally, it is possible that the relationship between poor outcome and serum phosphate is not causative but reflects an association observed between worse outcomes and declining renal function. Although studies have attempted to mathematically correct for renal function, most have done so by adjusting for eGFR. The accuracy of such calculations is reduced at eGFR values close to 60 ml/min per 1.73 m2 compared with true GFR measurements.
In summary, in patients with stage 3 CKD, the use of sevelamer carbonate for 40 weeks was not associated with any significant changes in LV mass, arterial stiffness, or any parameters of LV systolic and diastolic function. However, adherence to the study medication was low, and more rigorous testing of this hypothesis may be achieved with the introduction of a well tolerated one-time daily drug regimen that more effectively reduces cardiovascular phosphate exposure.
Concise Methods
Study Design
The protocol for the Chronic Renal Impairment in Birmingham- Phosphate study has been previously published.42 The study was a single-center, prospective, double-blind, randomized, placebo-controlled trial. The noncalcium-based phosphate binder sevelamer carbonate was chosen because of its lack of calcium content and proven efficacy in the treatment of hyperphosphatemia in advanced CKD.43 All subjects were administered 1600 mg sevelamer carbonate with meals during a 4-week open-label run-in phase. This dose was reduced to 800 mg with meals only if persistent adverse effects or hypophosphatemia occurred. Subjects that tolerated sevelamer then underwent 1:1 randomization to continue another 36 weeks of treatment with either sevelamer or placebo with meals. The study was approved by West Midlands Research Ethics Committee and adhered to the Declaration of Helsinki. Written informed consent was obtained from each participant.
Setting and Participants
Subjects were recruited from renal outpatient clinics at Queen Elizabeth Hospital, Birmingham, United Kingdom, from 2009 to 2011 with the following inclusion criteria: age=18–80 years with stage 3 CKD (eGFR=30–59 ml/min per 1.73 m2), total cholesterol<212 mg/dl, and resting BP controlled to <140/90 mmHg for ≥12 months before enrolment. Exclusion criteria were existing or previous treatment within the last 1 year with a phosphate binder or vitamin D analog; hyperphosphatemia (>5.6 mg/dl); hypophosphatemia (<2.5 mg/dl); uncontrolled secondary hyperparathyroidism (PTH>80 pg/ml); diabetes mellitus; pregnancy; and history of significant gastrointestinal pathology.
Subjects were assessed at baseline (week 0) and the end of study (week 40). At each time point, the following tests were performed: measurement of height, weight, hip, and waist circumference, 12-lead electrocardiogram (ECG), resting brachial BP, applanation tonometry for pulse wave analysis (PWA) and aortic PWV, dual-energy x-ray absorptiometry for assessment of lumbar spine and femoral bone density, collection of serum and plasma for biochemical and hematologic analysis, spot urine collection for albumin-creatinine ratio, 24-hour urine collection for phosphate excretion, lateral lumbar spine radiography to assess baseline aortic calcification as previously described,44 transthoracic echocardiography, CMR, and 24-hour ambulatory BP monitoring. At the end of the open-label run-in phase (week 4), assessments of BP, PWA, and PWV and serum, plasma, and urine collections were repeated. Serum, plasma, and urine were collected at the same time of day after an overnight fast. No dietary changes were enforced during the study, and no dietary counseling was provided. All participants were advised to take two tablets with each meal. Biochemical and safety monitoring was performed at baseline, week 2, week 4, and 4-week intervals until end of study. Compliance with study medication was assessed at 4-week intervals using pill counts of returned medication (taking into consideration the typical number of meals per day) to determine the percentage of prescribed pills taken. Participants were regularly encouraged to take tablets with meals as prescribed; recommendations to improve compliance, such as use of a pill diary and use of a portable medication container to facilitate drug administration away from home, were adopted as necessary. Treatment with vitamin D analogs and other phosphate binders was not permitted during study enrolment. Regular medications unrelated to mineral metabolism that were taken at study entry were continued; no changes were made to regular medications during the study period. Subjects were withdrawn if hypophosphatemia (serum phosphate<2.5 mg/dl on two sequential blood tests) or adverse effects related to treatment persisted despite halving of therapy to 800 mg three times daily. Subjects withdrawn after randomization were included in final intention-to-treat analyses where possible.
End Points
The primary end point was change in LV mass from baseline assessed using CMR after 40 weeks of treatment. Secondary end points included changes in the following measurements: serum FGF-23, arterial stiffness assessed using PWA and PWV, LV systolic and diastolic function assessed using CMR and echocardiography, and aortic distensibility measured by CMR and bone density on dual-energy x-ray absorptiometry scanning.
Cardiovascular Magnetic Resonance Imaging
A 1.5-T scanner (Symphony; Siemens, Erlangen, Germany) was used for CMR studies. Serial contiguous short-axis cines were piloted from vertical and horizontal long-axis images of the left and right ventricles (ECG R wave-gated, steady-state free precession imaging [TrueFISP]; temporal resolution=40–50 ms, repetition time=3.2 ms, echo time=1.6 ms, flip angle=60°, slice thickness=7 mm with 3-mm gap) in accordance with previously validated methodologies.45 Analysis was performed offline (Argus Software; Siemens, Erlangen, Germany) by a single blinded observer (C.D.C.) using manual planimetry for determination of ventricular volumes (end diastolic, end systolic, and stroke volumes), function (ejection fraction), and LV mass, which was indexed to body surface area. Left ventricular hypertrophy was defined as LV mass index>2 SDs above the mean for age and sex.45 Aortic distensibility was assessed at the ascending, proximal descending, and distal descending aorta and calculated using the validated formula46:
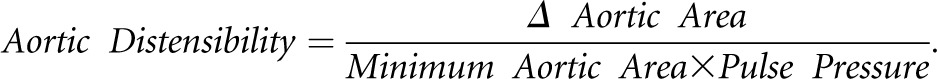 |
BP, PWA, and PWV
Resting brachial BP was measured in triplicate in the nondominant arm using a validated oscillometric sphygmomanometer (Dinamap Procare 200; GE Healthcare, United Kingdom) after 15 minutes of supine rest. All subjects underwent 24-hour ambulatory BP monitoring (ABPM-04; Meditech, Budapest, Hungary). PWA was performed on the radial artery using a high-fidelity micromanometer (SPC-301; Miller Instruments, Houston, TX). The peripheral arterial waveform was used to generate a central arterial waveform using a validated transfer function (SphygmoCor; AtCor Medical, Sydney, Australia). AIx and AIx adjusted to a heart rate of 75 beats/min were recorded. The same system was used to determine aortic PWV by sequentially recording ECG-gated carotid and femoral waveforms as previously described.47 All measurements were made in triplicate, and mean values were used in analysis.
Echocardiography
Transthoracic echocardiography (Vivid 7; GE Vingmed Ultrasound, Horten, Norway) was performed by an experienced echocardiographer (C.D.C.). All parameters were measured in triplicate according to American Society of Echocardiography recommendations48 and analyzed offline (EchoPAC; GE Vingmed Ultrasound, Horten, Norway) by a single blinded observer (C.D.C.). Resting LV diastolic function was assessed using standard techniques.49 Grayscale images for two-dimensional LV strain, strain rate, and twist were acquired in cine-loop format in triplicate from apical and short-axis views at end expiration at frame rates>70/s for offline analysis using commercially available EchoPAC software. The endocardial border was manually tracked at end systole, enabling frame-to-frame tracking of myocardial motion throughout the cardiac cycle. Peak longitudinal systolic velocities, strain, and strain rate were measured for each myocardial segment. Global peak longitudinal strain was calculated as the average peak systolic strain of all 18 myocardial segments. Peak systolic twist and twist rate were determined by subtracting the basal rotation curve from the apical rotation curve. Analyses were performed in triplicate, and results were averaged.
Biochemical Analyses
The four-variable Modified Diet in Renal Disease equation with serum creatinine recalibrated to be traceable to an isotope-derived mass spectroscopy method was used to determine eGFR.32 Serum calcium levels were adjusted for serum albumin. A two-site ELISA (Kainos Laboratories Inc., Tokyo, Japan) was used to measure FGF-23.50 Soluble α-klotho and 1,25-dihydroxyvitamin D were determined using immunoassays (Immuno-Biologic Laboratories Co., Japan; Immunodiagnostic Systems Ltd, Tyne and Wear, United Kingdom).51 Plasma 25-hydroxyvitamin D was measured by liquid chromatography tandem mass spectrometry. Plasma N-terminal pro-brain natriuretic peptide was measured using a validated immunoluminometric assay52; 24-hour urine collections were used to determine urinary phosphate concentration (milligrams per deciliter) and fractional excretion of phosphate ((urine phosphate × serum creatinine)/(serum phosphate × urine creatinine)×100%), which corrects for incomplete collections and standardizes phosphate excretion according to degree of renal impairment.13
Reproducibility
Variability for the primary end point of LV mass was assessed using intraclass correlation coefficients. Twelve scans (10% of the cohort) were randomly selected and analyzed by the same observer on two separate occasions to calculate intraobserver variability. For interstudy reproducibility, 12 subjects were chosen at random to undergo fully repeated CMR scanning on the same day; both scans were analyzed by the same observer.
Statistical Analyses
For >90% power to detect an 8-g difference between groups in the change in LV mass at 5% significance assuming an SD of change in LV mass of 12 g, 55 subjects were required in each arm. We planned to recruit 60 subjects to each group to allow for withdrawal or dropout. Data are expressed as mean ± SD, median (interquartile range), or frequency (%). The normality of distribution for continuous variables was determined using normality plots and Kolmogorov–Smirnov test. Variables not normally distributed were log-transformed before analysis to achieve normal distribution. Data were analyzed using t or chi-squared tests. Differences between groups after 40 weeks of treatment were assessed using repeated measures ANOVA. Analysis was by intention to treat. A two-tailed P value<0.05 was considered statistically significant.
Disclosures
The Chronic Renal Impairment in Birmingham-Phosphate study was funded by an unrestricted grant from Genzyme Corporation. Genzyme Corporation provided the study drug sevelamer carbonate and matching placebo. Genzyme Corporation had no role in study design or data analysis and interpretation. C.D.C. and C.J.F. have received lecture and advisory board fees from Genzyme Corporation.
Acknowledgments
We would like to thank Dr. Peter Nightingale for statistical support and Sister Helen Jones and Staff Nurse Theresa Brady for their help with running the study.
This work was presented as an abstract at the American Heart Association Scientific Sessions in Los Angeles, California, November 6, 2012.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “What Can We Learn from Null Randomized Controlled Trials?,” on pages 691–693.
References
- 1.Ellam TJ, Chico TJ: Phosphate: The new cholesterol? The role of the phosphate axis in non-uremic vascular disease. Atherosclerosis 220: 310–318, 2012 [DOI] [PubMed] [Google Scholar]
- 2.Dhingra R, Sullivan LM, Fox CS, Wang TJ, D’Agostino RB, Sr, Gaziano JM, Vasan RS: Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med 167: 879–885, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Ganesh SK, Stack AG, Levin NW, Hulbert-Shearon T, Port FK: Association of elevated serum PO(4), Ca x PO(4) product, and parathyroid hormone with cardiac mortality risk in chronic hemodialysis patients. J Am Soc Nephrol 12: 2131–2138, 2001 [DOI] [PubMed] [Google Scholar]
- 4.Block GA, Hulbert-Shearon TE, Levin NW, Port FK: Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: A national study. Am J Kidney Dis 31: 607–617, 1998 [DOI] [PubMed] [Google Scholar]
- 5.Eddington H, Hoefield R, Sinha S, Chrysochou C, Lane B, Foley RN, Hegarty J, New J, O’Donoghue DJ, Middleton RJ, Kalra PA: Serum phosphate and mortality in patients with chronic kidney disease. Clin J Am Soc Nephrol 5: 2251–2257, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kestenbaum B, Sampson JN, Rudser KD, Patterson DJ, Seliger SL, Young B, Sherrard DJ, Andress DL: Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol 16: 520–528, 2005 [DOI] [PubMed] [Google Scholar]
- 7.Chonchol M, Dale R, Schrier RW, Estacio R: Serum phosphorus and cardiovascular mortality in type 2 diabetes. Am J Med 122: 380–386, 2009 [DOI] [PubMed] [Google Scholar]
- 8.Tonelli M, Sacks F, Pfeffer M, Gao Z, Curhan G, Cholesterol And Recurrent Events Trial Investigators : Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation 112: 2627–2633, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Ix JH, De Boer IH, Peralta CA, Adeney KL, Duprez DA, Jenny NS, Siscovick DS, Kestenbaum BR: Serum phosphorus concentrations and arterial stiffness among individuals with normal kidney function to moderate kidney disease in MESA. Clin J Am Soc Nephrol 4: 609–615, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chue CD, Edwards NC, Moody WE, Steeds RP, Townend JN, Ferro CJ: Serum phosphate is associated with left ventricular mass in patients with chronic kidney disease: A cardiac magnetic resonance study. Heart 98: 219–224, 2012 [DOI] [PubMed] [Google Scholar]
- 11.Dhingra R, Gona P, Benjamin EJ, Wang TJ, Aragam J, D’Agostino RB, Sr, Kannel WB, Vasan RS: Relations of serum phosphorus levels to echocardiographic left ventricular mass and incidence of heart failure in the community. Eur J Heart Fail 12: 812–818, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foley RN, Collins AJ, Herzog CA, Ishani A, Kalra PA: Serum phosphate and left ventricular hypertrophy in young adults: The coronary artery risk development in young adults study. Kidney Blood Press Res 32: 37–44, 2009 [DOI] [PubMed] [Google Scholar]
- 13.Isakova T, Wahl P, Vargas GS, Gutiérrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, Hamm L, Gadegbeku C, Horwitz E, Townsend RR, Anderson CA, Lash JP, Hsu CY, Leonard MB, Wolf M: Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int 79: 1370–1378, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Craver L, Marco MP, Martínez I, Rue M, Borràs M, Martín ML, Sarró F, Valdivielso JM, Fernández E: Mineral metabolism parameters throughout chronic kidney disease stages 1-5—achievement of K/DOQI target ranges. Nephrol Dial Transplant 22: 1171–1176, 2007 [DOI] [PubMed] [Google Scholar]
- 15.Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, Gutiérrez OM, Aguillon-Prada R, Lincoln J, Hare JM, Mundel P, Morales A, Scialla J, Fischer M, Soliman EZ, Chen J, Go AS, Rosas SE, Nessel L, Townsend RR, Feldman HI, St John Sutton M, Ojo A, Gadegbeku C, Di Marco GS, Reuter S, Kentrup D, Tiemann K, Brand M, Hill JA, Moe OW, Kuro-O M, Kusek JW, Keane MG, Wolf M: FGF23 induces left ventricular hypertrophy. J Clin Invest 121: 4393–4408, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chue CD, Townend JN, Steeds RP, Ferro CJ: Arterial stiffness in chronic kidney disease: Causes and consequences. Heart 96: 817–823, 2010 [DOI] [PubMed] [Google Scholar]
- 17.Tonelli M, Wiebe N, Culleton B, House A, Rabbat C, Fok M, McAlister F, Garg AX: Chronic kidney disease and mortality risk: A systematic review. J Am Soc Nephrol 17: 2034–2047, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Schillaci G, Verdecchia P, Porcellati C, Cuccurullo O, Cosco C, Perticone F: Continuous relation between left ventricular mass and cardiovascular risk in essential hypertension. Hypertension 35: 580–586, 2000 [DOI] [PubMed] [Google Scholar]
- 19.Foley RN, Parfrey PS, Harnett JD, Kent GM, Martin CJ, Murray DC, Barre PE: Clinical and echocardiographic disease in patients starting end-stage renal disease therapy. Kidney Int 47: 186–192, 1995 [DOI] [PubMed] [Google Scholar]
- 20.Ferro CJ, Chue CD, Steeds RP, Townend JN: Is lowering phosphate exposure the key to preventing arterial stiffening with age? Heart 95: 1770–1772, 2009 [DOI] [PubMed] [Google Scholar]
- 21.Foley RN, Collins AJ, Herzog CA, Ishani A, Kalra PA: Serum phosphorus levels associate with coronary atherosclerosis in young adults. J Am Soc Nephrol 20: 397–404, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tuttle KR, Short RA: Longitudinal relationships among coronary artery calcification, serum phosphorus, and kidney function. Clin J Am Soc Nephrol 4: 1968–1973, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adeney KL, Siscovick DS, Ix JH, Seliger SL, Shlipak MG, Jenny NS, Kestenbaum BR: Association of serum phosphate with vascular and valvular calcification in moderate CKD. J Am Soc Nephrol 20: 381–387, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shuto E, Taketani Y, Tanaka R, Harada N, Isshiki M, Sato M, Nashiki K, Amo K, Yamamoto H, Higashi Y, Nakaya Y, Takeda E: Dietary phosphorus acutely impairs endothelial function. J Am Soc Nephrol 20: 1504–1512, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, Morii H, Giachelli CM: Phosphate regulation of vascular smooth muscle cell calcification. Circ Res 87: E10–E17, 2000 [DOI] [PubMed] [Google Scholar]
- 26.Guérin AP, London GM, Marchais SJ, Metivier F: Arterial stiffening and vascular calcifications in end-stage renal disease. Nephrol Dial Transplant 15: 1014–1021, 2000 [DOI] [PubMed] [Google Scholar]
- 27.McEniery CM, McDonnell BJ, So A, Aitken S, Bolton CE, Munnery M, Hickson SS, Yasmin, Maki-Petaja KM, Cockcroft JR, Dixon AK, Wilkinson IB, Anglo-Cardiff Collaboration Trial Investigators : Aortic calcification is associated with aortic stiffness and isolated systolic hypertension in healthy individuals. Hypertension 53: 524–531, 2009 [DOI] [PubMed] [Google Scholar]
- 28.Raggi P, Bellasi A, Ferramosca E, Islam T, Muntner P, Block GA: Association of pulse wave velocity with vascular and valvular calcification in hemodialysis patients. Kidney Int 71: 802–807, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Achinger SG, Ayus JC: Left ventricular hypertrophy: Is hyperphosphatemia among dialysis patients a risk factor? J Am Soc Nephrol 17[Suppl 3]: S255–S261, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Berndt T, Thomas LF, Craig TA, Sommer S, Li X, Bergstralh EJ, Kumar R: Evidence for a signaling axis by which intestinal phosphate rapidly modulates renal phosphate reabsorption. Proc Natl Acad Sci U S A 104: 11085–11090, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Block GA, Wheeler DC, Persky MS, Kestenbaum B, Ketteler M, Spiegel DM, Allison MA, Asplin J, Smits G, Hoofnagle AN, Kooienga L, Thadhani R, Mannstadt M, Wolf M, Chertow GM: Effects of phosphate binders in moderate CKD. J Am Soc Nephrol 23: 1407–1415, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D, Modification of Diet in Renal Disease Study Group : A more accurate method to estimate glomerular filtration rate from serum creatinine: A new prediction equation. Ann Intern Med 130: 461–470, 1999 [DOI] [PubMed] [Google Scholar]
- 33.Edwards NC, Steeds RP, Stewart PM, Ferro CJ, Townend JN: Effect of spironolactone on left ventricular mass and aortic stiffness in early-stage chronic kidney disease: A randomized controlled trial. J Am Coll Cardiol 54: 505–512, 2009 [DOI] [PubMed] [Google Scholar]
- 34.Myerson SG, Bellenger NG, Pennell DJ: Assessment of left ventricular mass by cardiovascular magnetic resonance. Hypertension 39: 750–755, 2002 [DOI] [PubMed] [Google Scholar]
- 35.Thadhani R, Appelbaum E, Pritchett Y, Chang Y, Wenger J, Tamez H, Bhan I, Agarwal R, Zoccali C, Wanner C, Lloyd-Jones D, Cannata J, Thompson BT, Andress D, Zhang W, Packham D, Singh B, Zehnder D, Shah A, Pachika A, Manning WJ, Solomon SD: Vitamin D therapy and cardiac structure and function in patients with chronic kidney disease: The PRIMO randomized controlled trial. JAMA 307: 674–684, 2012 [DOI] [PubMed] [Google Scholar]
- 36.Lee JK, Grace KA, Foster TG, Crawley MJ, Erowele GI, Sun HJ, Turner PT, Sullenberger LE, Taylor AJ: How should we measure medication adherence in clinical trials and practice? Ther Clin Risk Manag 3: 685–690, 2007 [PMC free article] [PubMed] [Google Scholar]
- 37.Muldoon MF, Manuck SB, Matthews KA: Lowering cholesterol concentrations and mortality: A quantitative review of primary prevention trials. BMJ 301: 309–314, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Russo D, Palmiero G, De Blasio AP, Balletta MM, Andreucci VE: Coronary artery calcification in patients with CRF not undergoing dialysis. Am J Kidney Dis 44: 1024–1030, 2004 [DOI] [PubMed] [Google Scholar]
- 39.Kakuta T, Tanaka R, Hyodo T, Suzuki H, Kanai G, Nagaoka M, Takahashi H, Hirawa N, Oogushi Y, Miyata T, Kobayashi H, Fukagawa M, Saito A: Effect of sevelamer and calcium-based phosphate binders on coronary artery calcification and accumulation of circulating advanced glycation end products in hemodialysis patients. Am J Kidney Dis 57: 422–431, 2011 [DOI] [PubMed] [Google Scholar]
- 40.Chertow GM, Burke SK, Raggi P, Treat to Goal Working Group : Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int 62: 245–252, 2002 [DOI] [PubMed] [Google Scholar]
- 41.Narang R, Ridout D, Nonis C, Kooner JS: Serum calcium, phosphorus and albumin levels in relation to the angiographic severity of coronary artery disease. Int J Cardiol 60: 73–79, 1997 [DOI] [PubMed] [Google Scholar]
- 42.Chue CD, Townend JN, Steeds RP, Ferro CJ: Evaluating the effects of sevelamer carbonate on cardiovascular structure and function in chronic renal impairment in Birmingham: The CRIB-PHOS randomised controlled trial. Trials 12: 30, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bleyer AJ, Burke SK, Dillon M, Garrett B, Kant KS, Lynch D, Rahman SN, Schoenfeld P, Teitelbaum I, Zeig S, Slatopolsky E: A comparison of the calcium-free phosphate binder sevelamer hydrochloride with calcium acetate in the treatment of hyperphosphatemia in hemodialysis patients. Am J Kidney Dis 33: 694–701, 1999 [DOI] [PubMed] [Google Scholar]
- 44.Kauppila LI, Polak JF, Cupples LA, Hannan MT, Kiel DP, Wilson PW: New indices to classify location, severity and progression of calcific lesions in the abdominal aorta: A 25-year follow-up study. Atherosclerosis 132: 245–250, 1997 [DOI] [PubMed] [Google Scholar]
- 45.Maceira AM, Prasad SK, Khan M, Pennell DJ: Normalized left ventricular systolic and diastolic function by steady state free precession cardiovascular magnetic resonance. J Cardiovasc Magn Reson 8: 417–426, 2006 [DOI] [PubMed] [Google Scholar]
- 46.Groenink M, de Roos A, Mulder BJ, Spaan JA, van der Wall EE: Changes in aortic distensibility and pulse wave velocity assessed with magnetic resonance imaging following beta-blocker therapy in the Marfan syndrome. Am J Cardiol 82: 203–208, 1998 [DOI] [PubMed] [Google Scholar]
- 47.Wilkinson IB, Fuchs SA, Jansen IM, Spratt JC, Murray GD, Cockcroft JR, Webb DJ: Reproducibility of pulse wave velocity and augmentation index measured by pulse wave analysis. J Hypertens 16: 2079–2084, 1998 [DOI] [PubMed] [Google Scholar]
- 48.Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ, Chamber Quantification Writing Group. American Society of Echocardiography’s Guidelines and Standards Committee. European Association of Echocardiography : Recommendations for chamber quantification: A report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 18: 1440–1463, 2005 [DOI] [PubMed] [Google Scholar]
- 49.Nagueh SF, Appleton CP, Gillebert TC, Marino PN, Oh JK, Smiseth OA, Waggoner AD, Flachskampf FA, Pellikka PA, Evangelista A: Recommendations for the evaluation of left ventricular diastolic function by echocardiography. J Am Soc Echocardiogr 22: 107–133, 2009 [DOI] [PubMed] [Google Scholar]
- 50.Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K, Tajima T, Takeuchi Y, Fujita T, Nakahara K, Yamashita T, Fukumoto S: Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab 87: 4957–4960, 2002 [DOI] [PubMed] [Google Scholar]
- 51.Yamazaki Y, Imura A, Urakawa I, Shimada T, Murakami J, Aono Y, Hasegawa H, Yamashita T, Nakatani K, Saito Y, Okamoto N, Kurumatani N, Namba N, Kitaoka T, Ozono K, Sakai T, Hataya H, Ichikawa S, Imel EA, Econs MJ, Nabeshima Y: Establishment of sandwich ELISA for soluble alpha-Klotho measurement: Age-dependent change of soluble alpha-Klotho levels in healthy subjects. Biochem Biophys Res Commun 398: 513–518, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Downie PF, Talwar S, Squire IB, Davies JE, Barnett DB, Ng LL: Assessment of the stability of N-terminal pro-brain natriuretic peptide in vitro: Implications for assessment of left ventricular dysfunction. Clin Sci (Lond) 97: 255–258, 1999 [PubMed] [Google Scholar]