

Background: KRAS is a known oncogene driving transformation in multiple tissues.
Results: We demonstrate a role for the transcription factor GLI1 in KRAS-induced transformation through regulation of the IL-6/STAT3 axis in the tumor microenvironment.
Conclusion: This study defines a novel oncogenic network downstream of KRAS modulating transformation.
Significance: This knowledge will contribute to the understanding of the pathogenesis of tumors driven by KRAS.
Keywords: Cytokine Induction, Gene Knock-out, Gene Regulation, Pancreas, Transcription Factors, Tumor Microenvironment
Abstract
Although the biological role of KRAS is clearly established in carcinogenesis, the molecular mechanisms underlying this phenomenon are not completely understood. In this study, we provide evidence of a novel signaling network regulated by the transcription factor GLI1 mediating KRAS-induced carcinogenesis. Using pancreatic cancer (a disease with high prevalence of KRAS mutations) as a model, we show that loss of GLI1 blocks the progression of KRAS-induced pancreatic preneoplastic lesions in mice with pancreas-specific Cre-activated oncogenic mutant kras. Mice lacking GLI1 develop only low-grade lesions at low frequency, and in most cases, the pancreata are histologically normal. Further characterization of the phenotype showed a decrease in the activation of STAT3 in pancreatic preneoplastic lesions; STAT3 is a transcription factor required for the development of premalignant lesions and their progression into pancreatic cancer. Analysis of the mechanisms revealed a key role for GLI1 in maintaining the levels of activated STAT3 through the modulation of IL-6 signaling. GLI1 binds to the IL-6 mouse promoter and regulates the activity and expression of this cytokine. This newly identified GLI1/IL-6 axis is active in fibroblasts, a known source of IL-6 in the tumor microenvironment. Sonic hedgehog induces GLI1 binding to the IL-6 promoter and increases IL-6 expression in fibroblasts in a paracrine manner. Finally, we demonstrate that mutant KRAS initiates this cascade by inducing the expression of Sonic hedgehog in cancer cells. Collectively, these results define a novel role for GLI1 in carcinogenesis acting as a downstream effector of oncogenic KRAS in the tumor microenvironment.
Introduction
The small GTPase KRAS plays a major role in the pathogenesis of numerous malignancies. Mutations in the KRAS gene appear early in tumor development and are found in multiple cancers, including pancreatic cancer, where KRAS mutations are present in >90% of cases (1–4). Most of these mutations result in amino acid substitutions at codons 12 and 13 (5–8). These missense mutations impair intrinsic and GTPase-activating protein-mediated GTP hydrolysis, thus increasing the levels of GTP-bound active KRAS. This in turn activates downstream signaling, resulting in increased cell growth and survival leading to neoplastic transformation (9–14). Although the biological role of KRAS during neoplastic transformation has been intensively studied, the detailed molecular mechanism underlying this phenomenon in cancer cells, especially the role of downstream nuclear effector molecules, remains to be established. Because direct targeting of KRAS or the use of mutant forms of KRAS as a biomarker for diagnosis of disease has been unsuccessful to date, a better understanding of the downstream effects of KRAS signaling may lead to novel strategies to overcome this clinical problem.
Here, using a combination of in vitro and in vivo models, we identified the transcription factor GLI1 as a key mediator of KRAS-induced pancreatic transformation. It was previously reported that KRAS can modulate GLI1 activity and requires this transcription factor to modulate transformation in vitro (15). However, the biological significance of these findings and the molecular mechanisms mediating this phenomenon remain elusive. To address these issues, we used the well established Lox-Stop-Lox (LSL)2-krasG12D mouse crossed with the pancreas-specific p48-Cre mouse model of pancreatic cancer (referred to as KC mice). In this model, KC mice develop the entire spectrum of precursor pancreatic intraepithelial neoplasia (PanIN) lesions, PanIN-1A to PanIN-3, with 100% penetrance and pancreatic cancer at an advanced age (13, 14). Mice with a total loss-of-function allele of gli1 (gli1 knock-out (GKO)) were crossed with the KC animals to generate the GKO/KC mouse. We show that GKO/KC mice do not develop tumors in the pancreas and present with fewer PanIN lesions that are only of low-grade compared with KC mice. Recent evidence suggests that activation of STAT3 in cancer cells is required for KRAS-induced PanIN progression (16, 17). Further analysis of the mechanism underlying this phenomenon shows that GLI1 regulates activation of STAT3 through modulation of IL-6 expression and promoter activity in pancreatic fibroblasts. Together, these findings define a novel signaling network in the tumor microenvironment required by KRAS to induce transformation that utilizes GLI1 as its central mediator.
EXPERIMENTAL PROCEDURES
Breeding
Mice were housed in pathogen-free conditions and maintained in facilities approved by the American Association for Accreditation of Laboratory Animal Care in accordance with current regulations and standards of the United States Departments of Agriculture and Health and Human Services and the National Institutes of Health Institutional Animal Care and Use Committee. LSL-kras mouse strain 1XJ6 B6.129-krastm4Tyj (LSL-krasG12D) and p48-Cre (13, 14) and GKO (18) mice were provided by F. C. and C. W. LSL-krasG12D and p48-Cre;GKO mice were crossed, ultimately producing three groups of mice: 1) GKO;LSL-krasG12D;p48-Cre (GKO/KC), 2) GHet;LSL-krasG12D;p48-Cre (GHet/KC), and WT gli1;LSL-krasG12D;p48-Cre (KC). Allele-specific PCR verified the presence of floxed LSL-krasG12D alleles in the tail and Cre-recombinase-dependent rearranged alleles in the pancreas, and two PCRs verified the presence of either the wild-type or β-gal allele in gli1Het mice (18). 15 animals per cohort were aged for survival. Mice were euthanized when they displayed signs of cachexia or weight loss or were not moving. Survival was analyzed by Kaplan-Meier curves, and statistics were measured using the log-rank (Mantel-Cox) test in Prism. This approach takes into account animals that had been withdrawn from the study due to causes other than those described above. Mice of each genotype were killed at 2, 4, 6, and 8 months, and selected tissues were collected for histopathological evaluation.
To determine that the phenotype seen in the GKO/KC cross was not due to loss of expression of the krasG12D mutant, pancreatic tissue from KC and GKO/KC mice was minced, and RNA was extracted using TRIzol reagent (Invitrogen) following the manufacturer's protocol. cDNA was generated using a High Capacity first-strand cDNA kit (Applied Biosystems) as recommended by the manufacturer. To amplify the 243-bp products from both the wild-type and mutant transcripts, the following primers were used: kras1, 5′-aggcctgctgaaaatgactg-3′; and kras7, 5′-ccctcccgagttctcatgta-3′. Verification of the krasG12D allele was performed using HindIII digestion of the PCR product, which yielded a 213- and 30-bp product present only in the mutant (but not wild-type) form of kras. Products were verified on 4% agarose gels and subsequently sequenced to detect the G→A mutation in the cDNA.
Genotyping
Tail DNA was extracted using the Qiagen DNeasy kit. The Qiagen Taq polymerase kit was used following the manufacturer's instructions. The primers used for WT gli1 and GKO (18, 19), krasG12D (NCI Mouse Repository), and p48-Cre were as follows: WT gli1, 5′-ccagtttctgagatgagggttaga-3′ (forward) and 5′-ttgaatggggaatacaggggcttac-3′ (reverse); GKO, 5′-gcatcgagctgggtaataagcgttggcaat-3′ (forward) and 5′-gacaccagaccaactggtaatggtagcgac-3′ (reverse); krasG12D, 5′-agctagccaccatggcttgagtaagtctgca-3′ (forward) and 5′-cctttacaagcgcacgcagactgtaga-3′ (reverse); and p48-Cre, 5′-agatgttcgcgattatcttc-3′ (forward) and 5′-agctacaccagagacgg-3′ (reverse). PCR cycling conditions were as follows: GKO and krasG12D, 95 °C for 5 min, 95 °C for 30 s, 63 °C for 45 s, and 72 °C for 1 min for 34 cycles; p48-Cre, 95 °C for 5 min, 95 °C for 30 s, 58 °C for 45 s, and 72 °C for 1 min for 34 cycles, followed by 72 °C for 10 min; and WT gli1, 95 °C for 5 min, 95 °C for 30 s, 61 °C for 45 s, and 72 °C for 1 min for 34 cycles, followed by 72 °C for 10 min. Amplified PCR products were run on 1% agarose gels with molecular weight markers, and amplified products were visualized under UV transillumination.
Syngeneic Model
Subconfluent cultures of cancer cells derived from the KC mouse were harvested using a 0.05% trypsin solution, washed twice with PBS, and resuspended as single-cell suspensions in PBS. Cell viability was tested using trypan blue exclusion. Mice were given a general anesthesia of ketamine hydrochloride (Sigma) and xylazine (Fort Dodge Animal Health, Fort Dodge, IA) at concentrations of 90 and 10 mg/kg injected intraperitoneally. A 1-cm incision was made in the left abdominal flank, exposing the spleen. The spleen was gently guided out to expose the pancreas. 5 × 105 cells in a volume of 50 μl were injected slowly into the tail of the pancreas using a 27-gauge needle. The pancreas was then replaced in the abdominal cavity. The abdominal cavity was closed with a 4-0 vicryl suture (Fisher).
Immunohistochemistry
Paraffin-embedded tissue sections were processed using 3× xylene for 5 min, 2× 100% alcohol for 5 min, 95% alcohol for 5 min, 75% alcohol for 5 min, and 2 changes of deionized water for 5 min. Antigen retrieval was performed using citrate buffer (pH 6.0) or 1 mm EDTA for 15 min in the microwave and cooled. Blocking was achieved using 10% hydrogen peroxide for 10 min, and then sections were blocked with 5% normal goat serum in Tris-buffered saline containing 1% Tween for 30 min. Primary antibody was applied to sections overnight at 4 °C, and anti-rabbit polymer secondary antibody (HRP-conjugated EnVision-labeled polymer) or SignalStain Boost IHC detection agent (Cell Signaling Technology, Danvers, MA) was applied for 15 min. Sections were developed using 3,3′-diaminobenzidine (Sigma D3939). Phospho-STAT3 (p-STAT3), amylase, and insulin (Cell Signaling Technology) were used as primary antibodies. Anti-Sonic hedgehog (SHH) antibody was purchased from R&D Systems (Minneapolis, MN). Images were taken with an Olympus BX51 microscope, an Olympus DP71 digital camera, and DP Controller software.
Cell Culture Conditions
Mouse embryonic fibroblasts were established using embryonic day 18.5 embryos and pancreatic fibroblasts from adult pancreata. Tissues were minced with a razor blade and homogenized with TrypLE Express (Invitrogen). Cells were placed at 37 °C for 10 min and homogenized by pipetting; this process was repeated four times. Fibroblast cells were seeded to 80–90% confluency in Iscove's modified Eagle's medium with 10% FBS and then starved by replacing the medium with Iscove's modified Eagle's medium with 0.5% of FBS. Sixteen h later, cells were treated with the recombinant mouse and human SHH N-terminal (R&D Systems) in 0.5% Iscove's modified Eagle's medium. The control group was treated with the vehicle (0.5% Iscove's modified Eagle's medium with 0.2% BSA in PBS) used to reconstitute the recombinant ligand. Cells were harvested for RNA at 24, 48, and 72 h after treatment.
Plasmids
The FLAG-tagged GLI1 plasmid was previously used by Elsawa et al. (20). The mouse IL-6-luciferase reporter was a gift from Dr. Gail A. Bishop (Department of Microbiology, University of Iowa, Iowa City, IA). The GLI1-luciferase reporter was kindly provided by Dr. Chi-chung Hui (University of Toronto, Ontario, Canada). The mouse SHH promoter was purchased from GeneCopoeia (Rockville, MD). The KRASG12D mutant was a gift from Dr. Daniel Billadeau (Mayo Clinic, Rochester, MN).
Transfection
Cells were transfected using Lipofectamine (Invitrogen) following the manufacturer's recommendations. Briefly, 1.5 × 105 cells were plated in 6-well plates with standard medium and transfected 24 h later. For each condition, 3 μg of IL-6 or 5 μg of SHH promoter-reporter constructs was used in 3 wells of a 6-well plate. In overexpression assays, 3 μg of gli1, and 5 μg of KRASG12D, or control vector was used.
Luciferase Reporter Assay
Cells were grown and transfected as described above. For luciferase reporter assays, cells were plated in triplicate in 6-well plates in medium containing 10% FBS. Samples were harvested and prepared for luciferase assays in accordance with the manufacturer's protocol (Promega, Madison, WI). Cells were harvested 36–48 h after transfection for overexpression studies and conditioned media experiments. The SHH-blocking antibody (5E1) was purchased from the Developmental Studies Hybridoma Bank (University of Iowa). To control for intersample variations in transfection efficiency, the total protein from samples in each well was quantitated using the Bio-Rad protein assay, and luciferase readouts were normalized to protein content.
Western Blotting
For conditioned media experiments, cancer cells were harvested 24 h after treatment with medium from pancreatic fibroblasts treated with recombinant mouse SHH for 48 h. The IL-6-blocking antibody was purchased from R&D Systems. Protein lysates were prepared in lysis buffer as described previously (20). A similar lysis protocol was followed to obtain whole pancreas lysates. Equal amounts of protein (50–100 μg/lane) were separated by electrophoresis on a 10% Tris-HCl gel and then transferred to PVDF membrane. Blots were probed with polyclonal antibodies against phosphorylated and total STAT3 or ERK (1:1000 and 1:2000, respectively; Cell Signaling Technology). Peroxidase-conjugated secondary antibodies were used, and immunoreactive proteins were detected by chemiluminescence (GE Healthcare).
Quantitative RT-PCR
RNA was extracted from tissues and cell lines using Trizol and RNeasy mini kit (Qiagen) according to the manufacturer's instructions. Reverse transcription reactions were conducted using the High Capacity cDNA reverse transcription kit (Applied Biosystems). Samples for quantitative PCR were prepared with 1× SYBR Green PCR Master Mix (Applied Biosystems) and various primers. All primers were optimized for amplification under the following reaction conditions: 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. GAPDH and cyclophilin was used as the housekeeping gene expression control. The primers used were as follows: GLI1, 5′-aggcactagagttgaggaatttgt-3′ (forward) and 5′-gtgcacagcagccccacactctc-3′ (reverse); IL-6, 5′-ctctgcaagagacttccatccagt-3′ (forward) and 5′-gaagtagggaaggccgtgg-3′ (reverse); human SHH, 5′-5′tgggtgaaagcagagaactc-3′ (forward) and 5′-tctcgatcacgtagaagacc-3′ (reverse); mouse SHH, 5′-aaatgccttggccatctc-3′ (forward) and 5′-tttcacagaggagtggatgc-3′ (reverse); GAPDH, 5′-ggcattgctctcaatgacaa-3′ (forward) and 5′-tgtgagggagatgctcagtg-3′ (reverse); and cyclophilin, 5′-tcacagaattattccaggattcatg-3′ (forward) and 5′-tgccgccagtgccatt-3′ (reverse). Amplification was performed using the LightCycler 480 system (Roche Applied Science), and experiments were performed in triplicates in three independent experiments. The results were calculated following the 2ΔCp method.
ELISA
Mouse IL-6 ELISA (PeproTech, Rocky Hill, NJ) was used to measure serum IL-6 levels in cell culture supernatants following the manufacturer's recommendations. Results were obtained with a 96-well plate reader (Molecular Devices, Palo Alto, CA) and analyzed using SoftMax Pro 5.2 software.
ChIP
24–36 h after recombinant SHH treatment or transfections, DNA/proteins were cross-linked with 1% formaldehyde, followed by cell lysis. DNA was sheared by 45 repeated cycles of 30 s of sonication followed by 30 s of rest. ChIP was performed with EZ-Magna ChIPTM G (Millipore, Billerica, MA) using 1 μg of rabbit IgG or anti-GLI1 antibody (Santa Cruz Biotechnology and Cell Signaling Technology). PCR primers were designed to amplify regions of the IL-6 promoter containing consensus GLI1-binding sites: IL-6, 5′-gcagtgggatcagcactaacagat-3′ (forward) and 5′-cctggacaacagacagtaatgttg-3′ (reverse). PCR products were run on 1% agarose gels and visualized by UV light. Quantitative SYBR PCR was performed in triplicate for each sample or control using the LightCycler 480 system. Experiments were repeated three times.
RESULTS
GLI1 Is Dispensable in the Developing Pancreas
Characterization of the GKO mice did not extend beyond the central nervous system and blood-related cellular compartment (18, 20). We initially sought to evaluate the pancreata of GKO mice in detail over an extended period of time to determine whether subtle developmental defects (or Cre-mediated effects) are present in the background of the colony in the absence of krasG12D, which could lead to an error in interpreting the results of our experiments. GKO and GKO;p48-Cre mice were viable and born at the expected Mendelian ratio. Neither weight nor size of these mice was altered compared with their WT littermates (data not shown). We analyzed the histology of the pancreata of five GKO and five wild-type mice at 2-month intervals. Pancreatic histology at 2, 4, 6, and 8 months of age appeared normal at all time points considered, with minimal cytoplasmic alterations or atrophy observed in both GKO and wild-type mice (Fig. 1A). Further histopathological analysis of organs that could be affected by pancreatic cancer pathogenesis, including the spleen (supplemental Fig. 1A), liver (supplemental Fig. 1B), kidney, and lung (data not shown), showed normal organ size, gross morphology, and histology for this cohort of transgenic mice. Finally, we examined pancreatic markers of differentiation and function by immunohistochemistry and observed similar staining patterns of amylase (exocrine pancreas) and insulin (endocrine pancreas) expression in control and GKO mice (Fig. 1B). Similar results were obtained in the GKO;p48-Cre mice (data not shown). Thus, we conclude that animals with GLI1 loss do not present apparent developmental abnormalities and have pancreata within normal limits similar to wild-type mice.
FIGURE 1.

GKO pancreatic development is within normal limits. A, histopathological analysis performed by two expert pathologists showed that GKO pancreatic development was within normal parameters compared with wild-type mice in the colony. Minor cytoplasmic vacuolation was observed in the exocrine pancreas at 6 months in both groups. Representative H&E pictures are shown in A at ×20 (left panels) and ×40 (right panels) magnifications. B, WT and GKO pancreata were evaluated by immunohistochemistry for markers of differentiation and function. Amylase and insulin showed no differences in either group in the localization and levels of expression of these two markers.
Loss of GLI1 Impairs PanIN Progression and Pancreatic Tumor Development
PanIN lesions are the most common noninvasive precursor lesions to pancreatic cancer. These lesions have many molecular alterations, including mutations in the KRAS gene, that are thought to be early events essential for pancreatic cell transformation (21). Furthermore, as the degree of atypia in ducts increases, these mutations become more prevalent, driving aberrant activation of otherwise quiescent signaling pathways (4, 22–26). Recent studies have demonstrated that GLI1 activity is regulated by KRAS and requires this transcription factor to modulate transformation in vitro (15, 27). However, the biological significance of these findings remains elusive. To analyze the role of GLI1 in KRAS-induced carcinogenesis, we used the LSL-krasG12D mouse model of pancreatic cancer (13, 14). In this model, tumor initiation is achieved by Cre-mediated removal of a transcriptional stop element and activation of an oncogenic allele of krasG12D under physiologic control. This is achieved by crossing the LSL-krasG12D mice with pancreas-specific p48-Cre transgenic mice. The krasG12D;p48-Cre (KC) mice show preneoplastic formation with a concomitant increase in the number and grade of PanIN lesions with age, as seen in humans. Mice with homozygous loss-of-function alleles of gli1 (GKO) were crossed with the KC animals to generate GKO/KC mice. A cohort of mice was evaluated for signs of disease progression and survival. Notably, loss of GLI1 prevented tumor development and resulted in a significantly prolonged median survival rate of 21 months compared to the 16 months of the KC mice (p = 0.0147) (Fig. 2A). In our study, the GKO/KC mice did not develop pancreatic tumors (Fig. 2B), yet the clinical course of KC mice was in agreement with the previous reports that ∼50% of mice developed pancreatic tumors (13, 28). Of note, the GKO/KC phenotype was not due to a change in the expression or activity of mutant krasG12D, as KC and GKO/KC mice showed similar expression of this mutant, as well as activation of its downstream target, ERK (data not shown).
FIGURE 2.

GKO/KC mice do not develop pancreatic cancer and have an increased survival advantage. A, Kaplan-Meier survival curves show a statistically significant difference in survival between the GKO/KC mice (21 months) and KC mice (16 months) (p = 0.147). B, KC mice developed pancreatic cancer at a rate of 56%, whereas GKO/KC mice did not develop tumors. Representative images of the KC tumors and GKO/KC pancreata are shown in the insets. ND, not detected.
Double-blinded histopathological analysis was performed by two expert pathologists. Seventy-eight percent of the KC pancreata between 13 and 16 months contained high-grade PanIN-3 lesions, characterized by complete loss of polarity, nuclear atypia, and destruction of the basement membrane (Fig. 3A) or pancreatic cancer with surrounding characteristic accumulation of stromata (Fig. 3B). Overall, the KC group had a moderate rate of high-grade acinar metaplasia and low-to-moderate lymphocytic infiltration that was not seen in the GKO/KC cohort. Evaluation of the GKO/KC pancreata revealed mixed phenotypes. Two of 10 animals exhibited focal PanIN-1A lesions, which are characterized by a change from normal cuboidal to columnar morphology with supranuclear cytoplasmic mucin and basally located nuclei (Fig. 3C), or PanIN-1B, in which the development of micropapillary lesions is present. Eighty percent of mice in this cohort were evaluated and diagnosed with pancreata within normal limits (Fig. 3D). Of note, none of the GKO/KC mice examined in this study presented with high-grade PanIN-3 lesions. Interestingly, comparison of GHet/KC mice (with heterozygous loss of GLI1) and KC mice showed similar gross morphology and survival (supplemental Fig. 2A and data not shown) and histological appearance (supplemental Fig. 2B), suggesting that one allele of gli1 is sufficient for tumor development.
FIGURE 3.

Loss of GLI1 blocks PanIN progression in KC mice. A–D, pancreata at ×20 (left panels) and ×40 (right panels) magnifications. A and B, KC pancreas showing developing PanIN-2 and PanIN-3 (A) and pancreatic cancer (B). C and D, examples of the GKO/KC pancreatic phenotypes. C, representative H&E image showing PanIN-1A. D, example of the most common phenotype of the GKO/KC mice. These animals present with pancreata containing normal acini and islet structures.
Recent reports have demonstrated a role for GLI1 in regulation of the pancreatic epithelial tumor compartment (27, 29); however, GLI1 is expressed in both pancreatic cancer and stromal cells (supplemental Fig. 3A). We sought to define the role of stromal GLI1 in KRAS-induced carcinogenesis. To this end, we used a syngeneic model in which murine cancer cells with WT levels of GLI1 were injected orthotopically into the pancreata of WT or GKO mice. The data included in Fig. 4 show that the average tumor volume in WT pancreas was 128.9 mm3 compared with 2.3 mm3 in GKO pancreas (Fig. 4, A and B). Pancreatic histopathology showed a >75% decrease in tumorigenesis in mice lacking GLI1 in the pancreatic microenvironment, whereas 100% of WT mice developed tumors that spread through at least 80% of the pancreas (Fig. 4C). Together, these findings provide evidence that this transcription factor plays a key role in the PanIN lesion progression through regulation of the pancreatic tumor microenvironment.
FIGURE 4.

Loss of GLI1 in the pancreatic microenvironment impairs tumorigenesis. Tumor volume (A) and macroscopic analysis (B) showed that the GKO mice did not develop detectable tumors, whereas most of the WT mice had palpable tumors in all cases. C, representative H&E image of the phenotypes present in these mice. Images were taken at ×1 and ×40 magnifications as indicated.
GLI1 Regulates Activation of IL-6/STAT3 Signaling in the Tumor Microenvironment
Recent reports show a central role for the STAT3 pathway in the regulation of preneoplastic lesion progression and development of pancreatic cancer. Furthermore, STAT3 activation is thought to be involved in driving early changes in the microenvironment promoting PanIN formation in the presence of oncogenic KRAS (16, 17). We examined the level of activity of the pathway by measuring the levels of p-STAT3, the active form of this transcription factor. We observed that p-STAT3 was widely expressed in the cancer cells of pancreatic tumor tissues and in the preneoplastic lesions of KC mice (Fig. 5A, left panel). In contrast, p-STAT3 was significantly reduced and, in some cases, absent in the pancreata of GKO/KC mice (Fig. 5A, middle and right panels). Similar results were obtained by Western blot analysis of pancreatic lysates from KC and GKO/KC mice (supplemental Fig. 3B). Activation of the STAT3 pathway is non-cell-autonomous and likely depends on the tumor microenvironment in pancreatic cancer. Recently, cytokines acting upstream of STAT3, specifically IL-6, have been shown to be important for KRAS-induced transformation (17, 30, 31). Here, we used one of the known IL-6-expressing cellular models in the tumor microenvironment (fibroblasts) to define the role of GLI1 in the regulation of the IL-6/STAT3 axis. IL-6 expression was 20-fold higher in fibroblasts than in tumor cells (supplemental Fig. 3C). Overexpression of GLI1 induced IL-6 mRNA expression compared with the control-transfected cells (Fig. 5B). The basal levels of IL-6 in the culture medium were undetectable. However, overexpression of GLI1 resulted in a robust 25-fold increase in IL-6 secretion (Fig. 5C), suggesting that GLI1 expression in fibroblasts may be one of the drivers of IL-6 expression in the stromal compartment, a component necessary for continued activation of downstream targets leading to carcinogenesis (16, 17, 32–34). To further understand the regulatory mechanism underlying GLI1 modulation of IL-6 expression, we transfected fibroblasts with GLI1 and a control vector along with the full-length mouse IL-6 promoter-luciferase reporter (35). Overexpression of GLI1 increased IL-6 promoter activity by 3–4-fold compared with the control cells (Fig. 5D). The IL-6 promoter sequence contains three candidate binding sites for GLI1 (Fig. 5E). ChIP assay confirmed binding of endogenous GLI1 to a region of the IL-6 promoter located −531 to −800 bp upstream of the first exon (Fig. 5F). Finally, using conditioned medium from pancreatic fibroblasts, we observed that the activation of STAT3 in pancreatic cancer cells was induced by IL-6. Western blot analysis showed a strong induction of p-STAT3 at 24 h, and this effect was decreased in the presence of conditioned medium treated with IL-6-neutralizing antibody (Fig. 5G).
FIGURE 5.

GLI1 modulates IL-6/STAT3 signaling activity. A, p-STAT3 immunohistochemical staining demonstrated increased expression of the activated form of this transcription factor in KC pancreas, but low or no staining was evident in the pancreatic cells of GKO/KC mice. B and C, GLI1 induced >2.5-fold IL-6 mRNA expression (B) and IL-6 secretion (C) as measured by ELISA in WT fibroblasts. abs, absorbance; ND, not detected. D, a 4-fold increase in full-length IL-6 promoter activity in fibroblasts overexpressing GLI1 was determined by luciferase assay. E, bioinformatics analysis showed three candidate GLI1-binding sites (G) in the mouse IL-6 promoter. F, to define IL-6 as the direct target of GLI1, we performed the ChIP assay as described under “Experimental Procedures.” The data show that endogenous GLI1 bound to a region located −531 to −800 bp upstream of the first exon in the promoter. IP, immunoprecipitate. G, p-STAT3 and total STAT3 Western blot analysis showed lower levels of the activated form of this transcription factor in cancer cells incubated with conditioned medium (CM) treated with IL-6-blocking antibody.
Furthermore, signaling within the tumor microenvironment has been implicated as the driver of GLI1 activity in tumors (36, 37). Immunocytochemical studies demonstrated that IL-6 protein expression increased with the addition of SHH ligand in a dose-dependent manner (Fig. 6A). As a control for activation of the pathway by SHH, the GLI1 mRNA levels were measured upon SHH treatment. GLI1 expression was induced in SHH-treated WT fibroblasts. No changes in expression were seen in the GKO fibroblasts (supplemental Fig. 3D). Furthermore, we observed that ligand-dependent activation of SHH signaling induced binding of GLI1 to the mouse IL-6 promoter (Fig. 6B). Next, we sought to determine whether the effect of SHH on the fibroblasts was paracrine. First, we observed that SHH was expressed only in the epithelial compartment. SHH was undetected in the pancreatic fibroblasts (Fig. 6C). Moreover, the expression of SHH was regulated by KRASG12D. Using human and mouse pancreatic cancer cells, we showed an increase in SHH expression and promoter activity upon overexpression of oncogenic mutant KRAS (Fig. 6, D and E). Finally, using a SHH-blocking antibody, we impaired GLI1 activation in fibroblasts treated with conditioned medium from cancer cells overexpressing KRASG12D (Fig. 6F). Together, these results suggest a novel tumor-promoting mechanism that requires GLI1 transcriptional activity in the microenvironment to induce IL-6 secretion and consequently STAT3 activation in the epithelial compartment (Fig. 7).
FIGURE 6.

SHH regulates the activity of the newly identified GLI1/IL-6 axis. A, IL-6 expression was induced in pancreatic fibroblasts treated with SHH in a dose- and time-dependent manner as shown by immunocytochemistry. B, GLI1 binding to the mouse IL-6 promoter was increased by >7-fold in fibroblasts treated with SHH ligand (1.5 μg/ml). IP, immunoprecipitate. C, real-time PCR showed SHH expression in pancreatic cancer cells. SHH was not expressed in fibroblasts. Immunohistochemical analysis confirmed the presence of the SHH ligand exclusively in the tumor compartment (inset, arrows). ND, not detected. D and E, overexpression of human KRASG12D in human (BxPC3) (right panels) and mouse (left panels) KC pancreatic cancer cell lines increased the expression (D) and promoter activity (E) of SHH. F, GLI1-reporter assay of fibroblasts treated with cancer cell-conditioned medium overexpressing KRASG12D. Activity was decreased in the presence of SHH-blocking antibody in the conditioned medium.
FIGURE 7.
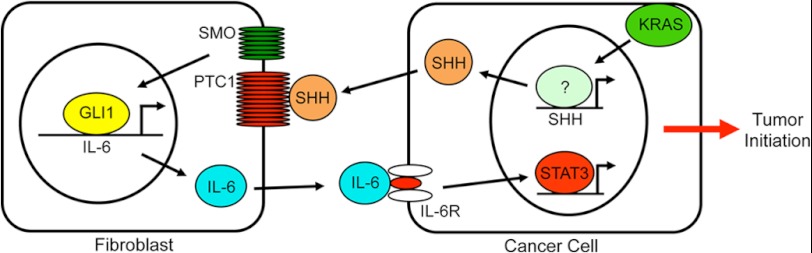
Schematic representation of the model of GLI1-mediated pancreatic carcinogenesis. SMO, Smoothened; IL-6R, IL-6 receptor.
DISCUSSION
KRAS and its downstream effector molecules play a central role in the development of several tumor types, including pancreatic cancer. Given the high frequency of oncogenic KRAS mutations in preneoplastic lesions and the recapitulation of the carcinogenic process in mouse models with an endogenous oncogenic kras allele, activation of this GTPase is believed to be the crucial step in the initiation of pancreatic tumors (38). Here, we identified GLI1 as a new effector of KRAS at early stages of pancreatic carcinogenesis. We have shown that loss of GLI1 impairs KRAS-induced carcinogenesis. KC pancreata show the entire spectrum of precursor lesions, with increasing PanIN stage as observed by others (13). Although GLI1 ablation did not result in complete loss of PanIN lesions, we did observe a marked attenuation of these lesions, and these mice did not develop pancreatic cancer (Fig. 2B). Moreover, we observed prolonged survival of GKO/KC mice relative to KC controls (16 months for KC mice and 21 months for in GKO/KC mice) (Fig. 2A). These results demonstrate an obligatory role for GLI1 in KRAS-induced PanIN progression and pancreatic tumor development.
Initiation of pancreatic carcinogenesis is accompanied by well described changes in epithelial cell morphology and in the surrounding microenvironment. A key step in the development of PanIN lesions is the induction of acinar-to-ductal metaplasia. The onset of acinar-to-ductal metaplasia was substantially reduced in gli1 knock-out (GKO/KC) mice at an early age (data not shown). GKO/KC mice did not develop PanIN-2 or PanIN-3 lesions, and in many cases, the pancreata were normal and lacked any lesions. These results suggest that GLI1 is necessary for the first metaplastic events in acinar cells and potentially for progression of the early lesions to high-grade neoplasia.
In parallel with the development of acinar-to-ductal metaplasia, the activation of stromata surrounding PanIN lesions has been reported in early stages of pancreatic cancer (13, 14). The use of a gli1 total knock-out allowed us to identify a novel signaling network explaining in part the mechanism underlying the cross-talk between the epithelial and stromal compartments during pancreatic cancer initiation. Recent reports established an activated STAT3 (p-STAT3) in pancreatic cancer biology to be essential for PanIN lesion progression (16, 17, 32). Furthermore, in vivo studies demonstrated that p-STAT3 is not dependent on the constitutively active form of mutant kras; rather, activation is dependent on sustained IL-6 secretion. In our studies, we detected high levels of p-STAT3 in KC mice; however, GKO/KC mice had lower levels of this activated form of STAT3, and in some cases, the pancreata showed no expression of p-STAT3. Here, we have provided evidence that GLI1 induces IL-6 expression, secretion, and promoter activity in pancreatic fibroblasts, and this effect was significantly reduced in GKO fibroblasts. This result is particularly important because it provides evidence that GLI1 in the microenvironment is a key protein involved in activating the cytokine that is essential for continued activation of the oncogenic transcription factor STAT3 (16, 17, 32–34). Loss of GLI1 in KC mice leads to fewer PanIN lesions, thus breaking the cycle of sustained IL-6 signaling.
Using a syngeneic model, we confirmed the importance of GLI1 expression in the microenvironment by injecting pancreatic cancer cell lines generated from KC pancreatic tumors (expressing WT GLI1) into the pancreata of WT and GKO mice, resulting in 100% tumor penetrance in WT mice compared with 25% in GKO mice as determined by histopathological analysis. These results complement recent work by Nolan-Stevaux et al. (27) and Rajurkar et al. (29) showing the importance of GLI1 in the epithelial tumor compartment. These reports, together with our studies, support a key role for GLI1 during pancreatic cancer initiation. Additionally, we have defined a model in which KRAS requires active GLI1 in both the epithelial and tumor compartments.
Furthermore, in this study, we have provided evidence of a novel role for SHH in the tumor microenvironment. Multiple reports have shown a crucial function for SHH in regulation of the desmoplastic reaction, a typical feature of pancreatic cancer playing a role in pancreatic carcinogenesis by regulating tumor maintenance and progression (36, 37). Here, we determined that the SHH signaling cascade regulates early stages of pancreatic development. The SHH/GLI1 axis modulates the progression of PanIN lesions by acting in the tumor stromata, regulating the IL-6/STAT3 pathway. In addition, we provided a mechanistic connection between mutant KRAS and activation of the SHH pathway. Oncogenic KRAS induces the expression of SHH in cancer cells through regulation of its promoter.
In summary, our study has defined a novel signaling cascade that identifies the transcription factor GLI1 as a central mediator of the IL-6 signaling network initiated in the tumor microenvironment, regulating the progression of precursor lesions and tumor formation (Fig. 7). When GLI1 is absent, IL-6 signaling from tumor-associated fibroblasts is diminished, and pancreatic precursor lesions do not progress to advanced stages. Future studies are aimed at defining the epigenetic mechanism used by GLI1 to control gene expression as well as the translational significance of targeting this transcription factor in pancreatic cancer as well as other tumors driven by oncogenic KRAS.
Acknowledgments
We thank Emily Porcher for excellent administrative assistance and members of the Fernandez-Zapico laboratory for kind support, constructive criticism, and reading the manuscript in preparation for publication. We express our gratitude to Dr. Menashe Bar-Eli for mentorship. We also thank Drs. Lucas Nacusi and Ben Stanger for insightful comments and discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant CA136526 from NCI. This work was also supported by Mayo Clinic Pancreatic SPORE Grant P50 CA102701 and Mayo Clinic Center for Cell Signaling in Gastroenterology Grant P30 DK84567 (to M. E. F.-Z.).

This article contains supplemental Figs. 1–3.
- LSL
- Lox-Stop-Lox
- KC
- krasG12D;p48-Cre
- PanIN
- pancreatic intraepithelial neoplasia
- GKO
- gli1 knock-out
- p-STAT3
- phospho-STAT3
- SHH
- Sonic hedgehog.
REFERENCES
- 1. Almoguera C., Shibata D., Forrester K., Martin J., Arnheim N., Perucho M. (1988) Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 53, 549–554 [DOI] [PubMed] [Google Scholar]
- 2. Hruban R. H., van Mansfeld A. D., Offerhaus G. J., van Weering D. H., Allison D. C., Goodman S. N., Kensler T. W., Bose K. K., Cameron J. L., Bos J. L. (1993) K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am. J. Pathol. 143, 545–554 [PMC free article] [PubMed] [Google Scholar]
- 3. Caldas C., Kern S. E. (1995) K-ras mutation and pancreatic adenocarcinoma. Int. J. Pancreatol. 18, 1–6 [DOI] [PubMed] [Google Scholar]
- 4. Lüttges J., Klöppel G. (2005) Pancreatic ductal adenocarcinoma and its precursors. Pathologe 26, 12–17 [DOI] [PubMed] [Google Scholar]
- 5. Grünewald K., Lyons J., Fröhlich A., Feichtinger H., Weger R. A., Schwab G., Janssen J. W., Bartram C. R. (1989) High frequency of Ki-ras codon 12 mutations in pancreatic adenocarcinomas. Int. J. Cancer 43, 1037–1041 [DOI] [PubMed] [Google Scholar]
- 6. Mariyama M., Kishi K., Nakamura K., Obata H., Nishimura S. (1989) Frequency and types of point mutation at the 12th codon of the c-Ki-ras gene found in pancreatic cancers from Japanese patients. Jpn. J. Cancer Res. 80, 622–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nagata Y., Abe M., Motoshima K., Nakayama E., Shiku H. (1990) Frequent glycine-to-aspartic acid mutations at codon 12 of c-Ki-ras gene in human pancreatic cancer in Japanese. Jpn. J. Cancer Res. 81, 135–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Van Laethem J. L., Vertongen P., Deviere J., Van Rampelbergh J., Rickaert F., Cremer M., Robberecht P. (1995) Detection of c-Ki-ras gene codon 12 mutations from pancreatic duct brushings in the diagnosis of pancreatic tumours. Gut 36, 781–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barbacid M. (1987) ras genes. Annu. Rev. Biochem. 56, 779–827 [DOI] [PubMed] [Google Scholar]
- 10. McCormick F. (1989) ras GTPase activating protein: signal transmitter and signal terminator. Cell 56, 5–8 [DOI] [PubMed] [Google Scholar]
- 11. Fan J., Bertino J. R. (1997) K-ras modulates the cell cycle via both positive and negative regulatory pathways. Oncogene 14, 2595–2607 [DOI] [PubMed] [Google Scholar]
- 12. Ellis C. A., Clark G. (2000) The importance of being K-Ras. Cell. Signal. 12, 425–434 [DOI] [PubMed] [Google Scholar]
- 13. Hingorani S. R., Petricoin E. F., Maitra A., Rajapakse V., King C., Jacobetz M. A., Ross S., Conrads T. P., Veenstra T. D., Hitt B. A., Kawaguchi Y., Johann D., Liotta L. A., Crawford H. C., Putt M. E., Jacks T., Wright C. V., Hruban R. H., Lowy A. M., Tuveson D. A. (2003) Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450 [DOI] [PubMed] [Google Scholar]
- 14. Tuveson D. A., Shaw A. T., Willis N. A., Silver D. P., Jackson E. L., Chang S., Mercer K. L., Grochow R., Hock H., Crowley D., Hingorani S. R., Zaks T., King C., Jacobetz M. A., Wang L., Bronson R. T., Orkin S. H., DePinho R. A., Jacks T. (2004) Endogenous oncogenic K-rasG12D stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 5, 375–387 [DOI] [PubMed] [Google Scholar]
- 15. Ji Z., Mei F. C., Xie J., Cheng X. (2007) Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 282, 14048–14055 [DOI] [PubMed] [Google Scholar]
- 16. Fukuda A., Wang S. C., Morris J. P., 4th, Folias A. E., Liou A., Kim G. E., Akira S., Boucher K. M., Firpo M. A., Mulvihill S. J., Hebrok M. (2011) Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 19, 441–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lesina M., Kurkowski M. U., Ludes K., Rose-John S., Treiber M., Klöppel G., Yoshimura A., Reindl W., Sipos B., Akira S., Schmid R. M., Algül H. (2011) Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 19, 456–469 [DOI] [PubMed] [Google Scholar]
- 18. Bai C. B., Auerbach W., Lee J. S., Stephen D., Joyner A. L. (2002) Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 129, 4753–4761 [DOI] [PubMed] [Google Scholar]
- 19. Park H. L., Bai C., Platt K. A., Matise M. P., Beeghly A., Hui C. C., Nakashima M., Joyner A. L. (2000) Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 127, 1593–1605 [DOI] [PubMed] [Google Scholar]
- 20. Elsawa S. F., Almada L. L., Ziesmer S. C., Novak A. J., Witzig T. E., Ansell S. M., Fernandez-Zapico M. E. (2011) GLI2 transcription factor mediates cytokine cross-talk in the tumor microenvironment. J. Biol. Chem. 286, 21524–21534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kanda M., Matthaei H., Wu J., Hong S. M., Yu J., Borges M., Hruban R. H., Maitra A., Kinzler K., Vogelstein B., Goggins M. (2012) Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 142, 730–733.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hruban R. H., Goggins M., Kern S. E. (1999) Molecular genetics and related developments in pancreatic cancer. Curr. Opin. Gastroenterol. 15, 404–409 [DOI] [PubMed] [Google Scholar]
- 23. Hruban R. H., Goggins M., Parsons J., Kern S. E. (2000) Progression model for pancreatic cancer. Clin. Cancer Res. 6, 2969–2972 [PubMed] [Google Scholar]
- 24. Furukawa T., Klöppel G., Volkan Adsay N., Albores-Saavedra J., Fukushima N., Horii A., Hruban R. H., Kato Y., Klimstra D. S., Longnecker D. S., Lüttges J., Offerhaus G. J., Shimizu M., Sunamura M., Suriawinata A., Takaori K., Yonezawa S. (2005) Classification of types of intraductal papillary-mucinous neoplasm of the pancreas: a consensus study. Virchows Arch. 447, 794–799 [DOI] [PubMed] [Google Scholar]
- 25. Brat D. J., Lillemoe K. D., Yeo C. J., Warfield P. B., Hruban R. H. (1998) Progression of pancreatic intraductal neoplasias to infiltrating adenocarcinoma of the pancreas. Am. J. Surg. Pathol. 22, 163–169 [DOI] [PubMed] [Google Scholar]
- 26. Brockie E., Anand A., Albores-Saavedra J. (1998) Progression of atypical ductal hyperplasia/carcinoma in situ of the pancreas to invasive adenocarcinoma. Ann. Diagn. Pathol. 2, 286–292 [DOI] [PubMed] [Google Scholar]
- 27. Nolan-Stevaux O., Lau J., Truitt M. L., Chu G. C., Hebrok M., Fernández-Zapico M. E., Hanahan D. (2009) GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 23, 24–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Olive K. P., Tuveson D. A. (2006) The use of targeted mouse models for preclinical testing of novel cancer therapeutics. Clin. Cancer Res. 12, 5277–5287 [DOI] [PubMed] [Google Scholar]
- 29. Rajurkar M., De Jesus-Monge W. E., Driscoll D. R., Appleman V. A., Huang H., Cotton J. L., Klimstra D. S., Zhu L. J., Simin K., Xu L., McMahon A. P., Lewis B. C., Mao J. (2012) The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 109, E1038–E1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chan K. S., Sano S., Kiguchi K., Anders J., Komazawa N., Takeda J., DiGiovanni J. (2004) Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J. Clin. Invest. 114, 720–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jenkins B. J., Grail D., Nheu T., Najdovska M., Wang B., Waring P., Inglese M., McLoughlin R. M., Jones S. A., Topley N., Baumann H., Judd L. M., Giraud A. S., Boussioutas A., Zhu H. J., Ernst M. (2005) Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF-β signaling. Nat. Med. 11, 845–852 [DOI] [PubMed] [Google Scholar]
- 32. Corcoran R. B., Contino G., Deshpande V., Tzatsos A., Conrad C., Benes C. H., Levy D. E., Settleman J., Engelman J. A., Bardeesy N. (2011) STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 71, 5020–5029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hodge D. R., Peng B., Cherry J. C., Hurt E. M., Fox S. D., Kelley J. A., Munroe D. J., Farrar W. L. (2005) Interleukin 6 supports the maintenance of p53 tumor suppressor gene promoter methylation. Cancer Res. 65, 4673–4682 [DOI] [PubMed] [Google Scholar]
- 34. Ancrile B., Lim K. H., Counter C. M. (2007) Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 21, 1714–1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baccam M., Woo S. Y., Vinson C., Bishop G. A. (2003) CD40-mediated transcriptional regulation of the IL-6 gene in B lymphocytes: involvement of NF-κB, AP-1, and C/EBP. J. Immunol. 170, 3099–3108 [DOI] [PubMed] [Google Scholar]
- 36. Bailey J. M., Mohr A. M., Hollingsworth M. A. (2009) Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene 28, 3513–3525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bailey J. M., Swanson B. J., Hamada T., Eggers J. P., Singh P. K., Caffery T., Ouellette M. M., Hollingsworth M. A. (2008) Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 14, 5995–6004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Löhr M., Klöppel G., Maisonneuve P., Lowenfels A. B., Lüttges J. (2005) Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia 7, 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]