

Background: c-di-AMP-degrading YybT homologs contain a PAS domain with heme binding capability.
Results: The solution structure of the PAS domain was determined to reveal a potential ligand-binding site.
Conclusion: PASYybT domains function as ligand-binding regulatory domains.
Significance: Cellular cyclic di-AMP levels are likely to be regulated by a small-molecule ligand.
Keywords: Antibiotic Resistance, Bacterial Signal Transduction, Cell Wall, Cyclic Nucleotides, Heme, GdpP, Heme, PAS Domain, YybT, Cyclic Di-AMP
Abstract
The Bacillus subtilis protein YybT (or GdpP) and its homologs were recently established as stress signaling proteins that exert their biological effect by degrading the bacterial messenger cyclic di-AMP. YybT homologs contain a small Per-ARNT-Sim (PAS) domain (∼80 amino acids) that can bind b-type heme with 1:1 stoichiometry despite the small size of the domain and the lack of a conserved heme iron-coordinating residue. We determined the solution structure of the PAS domain of GtYybT from Geobacillus thermodenitrificans by NMR spectroscopy to further probe its function. The solution structure confirms that PASGtYybT adopts the characteristic PAS fold composed of a five-stranded antiparallel β sheet and a few short α-helices. One α-helix and three central β-strands of PASGtYybT are noticeably shorter than those of the typical PAS domains. Despite the small size of the protein domain, a hydrophobic pocket is formed by the side chains of nonpolar residues stemming from the β-strands and α-helices. A set of residues in the vicinity of the pocket and in the C-terminal region at the dimeric interface exhibits perturbed NMR parameters in the presence of heme or zinc protoporphyrin. Together, the results unveil a compact PAS domain with a potential ligand-binding pocket and reinforce the view that the PASYybT domains function as regulatory domains in the modulation of cellular cyclic di-AMP concentration.
Introduction
YybT homologs are widely distributed among the phylum firmicutes, including such human pathogens as Staphylococcus aureus, Streptococcus mutans, and Listeria monocytogenes. Although early studies showed that the disruption of the LlYybT-encoding llmg_1816 gene rendered Lactococcus lactis more tolerant to acid stress (1), the biological function of YybT proteins remained obscure until the discovery that the DHH/DHHA1 domain of BsYybT from Bacillus subtilis possesses phosphodiesterase activity toward cyclic di-AMP (c-di-AMP)4 (2), a newly discovered messenger molecule found in Gram-positive bacteria (3, 4). Since then, the YybT family proteins have been established as c-di-AMP signaling proteins that play crucial roles in stress signaling and peptidoglycan biosynthesis in several bacterial species. It was shown that the ΔYybT strain of B. subtilis becomes more resistant to acid and nalidixic acid-caused DNA damage (2). The YybT homolog (SA0013) in S. aureus plays a role regulating the cross-linking process during peptidoglycan biosynthesis (5). The disruption of the SA0013 gene abolished the secretion of the virulence factor hemolysin for iron acquisition (6), suggesting that SA0013 could be crucial for the in vivo survival of S. aureus. More recent studies also revealed that c-di-AMP is an essential signaling molecule in B. subtilis and S. aureus cell wall homeostasis and antibiotic resistance (7, 8) and that the inactivation of homologous gene llmg_1816 led to temperature resistance and salt hypersensitivity in L. lactis (9). Given the importance of c-di-AMP and YybT proteins in cell wall formation and antibiotic resistance, YybT proteins may be exploited as novel targets for developing antimicrobial agents.
YybT family proteins are predicted to contain three protein domains in addition to the two N-terminal transmembrane helices (Fig. 1). The three domains include a Per-ARNT-Sim (PAS) domain, a GGDEF domain, and a DHH/DHHA1 domain. The C-terminal DHH/DHHA1 domain functions as a c-di-AMP-specific phosphodiesterase, with its biological role supported by both in vitro and in vivo studies (2, 5, 10). Unlike the orthodox GGDEF domains that function as diguanylate cyclases for synthesizing c-di-GMP (11), the GGDEFYybT domain lacks the GG(D/E)EF motif and exhibits residual ATPase activity (2). YybT family proteins also contain a putative PAS domain immediately following the second transmembrane helix. PAS domains are found in all kingdoms of life, and many of them are predicted to function as sensor domains for perceiving environmental cues (12). PAS domains are usually about 100 amino acids in length and are fused to a wide range of enzymatic or nonenzymatic effector domains. The capability of the PAS domains to bind a variety of cofactors or ligands is largely due to the plasticity of the ligand-binding pocket (13). By binding heme or flavin as cofactors or prosthetic groups, some PAS domains are known to sense signals such as gaseous ligands (14, 15), redox potential (16–19), or light (20–22). In addition, many of the PAS domains with structures deposited in the Protein Data Bank (PDB) contain hollow pockets that are likely used for binding ligands of diverse sizes and properties.
FIGURE 1.
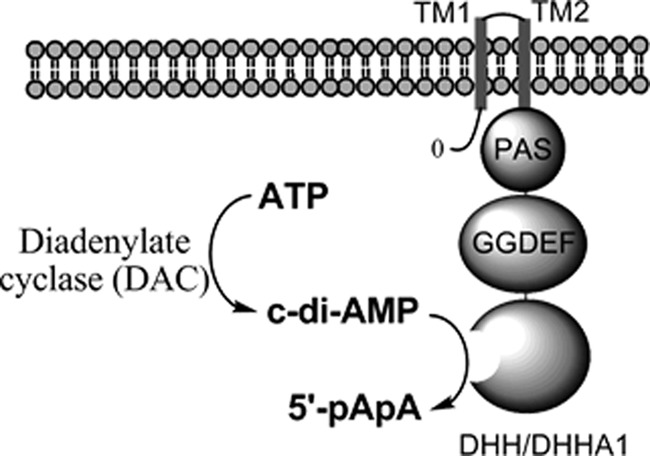
YybT family proteins and c-di-AMP signaling. The YybT protein contains two N-terminal transmembrane (TM) domains, a PAS domain, a GGDEF domain and a DHH/DHHA1 domain.
We previously reported the unexpected finding that two recombinant YybT proteins were associated with a substoichiometric amount of b-type heme (<5%) even after the protein purification by metal affinity and size-exclusion chromatography (23). By heme reconstitution, the PAS domains of the two YybT proteins were shown to bind b-type heme with 1:1 stoichiometry. These observations were surprising because of two reasons. First, the core of the PAS domain of YybT proteins contains 70–80 residues, which makes it a much smaller PAS domain than other heme-binding PAS domains that typically contain ∼100 residues (13, 14, 24, 25). Second, the PAS domains of YybT proteins do not seem to harbor any conserved residues for coordinating heme iron. In addition to the observed specificity for b-type heme, the binding of heme seems to be biologically relevant because the heme binding also alters the phosphodiesterase activity of the DHH/DHHA1 domain of YybT (23). To further probe the structure and function of the PASYybT domain, we determined the high-resolution NMR structure of the PAS domain (GtYybT) of the YybT protein from the thermophilic microorganism G. thermodenitrificans to reveal that the small PASGtYybT domain adopts a PAS fold with a putative hydrophobic ligand-binding site surrounded by shortened strands and loops. A model of the dimeric protein was constructed to reveal that the PAS domain dimerizes in an antiparallel fashion. Results obtained from the NMR titration with reconstituted heme and zinc protoporphyrin IX (ZnPPIX) suggest that the hydrophobic pocket is potentially involved in the binding of heme.
MATERIALS AND METHODS
Cloning, Expression, and Purification of Proteins
Cloning of the gene that encodes GtYybT55–162 was described previously (2). For the production of the 13C/15N-labeled protein, Escherichia coli were grown up in 1 liter of M9 medium supplemented with 1 g/liter [15N]ammonium chloride and 1.2 g/liter [13C]glucose to an optical density of 0.8 before inducing with 0.8 mm isopropyl-1-thio-β-d-galactopyranoside. The culture was kept shaking at 16 °C for ∼12 h before it was pelleted by centrifugation. The cells were lysed in 20 ml of lysis buffer (50 mm Tris (pH 8.0), 150 mm NaCl, 5% glycerol, 0.1% β-mercaptoethanol, and 1 mm PMSF). After centrifugation at 25,000 rpm for 30 min, the supernatant was filtered and then incubated with 2 ml of nickel-nitrilotriacetic acid resin (Qiagen) for 1 h at 4 °C. The resin was washed with 50 ml of W1 buffer (lysis buffer with 20 mm imidazole) and 20 ml of W2 buffer (lysis buffer with 50 mm imidazole). The proteins were eluted using a step gradient method with the elution buffer containing 50 mm Tris (pH 8.0), 150 mm NaCl, 5% glycerol and 200 mm, 300 mm, or 500 mm imidazole. The eluted protein was incubated with thrombin overnight at room temperature before size-exclusion chromatography at 4 °C using the ÄKTA FPLC system equipped with a Superdex 200 HR 16/60 column (Amersham Biosciences). The buffer used for gel filtration was composed of 50 mm phosphate buffer (pH 6.5), 200 mm NaCl and protease inhibitor mixture (Roche Applied Science). The protein was concentrated to the required NMR concentration (0.7 mm) and stored at −80 °C.
NMR Structure Determination
The structure of the PAS domain was calculated using the assignment of 1H, 13C, and 15N resonances and unassigned NOEs as input for the program CYANA (26, 27). Backbone dihedral angles were predicted using chemical shift data, with the website version of TALOS+ (spin.niddk.nih.gov/bax/nmrserver/talos/) (28). Hydrogen bond constraints for α-helices and β-strands were set for amide protons protected from D2O exchange. Structure calculations were started from 100 random conformers, using the standard simulated annealing protocol in CYANA. The statistics of meaningful NOE distance constraints in the final CYANA cycle showed a high density of structural constraints per amino acid. Seven cycles of NOE assignment and structure reconstruction resulted in a bundle of 20 conformers. All data sets used for CYANA calculation were reformatted for CNS using PDBstat 5.3 (29). The program CNS 1.2 (30) was used to refine the PAS structure by incorporating dihedral angles, hydrogen bonds, and upper limit distance constrains (from CYANA) and refined using residual dipolar coupling data obtained using pf1 filamentous bacteriophages. Structures were recalculated from an extended strand with random initial velocities using the default simulated annealing protocol of the CNS package. 100 conformers were calculated, of which the structure with the lowest energy structure was selected for the HADDOCK procedure.
To build the dimer model, the multibody interface of the HADDOCK web server-based calculation (31) were performed from two copies of monomeric structure. Center-of-mass restraints, C2 symmetry restraints, and noncrystallographic restraints were imposed on all sequential monomer pairs. The HADDOCK procedure was repeated twice with the truncated version GtYybTtrun (Glu-77–Thr-162) and GtYybTR50-T162 to test the robustness of the experimental intermonomer NOEs. To test the consistency of the data, and to prevent possible occlusion and steric hindrance during the docking process, residues of the A′α domain (Arg-50–Ser-76) were first removed from the monomer to GtYybT PAStrun. The NOE set was first used as ambiguous interaction restraints for preliminary building of the GtYybT PAStrun dimer model followed by refinement of these restraints to obtain an unambiguous restraint set resulting in the full atomic description of the dimer structure. Starting with two copies of the GtYybT PAStrun structure, HADDOCK produced a single cluster of dimer structures with an overall backbone r.m.s.d. from the lowest energy structure of 0.5 ± 0.3 Å. The docking procedure was then repeated for GtYybTR50-T162 using the same unambiguous restraint set.
NMR Spectroscopic Study of the Paramagnetic Tagged PASGtYybT
To probe the orientation of the PASGtYybT domain, we attached the paramagnetic tag (1-oxyl-2, 2, 5, 5-tetramethyl-Δ3-pyrroline-3-methyl) methanethiosulfonate (MTSL) (Toronto Research) to the native cysteine residue (Cys-106) of the PAS domain. To prepare the protein-MTSL conjugate, the purified protein was first added to a 10-fold excess volume of buffer at pH 8.5 (200 mm NaCl, 50 mm Tris, pH 8.5). 1.0 mg of MTSL (5-fold) was dissolved in 20 μl of acetonitrile and added to the protein solution. The mixture was incubated at 16 °C overnight. Unreacted MTSL was removed via buffer exchange with NMR-based buffer (200 mm NaCl, 50 mm phosphate, pH 6.5) using a concentrator. Fresh sodium dithionite (5-fold) was added 1 h before acquisition of NMR spectra to reduce the nitroxide probe.
NMR experiments were performed at 25 °C using Bruker Avance-II 700 and 600 NMR spectrometers equipped with cryogenic triple resonance (TXI) probes. Complete sequence-specific assignment of backbone was achieved using standard triple resonance experiments (HNCA, HN(CO)CA, CBCACONH, HN(CA)CB, HNCO, and HN(CA)CO), and side-chain and aromatic resonances were assigned using three-dimensional 15N-resolved NOESY-HSQC and three-dimensional 13C-resolved NOESY-HMQC. For long range intermonomer NOEs, three-dimensional 13C-resolved NOESY-HMQC and three-dimensional 15N-resolved NOESY-HSQC were used to determine the NOEs that could not be resolved as intramonomeric NOEs. Sodium 2,2-dimethyl-2-silapentane-5-sulfonate was used as the internal reference for 1H nuclei. The chemical shifts of 13C and 15N nuclei were calculated from the 1H chemical shifts (32). To identify residues protected from D2O exchange, the NMR sample was buffer-exchanged with the same buffer in 99% D2O and left to equilibrate for more than 6 h. 1H-15N heteronuclear steady-state NOEs were obtained by recording spectra with and without a 3-s 1H presaturation delay at 700-MHz 1H frequency. In addition, dilution series (0.16, 0.33, 0.5 mm) 1H-15N TROSY-HSQC spectra were measured. Numbers of scans were increased accordingly to maintain similar signal-to-noise ratios. Changes in the spectra were monitored by chemical shift perturbations.
Preparation of NMR Samples Weakly Aligned in Polarizing Magnetic Field
1H-15N residual dipolar couplings were determined using TROSY-anti-TROSY spectra in the absence/presence of 20 mg/ml Pf1 filamentous bacteriophage (ASLA Biotech). The acquired data were analyzed with MODULE (33).
Paramagnetic Reagent Titration
To delineate the solvent-exposed surfaces and the buried dimeric interface of the PAS domain, gadodiamide (Omniscan, GE Healthcare) was used. The paramagnetic relaxation rate was determined by measuring the decay in signal intensity in 1H-15N TROSY-HSQC spectra in the presence of increasing gadodiamide concentration (1–5 mm) (34–36).
Reconstitution of PASGtYybT with Heme or Zinc Protoporphyrin IX
Heme stock (Sigma) or ZnPPIX (Alfa Aesar) was first dissolved in D6-DMSO (Cambridge Isotope Laboratories) for the incorporation of heme into the PAS protein sample. The pH of the protein sample was first increased to pH 8.5 before slowly titrating with the ZnPPIX stock in DMSO, with gentle agitation for ∼4 h (4 °C) to a final ZnPPIX/protein ratio of 5:1. The free ZnPPIX was separated from the reconstituted protein solution by gel filtration. The protein solution was then exchanged back to the original buffer (50 mm phosphate buffer, pH 6.5, 200 mm NaCl) to ensure removal of any unreacted chemicals and precipitated protein. To reconstitute the protein with ferric heme, heme hydrochloride was first dissolved in D6-DMSO and titrated into purified protein solution at pH 8.0 with gentle agitation for 4 h with the final heme/protein ratio of 4:1. The unbound heme was then separated by gel filtration and exchanged back into the NMR buffer via buffer exchange concentrator.
NMR Spectroscopic Study of the Reconstituted PASGtYybT
Two different approaches were used to probe the heme-binding pocket of the PAS domain. In the first approach, chemical shift perturbations/signal attenuation were monitored using 1H-15N TROSY-HSQC spectra, and the magnitude of the perturbation by ZnPPIX binding was calculated using the weighted average chemical shifts for each amino acid residue. The scale factor α was established from atom-specific chemical shift ranges for proteins. The second approach involved using PAS domain reconstituted with heme. The TROSY-HSQC pulse sequence was modified with the super-WEFT (water-edited Fourier transform) element (d1 − 180° − Δ − 90° − ta) with delays set at Δ = 187 ms and ta + d1 = 302 ms, which allows the suppression of slow relaxing signals while monitoring fast relaxing signals (37). Resonances showing opposite amplitude signal intensity in comparison with the resonances stemming from residues remote to the heme-binding pocket were interpreted as being in the vicinity of the heme-binding site.
Heme Sensitivity Assay
A disk diffusion assay similar to that described previously(9, 38) was used to determine heme sensitivity of different L. lactis strains. The heme stock (porcine heme; Sigma) was prepared in 0.23 m sterile NaOH at 7.5 mg/ml. Strains tested included wild-type L. lactis MG1363, the ΔLlYybT mutant (Δ1816), and a wild-type reverted strain that was created by excision of the integration plasmid from Δ1816 (1816Rwt) (9). 100 μl of late-log phase grown cells (A600 nm ∼1.4) was added to 5 ml of soft M17 agar (0.75% agar) containing 0.5% glucose (GM17) and poured on a set 15-ml base of GM17 agar (1.2% agar). In the case of the Δ1816 mutant, erythromycin (Sigma) was added at 2 μg/ml final concentration. A sterile 8-mm filter disk (Advantec) was placed on the surface, and 20 μl of heme (230 nmol) was added. Plates were incubated overnight aerobically at 30 °C and then photographed. The experiment was repeated on three separate occasions with comparable results. Statistical analysis was carried out on triplicate assays using the Student's t test.
RESULTS
PASGtYybT Domain Adopts a PAS Fold with Shortened β-Strands
Using the standard triple resonance NMR experiments with uniformly 13C/15N-labeled protein, we determined the high resolution structure of the PAS domain (PASGtYybT or GtYybT55–162) of GtYybT. 1H-15N TROSY-HSQC spectra showed good dispersion of the cross-peaks (data now shown), indicating that the protein is well folded. In the buffer that contains 99% D2O, most 1H-15N correlation cross-peaks retain significant intensity in the 1H-15N HSQC spectrum, suggesting high protection of the amide protons from the exchange with bulk solvent. The structure of the PAS domain was solved using more than 2000 structural constrains that include interproton NOEs, hydrogen bonds, dihedral angles, and residual dipolar couplings. The overall r.m.s.d. in the final NMR ensemble of the PAS conformers is 0.46 Å for the backbone atoms and 1.04 Å for the heavy side-chain atoms. The core of the PAS domain (Pro-84–Thr-162) adopts the typical α/β-PAS domain fold that consists of several short and distorted α-helices and one five-stranded antiparallel β-sheet (Fig. 2, A and B). Notably, the structure distinguishes itself from other known PAS domains by containing a few short structural motifs to form the twisted β-sheet scaffold. When compared with the central β-strands of other PAS domains that contain 35–50 residues, the β-strands of PASGtYybT only contain 27 residues. The strands Gβ, Hβ, and Iβ are noticeably shorter than those of most known PAS domains (Fig. 2C). In addition, one of the helices (Fα) and the downstream loop are significantly shorter than those of other PAS domains. We also noticed that helix Dα appears to be disordered in our PASGtYybT structure. To ensure consistent naming convention with other PAS domains, we named this region the D-loop. With the shortened strands and loops, PASGtYybT is one of the most compact PAS domains for which the structure has been determined.
FIGURE 2.

Overall structure of PASGtYybT. A, front and back view of the PAS domain (Pro-84–Thr-162) showing a mixed α/β-domain fold, consisting of α-helices flanked by a five-stranded antiparallel β-sheet scaffold. Two loop regions are indicated. The figure was generated using MOLMOL (47). B, secondary structure plot for PASGtYybT with the NOE intensities depicted as bands of varying thickness. Sequential and medium range NOE connectivities are shown below the primary sequence. C, sequence alignment of seven PASGtYybT homologs with the two typical heme-binding PAS domains from BjFixL and EcDos. Ba, B. anthracis; Bs, B. subtilis; Gt, G. thermodenitrificans; Sm, S. mutans; Ll, L. lactis; Lm, L. monocytogenes; Sa, S. aureus; Ec, E. coli; Bj, Bradyrhizobium japonicum.
The PASGtYybT construct used for the NMR study also includes a short stretch of N-terminal residues (Arg-55–Met-83) that is important for maintaining the stability of the construct because its absence resulted in severe protein aggregation during protein purification and NMR measurement. The N-terminal portion consists of two helices connected by an unstructured loop region, in contrast to a continuous long helix as predicted by PSIPRED V3.0. Despite the presence of the unstructured region, the overall r.m.s.d. of this fragment was about 1.75 Å for backbone atoms and 2.79 Å for heavy side-chain atoms. The large r.m.s.d. values obtained are primarily due to the lack of secondary structure and medium range NOEs within the region of Ile-65–Met-83, which was further corroborated by the overall lower number of long range NOEs (Fig. 2B).
To characterize the dynamic properties of the PASGtYybT domain in solution, we measured the 1H-15N steady-state NOE values for the backbone amide groups. The N-terminal region and the PAS domain core exhibit an average heteronuclear NOE of 0.67 and 0.77, respectively, suggesting that both the N-terminal portion and the core regions are relatively rigid. Residues that exhibit the lowest heteronuclear NOE values (below 0.5) were observed at the dimeric interface between the GH-loop and the N-terminal fragment (Met-81, Gln-82, Arg-151, and Thr-162) or within or near the A′α domain (Ser-56, Ile-65, Arg-101, Arg-115, and Arg-154).
PASGtYybT Dimerizes through the Central β-Sheet in an Antiparallel Fashion
Size-exclusion chromatography indicates that PASGtYybT is present as a dimer in solution. At low and high protein concentrations (0.16 and 0.5 mm), the 1H-15N TROSY-HSQC spectra showed very small differences in chemical shift for all residues, indicating strong interaction between the two monomers (data not shown). PAS domains are known to dimerize in a number of different ways featuring either parallel or antiparallel arrangements. We used the gadolinium-based chelate compound (Omniscan, a commonly used contrast agent in MRI studies) to probe the dimer structure (36). We gradually added the agent into the protein solution and monitored changes in the 1H-15N TROSY-HSQC spectrum. Cross-peaks stemming from the solvent-exposed residues are expected to experience stronger relaxation attenuation when compared with to residues buried within the hydrophobic core or within the monomer-monomer interface. We found that the central β-strands of the PAS domain are inaccessible to the paramagnetic probe, suggesting that this region is most likely involved in dimerization (Fig. 3, A–C). On the opposite ends of the PAS domain, the strands Bβ and Gβ exhibit significant differences in the extent of relaxation enhancement. Strand Gβ exhibits an alternating pattern that is consistent with a solvent-exposed β-strand structure. Conversely, the strand Bβ and the D-loop located at the opposite side show only minor relaxation enhancement. This phenomenon is further confirmed by the different degree of solvent exposure in the D2O-containing buffer, where amides of Bβ showed little or no exchange with D2O, indicating that this region is protected against solvent exchange (data not shown). The data also suggest that the distorted helices in the N-terminal stretch are likely to be located next to strand Bβ and undergo increased relaxation enhancement, thus providing the protective effect against the solvent soluble paramagnetic agent.
FIGURE 3.

Determination of the dimer structure of PASGtYybT. A, front and back view of the PASGtYybT domain colored according to the extent of relaxation enhancement. Green, 0–5 s−1 mm−1; yellow, 5–10 s−1 mm−1; orange, 10−20 s−1 mm−1; red, above 20 s−1 mm−1; gray, data unavailable. B, plot of the selected residues showing signal attenuation in the presence of gadodiamide. Normalized relative intensities are fitted to the equation Ii(c)/Iref = exp(−ϵicT), where c is the concentration of paramagnetic reagent, T is the constant-time relaxation delay (T = 16.5 ms), and ϵ is the relaxation enhancement (39). C, plot of paramagnetic relaxation enhancement ϵ, with respect to protein sequence. Dotted lines represent the respective cutoffs according to the extent of relaxation enhancement presented above. D, front and back view of the PASGtYybT domain colored according to the extent of signal attenuation caused by the paramagnetic MTSL tag. Green, above 0.9; yellow, 0.5–0.9; orange, 0.1–0.5; red, 0–0.1; gray, data unavailable. E, 15N TROSY-HSQC spectrum of PASGtYybT in the presence of oxidized MTSL (red) and reduced MTSL (black) in the presence of 5-fold sodium dithionite. F, plot of signal attenuation Iox/Ired (where ox and red indicate oxidized and reduced) with respect to protein sequence. Dotted lines represent the respective cutoffs according to the extent of relaxation enhancement presented above. G, top scoring structure of GtYybTtrun from HADDOCK (r.m.s.d. from overall lowest energy structure = 0.5 ± 0.3 Å) with a total buried surface area of around 1500 Å2. H, top scoring structure of PASGtYybT from HADDOCK (r.m.s.d. from overall lowest energy structure = 0.8 ± 0.5 Å) with a total buried surface area of around 1900 Å2. I, electrostatic potential mapping of PAS monomers showing complementary electrostatic charges of Glu-78/Lys-59, Glu-135/Lys-146, and Arg-151/Thr-162, whereas the side chains of the central β-strands are primarily hydrophobic in nature.
To probe the relative orientation of the PAS subunits, we attached the paramagnetic tag MTSL to the cysteine residue (Cys-106). Cys-106 is positioned at the end of the helix Cα and within the proximity of a β-hairpin loop formed between strands Gβ and Hβ (GH-loop). The use of the paramagnetic MTSL tag allowed us to distinguish the parallel and antiparallel dimerization by selective broadening of the resonances on either end of the PAS domain. A parallel orientation would result in selective broadening of the resonances within the vicinity of the tag, and not across the monomer, whereas an antiparallel orientation would cause broadening at both ends of the PAS domain. Comparison of the 1H-15N TROSY-HSQC spectra for the samples taken before and after the treatment with the paramagnetic tag showed that most resonances were not affected by MTSL. However, the peak intensity of a subset of resonances from the residues located near Cys-106 was reduced in the presence of the paramagnetic tag (Fig. 3, D–F). The majority of residues in the N-terminal fragment were shown to be unperturbed by the presence of the MTSL tag, with the exception of several residues from the N-terminal fragment (residues between Met-54 and His-58). Two resonances corresponding to residues Arg-151 and His-152 from the HI-loop were also broadened, suggesting that these residues are in the vicinity of the MTSL tag at the opposite end of the protein. Considering that the two sets of residues from the N-terminal fragment and GH-loop are located within spatial proximity of each other, these results strongly suggest that the PAS monomers are arranged in an antiparallel fashion within the dimer.
A model of the dimeric PASGtYybT was constructed by using the HADDOCK web server by using two copies of monomers (31). The generated model features an antiparallel arrangement that was further corroborated by the presence of nine experimental NOEs that exhibit clear, detectable, and unique connectivities. The final model with the largest buried surface area of about 1900 Å also showed the lowest overall backbone r.m.s.d. from the lowest energy structure of 0.8 ± 0.5 Å (Fig. 3, G–I). Besides the electrostatics matching of residues between Glu-135/Lys-146 and Arg-151/Thr-162, an additional set of interactions between Glu-78/Lys-59 of the A′α domain was observed. A total of nine intermolecular NOEs were found primarily between the HI-loop (Arg-151–Leu-155) and a stretch of residues from the N-terminal fragment (Ala-79–Met-83). Overall, the dimeric structure of PASGtYybT bears some resemblance to the structures of the ARNT PAS-B homodimer (PDB ID: 2HV1) and HIF-2a/ARNT PAS-B heterodimer (PDB ID: 2A24) (40), which also form homo- or heterodimer interface through the central β-sheet in a parallel or antiparallel manner (40).
PASGtYybT Contains a Hydrophobic Pocket Formed between the β-Sheet and the D-loop
Inspection of the PASGtYybT monomer structure revealed a hydrophobic cavity formed by backbone and side chains of predominately nonpolar residues Leu-88, Leu-103, Ala-104, Phe-107, Glu-109, Leu-112, Leu-117, Leu-120, Ser-121, Leu-124, Leu-145, and Phe-158 (Fig. 4, A and B). The lack of suitable heme coordinating axial ligand (e.g. histidine or cysteine) indicates that if heme is bound in this pocket, the binding is likely to be driven by hydrophobic interactions. When compared with the heme-binding PAS domains of FixL and DosP (25, 41), where the heme-binding pocket is located between the β-sheet and the Fα-helix, the putative ligand-binding pocket of PASGtYybT is located between the β-sheet and the D-loop. In fact, the position of the pocket in PASGtYybT appears to resemble these of DucS, CitA, and DctB (42–44). For these PAS domains, the ligand-binding site is found to be within the vicinity of Fα helix and the GH-loop with various charged side-chain groups and backbone amides used to bind the ligands such as malate, fumarate, citrate, or succinate.
FIGURE 4.

PASGtYybT contains a hydrophobic pocket covered by the dynamic D-loop. A, surface representation of the PASGtYybT showing a hydrophobic pocket as the potential ligand-binding pocket. B, Thr-110- and Ile-112-containing D-loop forms a lid over the hydrophobic pocket. C and D, 1H-13C HSQC spectra indicating that the methyl groups of Thr-111 and Ile-113 adopt two conformations. E and F, 13C NOESY-HMQC strips for the major and minor conformations showing the presence/absence of water NOEs, indicating that the side chains of Thr-111 and Ile-113 can adopt solvent-exposed (major) and buried (minor) conformations.
When compared with other heme-binding PAS domains, the binding pocket of PASGtYybT seems to be relatively small with a narrow entrance (Fig. 4, A and B). To bind heme, significant conformational changes are required to enlarge the pocket and broaden the entrance. The side-chain methyl resonance of Thr-110 and Thr-132, which are located on the D-loop, was observed to undergo slow conformational exchange between major and minor conformations with the population ratio of 6:1 (Fig. 4C). Slow conformational exchange was also observed for the neighboring Leu-112 with two subpopulations indicated by the NMR spectra (Fig. 4D). In the presence of reduced/oxidized paramagnetic tag, the minor population appears to be within the vicinity of Cys-106 within the hydrophobic core of the protein. The presence of the alternate conformations is corroborated by matching Hα and Hβ NOEs, whereas in each population, the presence/absence of water NOEs suggests alternating conformations of solvent-exposed and shielded Thr-111 and Ile-113 (Fig. 4E). Similar protein dynamics in the D-loop region were also detected in other PAS domains (FixL, Phy3, and human PAS kinase (hPASK)), and this flexibility has been suggested to be important for ligand binding (41, 45, 46). These observations suggest that the D-loop may adopt an open conformation in the presence of a ligand such as heme and that the flexibility in the D-loop region may be critical for the opening of the ligand-binding pocket.
Probing the Heme Binding Mode by Studying PASGtYybT Reconstituted with Heme and Zinc Protoporphyrin IX
To further probe the mode of heme binding in PASGtYybT, we used the 1H-15N HSQC-based method to monitor the structural changes in the backbone amide moiety upon the binding of heme or ZnPPIX. We previously found that PASGtYybT can be reconstituted with either heme or ZnPPIX (23). However, unlike the BsYybT or GtYybT proteins that can be readily reconstituted with heme with close to 1:1 stoichiometry, the stand-alone PASGtYybT domain could only be partially reconstituted with heme or ZnPPIX with ∼30% occupancy. The reduced heme occupancy of the stand-alone PAS domain rendered the identification of the residues interacting with the heme challenging.
Despite the reduced protein stability and partial heme occupancy, we conducted the NMR experiments with the reconstituted PASGtYybT in an effort to identify the heme-binding site. When compared with the apo-protein, the ZnPPIX-reconstituted PASGtYybT exhibited both chemical shift perturbations and conformational exchange-induced resonance line-broadening effect. The perturbed residues can be grouped into two clusters, with the first cluster situated around strand Aβ, helix Cα, and the D-loop (Val-72, Ile-85, Gly-86, Leu-88, Leu-89, Trp-98, Asn-100, Cys-106, Phe-107, Gln-110, Thr-111, Ile-113, Leu-156, and Tyr-157) (Fig. 5A) and the second cluster located around the dimeric interface between the HI-loop and the N-terminal fragment (Glu-78, Ala-79, Leu-80, Met-81, Gln-82, Met-83, Val-149, Arg-151, NϵArg-151, Glu-153, and Arg-154) (Fig. 5B). The extent of perturbation varies among residues (Fig. 5C), with residue Met-81 showing the greatest change with a resonance peak that almost completely disappeared. Other peak resonances appear to undergo attenuation or peak doubling, which may be attributed to slow conformational change or slow exchange dynamics in the presence of ZnPPIX. Due to the cross-peak overlap or low intensities of some cross-peaks, the number of perturbed residues may be slightly underestimated. Despite the repeated efforts, unambiguous NOE signals between the protein and ligand could not be established due to low ligand binding stoichiometry of the protein sample.
FIGURE 5.

Perturbations caused by the binding of ZnPPIX or heme. A and B, residues (indicated by the black dots) affected by ZnPPIX reconstitution. Perturbed residues clustered around strand Aβ, helix Cα, and the D-loop are shown in panel A. Perturbed residues clustered around the dimeric interface between the HI-loop and the A′α domain are shown in panel B. C, partial 1H-15N HSQC spectra before (left panels) and after (right panels) ZnPPIX reconstitution. D, residues affected in the WEFT-HSQC experiment upon heme reconstitution. A total of 12 residues (indicated by the black dots) are affected upon the reconstitution with heme. The residues are within the vicinity of helix Cα, the D-loop, the GH-loop, the HI-loop, and the A′α domain. E, 1H-15N WEFT HSQC spectra before heme reconstitution (left panel) and after heme reconstitution (right panel) are shown.
Partial reconstitution of the PAS domain with the paramagnetic heme (ferrous protoporphyrin IX) yielded no significant shifts in the 1H-15N HSQC spectra, despite the protein solution turning dark brown. We conducted a modified 1H-15N HSQC experiment by using the WEFT-HSQC method (37). This experiment allows the suppression of slow relaxing signals while monitoring fast relaxing signals in the presence of a paramagnetic ferrous heme. The measurements were performed on the free protein and heme-reconstituted protein. The resonances showing opposite amplitude signal intensity were interpreted as being in the vicinity of the heme-binding site. Overall, a total of 12 residues were observed to be affected by the presence of heme: Asn-67, Val-72, Ala-79, Gln-82, Phe-102, Cys-106, Leu-112, Ile-113, Gly-142, Gln-144, Arg-154, and Thr-162 (Fig. 5, D and E). These residues are distributed around the hydrophobic pocket and from helix Cα (Phe-102, Cys-106, and Phe-107), the D-loop (Thr-111, Leu-112, and Ile-113),and the GH-loop, HI-loop, or N-terminal fragment (Ala-79–Gln-82). By site-directed mutagenesis, we evaluated the roles of the residues in the putative binding pocket in heme binding by alanine replacement. Although none of the mutations completely abolished the heme binding capability for the B. subtilis YybT55–651 construct as assessed by the heme reconstitution method, replacement of several residues caused various extents of reduction in the yield of heme reconstruction. The most significant reduction was observed for the F107A mutant with a 1:0.3 protein to heme ratio, relative to the 1:1 ratio for the wild-type protein (data not shown).
Disruption of LlYybT Gene Affects Heme Resistance in L. lactis
To test whether the observed in vitro heme binding by the PAS domain is biologically relevant, we tested the heme resistance of the wild type and ΔLlYybT mutant of L. lactis, which was generated by integration of a suicide plasmid by homologous recombination as described previously (9). The parental and mutant strains were examined for heme sensitivity on solid medium under aerobic conditions. We found that the disruption of LlYybT gene resulted in greater sensitivity toward heme treatment, as indicated by the greater growth inhibition zone for the mutant strain (Fig. 6). A wild-type reverted strain derived from the ΔLlYybT mutant but with the plasmid excised had the expected equivalent level of heme resistance as the parent. This observation suggests that the YybT protein-containing wild-type strain is more equipped to cope with heme stress, likely by adjusting the c-di-AMP level to elicit cellular responses to alleviate the heme toxicity.
FIGURE 6.

Assay of heme sensitivity for the wild-type L. lactis and ΔLlYybT mutant strains. Late-log cultures of L. lactis wild-type strain, ΔLlYybT mutant (Δ1816), and revertant 1816Rwt strain were plated on GM17 agar plates, and 230 nmol of heme was added to a filter disk. Diameter measurements of growth inhibition zones from triplicate assays are provided below each image with standard deviations in millimeters. Different superscript letters indicate significant differences in zone sizes (p < 0.001), whereas the same superscript letters indicate zones that were not significantly different (p > 0.05). Inactivation of LlYybT gene in the Δ1816 mutant resulted in increased heme sensitivity when compared with the wild type (wt) as determined by a larger diameter of growth inhibition. As expected the revertant strain (1816Rwt), which is derived from Δ1816, but had the inactivation plasmid removed, had an equivalent growth inhibition zone as the wild type. The ring of enhanced growth outside the growth inhibition zone is due to respiration growth in the presence of subinhibitory levels of heme (39).
DISCUSSION
This work was motivated by the surprising observation that the small PASYybT domains are able to bind b-type heme with 1:1 stoichiometry. The NMR study established the PAS domain fold and revealed some unique features of the PASGtYybT domain. Although the PASGtYybT domain adopts the conserved PAS structural fold that contains a single antiparallel, five-stranded β-sheet with a 2-1-5-4-3 order (13, 48), three of the strands and one of the helices are shorter than those found in other typical PAS domains. With only 80 residues forming the core of the PAS domain, PASGtYybT is one of the smallest PAS domains with three-dimensional structure determined. Despite its small size, it is remarkable that a hydrophobic pocket can still be formed between the β-sheet and the D-loop. In conjunction with previous biochemical data (2, 23), the presence of the hydrophobic pocket suggests that the PAS domain most likely acts as a regulatory domain by binding a small-molecule ligand. Together with the sensor domains that putatively mediate the enzymatic activity of the c-di-AMP-synthesizing diadenylate cyclase (DAC) domains (3), the PAS domain of the YybT homologs would enable the bacteria to modulate cellular c-di-AMP levels in response to intra- and extracellular stimuli.
The results establish that the PASGtYybT domain dimerizes in an antiparallel fashion through the central β-sheet. The use of the β-sheet is rather common for the homo- or heterodimerization of PAS domains (49, 50). The directionality of the monomers in the dimer is rather diverse for PAS domain proteins, with structures showing parallel (45), antiparallel, orthogonal (24), or both parallel and antiparallel orientations (41). Moreover, in the blue-light photoreceptors phototropin1 (AsLOV2) (51) and photoactive yellow protein (52), the β-sheet interface appears to be crucial for signal transduction. The involvement of the β-sheet interface in signal transduction is further supported by the studies on the PAS domains of human PAS kinase (46), hypoxia-inducible factor/aryl hydrocarbon receptor nuclear translocator (HIF/ARNT) (40, 53), KinA histidine kinase (54), and the hERG (human ether-à-go-go-related gene) potassium channel (55, 56). The similarity shared by PASGtYybT in dimerization with ARNT PAS-B homodimer and HIF-2a/ARNT PAS-B heterodimer (40) indicates that PASGtYybT may share a similar mechanism in signal propagation. YybT proteins contain two predicted transmembrane helices (Pro-12–Phe-29 and Met-33–Trp-52) located upstream of the PAS domain. The C terminus of the transmembrane domain (ending at Trp-52) is predicted to be connected to the disjointed α-helix of the A′α domain starting at residue Lys-59 through a short linker of six residues. We speculate that the signal initiated by the binding of ligand to the PAS domain may be transduced to the C-terminal helices and the catalytic domains through the dimerization interface.
Early studies suggested that the PASGtYybT domain may bind b-type heme as ligand or cofactor for the regulation of C-terminal phosphodiesterase domain. The NMR structure presented here reveals a mostly hydrophobic pocket located in the center of the PAS domain. The comparative NMR studies with apo- and heme- or ZnPPIX-reconstituted PASGtYybT showed perturbations in the regions surrounding the pocket and the N-terminal region located at the dimer interface. The current data are supportive of the model that a portion of the macrocycle of the heme or ZnPPIX can be inserted into the hydrophobic pocket and that the binding event is accompanied by concomitant conformational changes in the flexible D-loop and the N-terminal fragment at the dimer interface. Because only a small portion (∼30%) of the PASGtYybT construct could be reconstituted with heme or ZnPPIX, the NMR signals were not strong enough for us to unambiguously identify all the residues that undergo perturbations upon heme or ZnPPIX binding. Nonetheless, the use of the sensitive TROSY-HSQC method allowed us to detect perturbations for a dozen residues in the vicinity of the nonpolar pocket and C-terminal region. These observed perturbations lend support to the claim that the nonpolar pocket of the PASGtYybT domain is involved in the binding of heme. Meanwhile, the dynamic properties of the D-loop indicated by the NMR data could be crucial for the widening of the binding pocket for heme binding.
It has been recently established that c-di-AMP and YybT proteins regulate peptidoglycan biosynthesis and antibiotic resistance in several bacteria. The heme sensitivity assay with the L. lactis ΔYybT mutant strain demonstrated that the LlYybT protein also plays a role in mediating heme resistance (in addition to acid resistance), most likely through the modulation of cellular c-di-AMP concentration. In conjunction with the in vitro heme binding capability exhibited by the PASYybT domains and the importance of heme homeostasis in bacterial growth and infection, it is tempting to suggest that the YybT protein can bind directly to the imported heme to initiate cellular response through the c-di-AMP signaling pathways. However, because the exact binding mode of heme for the PASYybT domains remains to be fully revealed and the altered heme resistance for the L. lactis ΔYybT mutant may be caused by the loss of the c-di-AMP-degrading DHH/DHHA1 domain, the current data do not unequivocally establish the PASGtYybT domain as a direct heme sensor. The possibility for the PASGtYybT domain to bind another small-ligand molecule with higher affinity than heme in cellular environment cannot be ruled out. Hence, the direct involvement of YybT proteins in heme sensing and response in bacterial cells requires further evidence. The structural data presented here nonetheless heighten the structural and functional diversity of the versatile PAS domain and constitute another step toward a full understanding of the biological function of YybT proteins in c-di-AMP signaling.
This work was supported by a Tier II Academic Research Council (ARC) grant (to Z.-X. L.) from the Ministry of Education (MOE) of the Republic of Singapore and grants from the Australian Research Council (LP1210282) and Dairy Innovation Australia Limited (to M. S. T.).
The atomic coordinates and structure factors (code 2M1C) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- c-di-AMP
- cyclic di-AMP
- PAS domain
- Per-ARNT-Sim domain
- ARNT
- aryl hydrocarbon receptor nuclear translocator
- HIF
- hypoxia-inducible factor
- ZnPPIX
- zinc protoporphyrin IX
- r.m.s.d.
- root mean square deviation
- MTSL
- (1-oxyl-2, 2, 5,5-tetramethyl-Δ3-pyrroline-3-methyl) methanethiosulfonate
- DMSO
- dimethyl sulfoxide
- HSQC
- heteronuclear single quantum correlation
- HMQC
- heteronuclear multiple-quantum correlation
- TROSY
- transverse relaxation optimized spectroscopy
- WEFT
- water-edited Fourier transform
- trun
- truncated.
REFERENCES
- 1. Rallu F., Gruss A., Ehrlich S. D., Maguin E. (2000) Acid- and multistress-resistant mutants of Lactococcus lactis: identification of intracellular stress signals. Mol. Microbiol. 35, 517–528 [DOI] [PubMed] [Google Scholar]
- 2. Rao F., See R. Y., Zhang D., Toh D. C., Ji Q., Liang Z. X. (2010) YybT Is a signaling protein that contains a cyclic dinucleotide phosphodiesterase domain and a GGDEF domain with ATPase activity. J. Biol. Chem. 285, 473–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Römling U. (2008) Great times for small molecules: c-di-AMP, a second messenger candidate in Bacteria and Archaea. Sci. Signal. 1, pe39. [DOI] [PubMed] [Google Scholar]
- 4. Witte G., Hartung S., Büttner K., Hopfner K.-P. (2008) Structural biochemistry of a bacterial checkpoint protein reveals diadenylate cyclase activity regulated by DNA recombination intermediates. Mol. Cell 30, 167–178 [DOI] [PubMed] [Google Scholar]
- 5. Corrigan R. M., Abbott J. C., Burhenne H., Kaever V., Gründling A. (2011) c-di-AMP is a new second messenger in Staphylococcus aureus with a role in controlling cell size and envelope stress. Plos Pathogens 7, e1002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burnside K., Lembo A., de Los Reyes M., Iliuk A., Binhtran N.-T., Connelly J. E., Lin W.-J., Schmidt B. Z., Richardson A. R., Fang F. C., Tao W. A., Rajagopal L. (2010) Regulation of hemolysin expression and virulence of Staphylococcus aureus by a serine/threonine kinase and phosphatase. PLoS ONE 5, e11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Luo Y., Helmann J. D. (2012) Analysis of the role of Bacillus subtilis σM in β-lactam resistance reveals an essential role for c-di-AMP in peptidoglycan homeostasis. Mol. Microbiol. 83, 623–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pozzi C., Waters E. M., Rudkin J. K., Schaeffer C. R., Lohan A. J., Tong P., Loftus B. J., Pier G. B., Fey P. D., Massey R. C., O'Gara J. P. (2012) Methicillin resistance alters the biofilm phenotype and attenuates virulence in Staphylococcus aureus device-associated infections. PLoS Pathog. 8, e1002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith W. M., Pham T. H., Lei L., Dou J., Soomro A. H., Beatson S. A., Dykes G. A., Turner M. S. (2012) Heat resistance and salt hypersensitivity in Lactococcus lactis due to spontaneous mutation of llmg_1816 (gdpP) induced by high-temperature growth. Appl. Environ. Microbiol. 78, 7753–7759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oppenheimer-Shaanan Y., Wexselblatt E., Katzhendler J., Yavin E., Ben-Yehuda S. (2011) c-di-AMP reports DNA integrity during sporulation in Bacillus subtilis. EMBO Rep. 12, 594–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chan C., Paul R., Samoray D., Amiot N. C., Giese B., Jenal U., Schirmer T. (2004) Structural basis of activity and allosteric control of diguanylate cyclase. Proc. Natl. Acad. Sci. U.S.A. 101, 17084–17089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ulrich L. E., Koonin E. V., Zhulin I. B. (2005) One-component systems dominate signal transduction in prokaryotes. Trends Microbiol. 13, 52–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Möglich A., Ayers R. A., Moffat K. (2009) Structure and signaling mechanism of Per-ARNT-Sim domains. Structure 17, 1282–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gilles-Gonzalez M. A., Gonzalez G., Perutz M. F., Kiger L., Marden M. C., Poyart C. (1994) Heme-based sensors, exemplified by the kinase FixL, are a new class of heme protein with distinctive ligand binding and autoxidation. Biochemistry 33, 8067–8073 [DOI] [PubMed] [Google Scholar]
- 15. Tuckerman J. R., Gonzalez G., Sousa E. H. S., Wan X., Saito J. A., Alam M., Gilles-Gonzalez M.-A. (2009) An oxygen-sensing diguanylate cyclase and phosphodiesterase couple for c-di-GMP control. Biochemistry 48, 9764–9774 [DOI] [PubMed] [Google Scholar]
- 16. Purcell E. B., McDonald C. A., Palfey B. A., Crosson S. (2010) An analysis of the solution structure and signaling mechanism of LovK, a sensor histidine kinase integrating light and redox signals. Biochemistry 49, 6761–6770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Qi Y., Rao F., Luo Z., Liang Z.-X. (2009) A flavin cofactor-binding PAS domain regulates c-di-GMP synthesis in AxDGC2 from Acetobacter xylinum. Biochemistry 48, 10275–10285 [DOI] [PubMed] [Google Scholar]
- 18. Söderbäck E., Reyes-Ramirez F., Eydmann T., Austin S., Hill S., Dixon R. (1998) The redox- and fixed nitrogen-responsive regulatory protein NIFL from Azotobacter vinelandii comprises discrete flavin and nucleotide-binding domains. Mol. Microbiol. 28, 179–192 [DOI] [PubMed] [Google Scholar]
- 19. Ukaegbu U. E., Henery S., Rosenzweig A. C. (2006) Biochemical characterization of MmoS, a sensor protein involved in copper-dependent regulation of soluble methane monooxygenase. Biochemistry 45, 10191–10198 [DOI] [PubMed] [Google Scholar]
- 20. Christie J. M., Salomon M., Nozue K., Wada M., Briggs W. R. (1999) LOV (light, oxygen, or voltage) domains of the blue-light photoreceptor phototropin (nph1): Binding sites for the chromophore flavin mononucleotide. Proc. Natl. Acad. Sci. U.S.A. 96, 8779–8783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Purcell E. B., Siegal-Gaskins D., Rawling D. C., Fiebig A., Crosson S. (2007) A photosensory two-component system regulates bacterial cell attachment. Proc. Natl. Acad. Sci. U.S.A. 104, 18241–18246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Swartz T. E., Tseng T.-S., Frederickson M. A., Paris G., Comerci D. J., Rajashekara G., Kim J.-G., Mudgett M. B., Splitter G. A., Ugalde R. A., Goldbaum F. A., Briggs W. R., Bogomolni R. A. (2007) Blue-light-activated histidine kinases: two-component sensors in bacteria. Science 317, 1090–1093 [DOI] [PubMed] [Google Scholar]
- 23. Rao F., Ji Q., Soehano I., Liang Z. X. (2011) Unusual heme-binding PAS domain from YybT family proteins. J. Bacteriol. 193, 1543–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kurokawa H., Lee D.-S., Watanabe M., Sagami I., Mikami B., Raman C. S., Shimizu T. (2004) A redox-controlled molecular switch revealed by the crystal structure of a bacterial heme PAS sensor. J. Biol. Chem. 279, 20186–20193 [DOI] [PubMed] [Google Scholar]
- 25. Delgado-Nixon V. M., Gonzalez G., Gilles-Gonzalez M.-A. (2000) Dos, a heme-binding PAS protein from Escherichia coli, is a direct oxygen sensor. Biochemistry 39, 2685–2691 [DOI] [PubMed] [Google Scholar]
- 26. Herrmann T., Güntert P., Wüthrich K. (2002) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227 [DOI] [PubMed] [Google Scholar]
- 27. Güntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–378 [DOI] [PubMed] [Google Scholar]
- 28. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bhattacharya A., Tejero R., Montelione G. T. (2007) Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795 [DOI] [PubMed] [Google Scholar]
- 30. Brunger A. T. (2007) Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2, 2728–2733 [DOI] [PubMed] [Google Scholar]
- 31. de Vries S. J., van Dijk M., Bonvin A. M. J. J. (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 [DOI] [PubMed] [Google Scholar]
- 32. Cavanagh J., Fairbrother W. J., Palmer A. G., Skelton N. J. (1996) Protein NMR Spectroscopy: Principles and Practice, Academic Press, New York [Google Scholar]
- 33. Dosset P., Hus J.-C., Marion D., Blackledge M. (2001) A novel interactive tool for rigid-body modeling of multi-domain macromolecules using residual dipolar couplings. J. Biomol. NMR 20, 223–231 [DOI] [PubMed] [Google Scholar]
- 34. Petros A. M., Mueller L., Kopple K. D. (1990) NMR identification of protein surfaces using paramagnetic probes. Biochemistry 29, 10041–10048 [DOI] [PubMed] [Google Scholar]
- 35. Zuiderweg E. R. P. (2002) Mapping protein-protein interactions in solution by NMR spectroscopy. Biochemistry 41, 1–7 [DOI] [PubMed] [Google Scholar]
- 36. Pintacuda G., Otting G. (2002) Identification of protein surfaces by NMR measurements with a paramagnetic Gd(III) chelate. J. Am. Chem. Soc. 124, 372–373 [DOI] [PubMed] [Google Scholar]
- 37. Jensen M. R., Hansen D. F., Ayna U., Dagil R., Hass M. A. S., Christensen H. E. M., Led J. J. (2006) On the use of pseudocontact shifts in the structure determination of metalloproteins. Magn. Reson. Chem. 44, 294–301 [DOI] [PubMed] [Google Scholar]
- 38. Pedersen M. B., Garrigues C., Tuphile K., Brun C., Vido K., Bennedsen M., Møllgaard H., Gaudu P., Gruss A. (2008) Impact of aeration and heme-activated respiration on Lactococcus lactis gene expression: Identification of a heme-responsive operon. J. Bacteriol. 190, 4903–4911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hilty C., Wider G., Fernández C., Wüthrich K. (2004) Membrane protein-lipid interactions in mixed micelles studied by NMR spectroscopy with the use of paramagnetic reagents. ChemBioChem. 5, 467–473 [DOI] [PubMed] [Google Scholar]
- 40. Card P. B., Erbel P. J. A., Gardner K. H. (2005) Structuralbasis of ARNT PAS-B dimerization: use of a common β-sheet interface for hetero- and homodimerization. J. Mol. Biol. 353, 664–677 [DOI] [PubMed] [Google Scholar]
- 41. Gong W., Hao B., Mansy S. S., Gonzalez G., Gilles-Gonzalez M. A., Chan M. K. (1998) Structure of a biological oxygen sensor: A new mechanism for heme-driven signal transduction. Proc. Natl. Acad. Sci. U.S.A. 95, 15177–15182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cheung J., Bingman C. A., Reyngold M., Hendrickson W. A., Waldburger C. D. (2008) Crystal structure of a functional dimer of the PhoQ sensor domain. J. Biol. Chem. 283, 13762–13770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou Y.-F., Nan B., Nan J., Ma Q., Panjikar S., Liang Y.-H., Wang Y., Su X.-D. (2008) C4-dicarboxylates sensing mechanism revealed by the crystal structures of DctB sensor domain. J. Mol. Biol. 383, 49–61 [DOI] [PubMed] [Google Scholar]
- 44. Cheung J., Hendrickson W. A. (2008) Crystal structures of C4-dicarboxylate ligand complexes with sensor domains of histidine kinases DcuS and DctB. J. Biol. Chem. 283, 30256–30265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Miyatake H., Mukai M., Park S.-Y., Adachi S.-i., Tamura K., Nakamura H., Nakamura K., Tsuchiya T., Iizuka T., Shiro Y. (2000) Sensory mechanism of oxygen sensor FixL from Rhizobium meliloti: crystallographic, mutagenesis and resonance raman spectroscopic studies. J. Mol. Biol. 301, 415–431 [DOI] [PubMed] [Google Scholar]
- 46. Amezcua C. A., Harper S. M., Rutter J., Gardner K. H. (2002) Structure and interactions of PAS kinase N-terminal PAS domain: model for intramolecular kinase regulation. Structure 10, 1349–1361 [DOI] [PubMed] [Google Scholar]
- 47. Koradi R., Billeter M., Wüthrich K. (1996) MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55 [DOI] [PubMed] [Google Scholar]
- 48. Zoltowski B. D., Gardner K. H. (2011) Tripping the light fantastic: blue-light photoreceptors as examples of environmentally modulated protein-protein interactions. Biochemistry 50, 4–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ma X., Sayed N., Baskaran P., Beuve A., van den Akker F. (2008) PAS-mediated dimerization of soluble guanylyl cyclase revealed by signal transduction histidine kinase domain crystal structure. J. Biol. Chem. 283, 1167–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Neiditch M. B., Federle M. J., Pompeani A. J., Kelly R. C., Swem D. L., Jeffrey P. D., Bassler B. L., Hughson F. M. (2006) Ligand-induced asymmetry in histidine sensor kinase complex regulates quorum sensing. Cell 126, 1095–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harper S. M., Neil L. C., Gardner K. H. (2003) Structural basis of a phototropin light switch. Science 301, 1541–1544 [DOI] [PubMed] [Google Scholar]
- 52. Borgstahl G. E. O., Williams D. R., Getzoff E. D. (1995) 1.4-Ä structure of photoactive yellow protein, a cytosolic photoreceptor: Unusual fold, active site, and chromophore. Biochemistry 34, 6278–6287 [DOI] [PubMed] [Google Scholar]
- 53. Erbel P. J. A., Card P. B., Karakuzu O., Bruick R. K., Gardner K. H. (2003) Structural basis for PAS domain heterodimerization in the basic helix–loop–helix-PAS transcription factor hypoxia-inducible factor. Proc. Natl. Acad. Sci. U.S.A. 100, 15504–15509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee J., Tomchick D. R., Brautigam C. A., Machius M., Kort R., Hellingwerf K. J., Gardner K. H. (2008) Changes at the KinA PAS-A dimerization interface influence histidine kinase unction. Biochemistry 47, 4051–4064 [DOI] [PubMed] [Google Scholar]
- 55. Li Q., Gayen S., Chen A. S., Huang Q., Raida M., Kang C. (2010) NMR solution structure of the N-terminal domain of hERG and its interaction with the S4–S5 linker. Biochem. Biophys. Res. Comm. 403, 126–132 [DOI] [PubMed] [Google Scholar]
- 56. Morais Cabral J. H., Lee A., Cohen S. L., Chait B. T., Li M., Mackinnon R. (1998) Crystal structure and functional analysis of the HERG potassium channel N-terminus: a eukaryotic PAS domain. Cell 95, 649–655 [DOI] [PubMed] [Google Scholar]