

Background: Up-regulation of survivin and repression of the tumor suppressor p16INK4a often occur during cellular immortalization.
Results: p16INK4a repression increased E2F1 binding at the survivin gene promoter, up-regulated survivin, and rendered immortal cells resistant to oxidative stress.
Conclusion: Up-regulation of survivin is intrinsically linked to silencing of p16INK4a.
Significance: Resistance to oxidative stress may facilitate oncogenic transformation of immortal cells.
Keywords: Cancer Biology, E2F Transcription Factor, Oxidative Stress, Survivin, Telomerase, TERT, Immortalization, p16INK4a
Abstract
Survivin is an essential component of the chromosomal passenger complex and a member of the inhibitor of apoptosis family. It is expressed at high levels in a large variety of malignancies, where it has been implicated in drug resistance. It was also shown previously that survivin is up-regulated during telomerase-mediated immortalization, which occurs at a relatively early stage during carcinogenesis. This study shows that up-regulation of survivin during immortalization of human myofibroblasts is an indirect consequence of the repression of p16INK4a. Survivin and p16INK4a were functionally linked by assays that showed that either the up-regulation of survivin or repression of p16INK4a rendered telomerase-transduced MRC-5 myofibroblasts resistant to oxidative stress. Conversely, siRNA-mediated down-regulation of survivin activated caspases and enhanced the sensitivity of immortal MRC-5 cells to oxidative stress. The E2F1 transcription factor, which is negatively regulated by the pRB/p16INK4a tumor suppressor pathway, was implicated in the up-regulation of survivin. Using the ChIP assay, it was shown that E2F1 directly interacted with the survivin gene (BIRC5) promoter in cells that spontaneously silenced p16INK4a during telomerase-mediated immortalization. E2F1 binding to the BIRC5 was also enhanced in telomerase-transduced cells subjected to shRNA-mediated repression of p16INK4a. Together, these data show that repression of p16INK4a contributes to the up-regulation of survivin and thereby provides a survival advantage to cells exposed to oxidative stress during immortalization. The up-regulation of survivin during immortalization likely contributes to the vulnerability of immortal cells to transformation by oncogenes that alter intracellular redox state.
Introduction
Cellular immortalization is an essential step in the development of most human cancers and a defining property of cancer stem cells (1). In vitro models have demonstrated the crucial role of telomere maintenance mechanisms in the process of immortalization (2–4). It has also been established that inactivation of tumor suppressor pathways governed by the retinoblastoma protein (pRB) and p16INK4a is required for the immortalization of a variety of epithelial, epidermal, and mesenchymal cell types (5–7). The very high frequency with which telomere maintenance mechanisms are activated and the p16INK4a/pRB pathway is disabled in human cancers attests to the relevance of these in vitro models of immortalization to the study of fundamental aspects of cancer cell biology (8, 9).
In normal cells that lack a telomere maintenance mechanism, telomere length shortens with each round of cell replication (10). When telomeres reach a critically short length, a DNA damage response is elicited. This involves the activation of p53, up-regulation of p16INK4a, and hypophosphorylation of pRB, which induces an irreversible proliferative arrest, referred to as senescence (11). Excessive exposure to oxidative stress hastens senescence by damaging telomeric, genomic and/or mitochondrial DNA, resulting in the activation of tumor suppressor pathways (12–15). Conversely, limiting exposure to oxidative stress has been shown to favor the replication and immortalization of human cells (16–18).
Our previous studies and numerous others have shown that reconstitution of telomerase activity by overexpression of human telomerase reverse transcriptase (hTERT),3 elongates telomeres and extends the replicative life span of normal human cells (4, 19–21). However, the overexpression of hTERT is insufficient for in vitro immortalization of many different cell strains, which eventually succumb to a growth crisis or delayed senescence when cultured under standard growth conditions (6, 18, 20, 22). Down-regulation of p16INK4A is thought to be required for these cell types to overcome the telomere-independent stresses that impede immortalization.
In addition to the frequent inactivation of p16INK4a, the inhibitor of apoptosis protein family member survivin is up-regulated during immortalization of human MRC5 and WI38 myofibroblasts (23). The up-regulation of survivin during immortalization poses a likely explanation for the abundance of survivin in virtually all cancers (24). In tumor cells, high expression of survivin protects against apoptotic cell death through direct interactions with other inhibitor of apoptosis proteins that bind and quench caspase activity (25, 26). Survivin has been shown to be of prognostic value in certain cancers and was specifically implicated in drug resistance (27). However, the functional significance of the up-regulation of survivin during the immortalization process and in premalignant cells is less clear. In this study, it is shown that the up-regulation of survivin in hTERT-immortalized myofibroblasts is intrinsically linked to repression of p16INK4a and underpins the resistance of immortal cells to oxidative stress, which may be advantageous during malignant transformation.
EXPERIMENTAL PROCEDURES
Cell Culture
MRC-5 human fetal lung fibroblasts were purchased from the ATCC. hTERT-immortalized WI-38 clones were provided by Prof. Varda Rotter (Weizmann Institute of Science, Israel). MRC5hTERT-1 was established by retroviral transduction of MRC5 cells with hTERT and then subcloned by limiting the dilution to establish MRC5hTERT-24, MRC5hTERT-30, and MRC5hTERT-36 (20, 28). MRC-5 and genetically modified derivative cell lines were grown in α minimal essential medium with 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen) plus 10% FBS (ThermoTrace, Noble Park, Australia) and grown in a humidified incubator at 37 °C with 5% CO2. To assess the effects of alleviating oxidative stress, MRC5hTERT clones were cultured in 5% O2 in a PRO OX model 110 C chamber fitted with a PRO OX oxygen controller (BioSpherix, Lacona, NY). Cells were treated with the oxidant tert-butylhydroperoxide (tBOOH) (Sigma-Aldrich, St. Louis, MO) as indicated in the text. Cell expansion and viability was assessed by trypan blue exclusion, with expansion defined as the number of viable cells harvested divided by the initial number of cells seeded. Population doublings (PD) were calculated using the formula PD = [log (expansion)/log 2], where expansion was the number of cells harvested divided by the initial number of cells seeded.
Retroviral Transductions and siRNA Transfection
The retroviral vectors MIGR1-survivin, MIGR1, Babepuro-shRNAp53, Babepuro-shRNAGFP, RetroSuper-16shRNA, and control RetroSuper were described previously (29–32). Transductions were performed using Phoenix A packaging cells according to our published method (28). The design and validation of siRNA targeting survivin (S1) and control oligonucleotide (Sc) were also published previously (23). Cells were transfected with 100 nm siRNA using LipofectamineTM RNAiMAX reagent (Invitrogen) according to the protocol of the manufacturer.
Apoptosis Assays
The proportion of apoptotic cells was assessed by costaining with allophycocyanin (APC)-labeled annexin V antibody (BD Biosciences) and propidium iodide (PI) (BD Biosciences) according to the instructions of the manufacturer. Annexin V-positive and PI-negative cells were quantified using a FACSCalibur flow cytometer (BD Biosciences). Caspase activity was measured using the ApolertTM caspase assay plate (BD Biosciences) according to the instructions of the manufacturer. Briefly, cells were suspended at 5 × 106/ml in cell lysis buffer and then incubated on ice for 10 min. The lysate was cleared of debris by centrifugation, and then 50 μl of lysate was added to 50 μl of 2× reaction buffer in a 96-well plate. After 2 h of incubation at 37 °C, caspase activity was quantified by measuring fluorescence using a VictorTM plate reader (PerkinElmer Life Sciences) with a 380 nm excitation filter and 460 nm emission filter.
Immunoblotting
Immunoblotting was performed as described previously (28). The antibodies used were rabbit anti-survivin NB500-201 (Novus Biologicals, Littleton, CO), rabbit anti-actin (Sigma-Aldrich), mouse anti-p16INK4a (BD Biosciences), mouse anti-p53 clone D0–1 (Santa Cruz Biotechnology, Inc.), mouse anti-p21cip1 (BD Biosciences). Membranes were stripped with 25 mm glycine-HCl (pH 2) and 1% SDS buffer.
Senescence-associated β-Galactosidase Staining
Cells were assayed for senescence-associated β-galactosidase activity at pH 6.0 as described previously (33). At least three fields of 50–300 cells were scored for each sample assayed.
Real-time Reverse Transcriptase PCR (qRT-PCR)
RNA was extracted using the Qiagen RNeasy mini kit (Qiagen, Australia) followed by reverse transcription using SuperScript® III reverse transcriptase (Invitrogen) according to the instructions of the manufacturer. qRT-PCR was performed using iQTM SYBR Green (Bio-Rad). PCR conditions and primers for quantitation of p16INK4a and survivin mRNA were as described previously (23, 34). β2-microglobin mRNA was used as a normalization control. All samples were assayed in duplicates, and average values were calculated from two to five assays.
ChIP Assay
ChIP assays were performed as described previously using an anti-E2F1 antibody or control IgG antibody (Santa Cruz Biotechnology, Inc.) and real-time PCR primers targeting an E2F1 binding site or upstream region of the BIRC5 gene promoter as a control (35). E2F1 binding was quantified as the fold enrichment of the E2F1 target region in real-time PCR products relative to the PCR amplification of the upstream region. PCR primers targeting the E2F1 binding site were as follows: 5′-AGCCCCTTCTGGTCCTAACTT-3′ (forward) and CCG GCCTAACTCCTTTTCACTTCT (reverse). PCR primers targeting the upstream region were as follows: 5′-AAGATCTGTTCGCCTGACATCCT-3′ (forward) and 5′-CCCACCTCCTTTCTCCTTACCTT-3′ (reverse).
RESULTS
TERT-transduced MRC5 Cells Acquire Resistance to Oxidative Stress during Immortalization
hTERT-transduced myofibroblasts (MRC5hTERT cells) cultured in atmospheric oxygen (∼21% O2) proliferate beyond the point at which normal MRC5 cells senesce (∼62 PD) but are subsequently subject to a telomere length-independent growth crisis at ∼110–120 PD (20, 28, 36). Some clones do not proliferate beyond crisis (e.g. MRC5hTERT-30 and MRC5hTERT-24, Fig. 1A), whereas others recover spontaneously from crisis after a significant growth delay to become immortal (e.g. MRC5hTERT-36, Fig. 1A).
FIGURE 1.

MRC5 cells acquire resistance to oxidative stress during immortalization. A, precrisis MRC5hTERT clones were thawed and propagated in either 5% O2 or 21% O2. The dashed line indicates the beginning of crisis under control (21% O2) conditions. B and C, MRC5, early-passage MRC5hTERT, and immortal MRC5hTERT cells were treated with tBOOH at the indicated concentrations for 48 h. B, cell expansion determined by trypan blue exclusion. C, quantification of apoptotic cells by annexin V-APC and PI staining. Values are presented as means ± S.E. relative to untreated cells and calculated from four independent experiments. The p value indicates the overall difference between cell lines determined by two-way ANOVA. *, p < 0.05 in Bonferroni post-test comparisons with EP-MRC5hTERT-1 cells at the indicated tBOOH concentrations.
Our previous studies demonstrated that p16INK4a is expressed and p53 is functional in precrisis MRC5hTERT cells (28). However, MRC5hTERT-1 cells and most subclones down-regulated p16INK4a as they escaped crisis and became immortal. p53 function was retained in the immortal cells until at least 398 PD. There is a mixture of senescent-like cells and apoptotic cells in MRC5hTERT cultures in crisis, which is consistent with the reported effects of oxidative stress on diploid human fibroblasts (28, 37). To determine whether oxidative stress contributes to the proliferative impairment observed during the crisis period, the proliferation of MRC5hTERT clones was compared in 21% and 5% oxygen (Fig. 1A). The growth curves of three MRC5hTERT clones cultured under these alternate conditions for ∼200 days clearly demonstrate that crisis was lessened and immortalization facilitated when oxidative stress was alleviated by growth in 5% oxygen.
We tested whether MRC5hTERT cells that escaped crisis at 21% oxygen acquired resistance to oxidative stress by comparing the response of immortal MRC5hTERT-1, early-passage MRC5hTERT-1 (EP MRC5hTERT-1) and normal MRC-5 cells to increasing concentrations of the model organic peroxide tBOOH (38). Cell counts revealed that 48-hour treatment with tBOOH caused a dose-dependent reduction in cell expansion, with an overall greater impact on proliferation observed in EP MRC5hTERT-1 cultures compared with cultures of immortal MRC5hTERT-1 cells (Fig. 1B) (p < 0.01, two-way ANOVA). There was no significant difference in expansion of normal MRC5 versus EP MRC5hTERT-1 cells, indicating that the enhanced proliferation of immortal MRC5hTERT-1 cells was a consequence of changes that occurred during the immortalization process rather than a direct result of hTERT transduction.
Annexin V/PI staining demonstrated an increased rate of apoptosis in each of the cultures treated with tBOOH, with the rate of apoptosis significantly higher in EP MRC5hTERT-1 cells than in immortal MRC5hTERT-1 cells (Fig. 1C, p < 0.01, two-way ANOVA). Together, these results confirm that oxidative stress presents a significant replicative barrier to myofibroblasts and demonstrates that these cells acquired resistance to oxidative stress during telomerase-mediated immortalization.
Survivin Confers Resistance to Oxidant-induced Cell Death
Because survivin is up-regulated during telomerase-mediated immortalization of myofibroblasts (Fig. 2A, left panel, and Ref. 23), we investigated whether high levels of survivin contributed to the resistance of immortal MRC5hTERT-1 cells to oxidative stress. For these investigations, EP MRC5hTERT-1 cells were transduced with a retroviral vector encoding survivin and GFP (SURV), or a control vector encoding GFP alone (GFP) (30). Following confirmation of survivin overexpression (Fig. 2A, right panel), the transduced cells were treated with tBOOH for 48 h and assayed for proliferation and apoptosis (Fig. 2, B and C). Although the proliferative rate of MRC5hTERT-1 cells cultured under control conditions (no tBOOH) was unaltered by overexpression of survivin, SURV-transduced cells exhibited a higher rate of proliferation (Fig. 2B, left panel) and a lower rate of apoptotic cell death compared with GFP-transduced cells when exposed to tBOOH (p < 0.05 and p < 0.01 respectively, two-way ANOVA).
FIGURE 2.
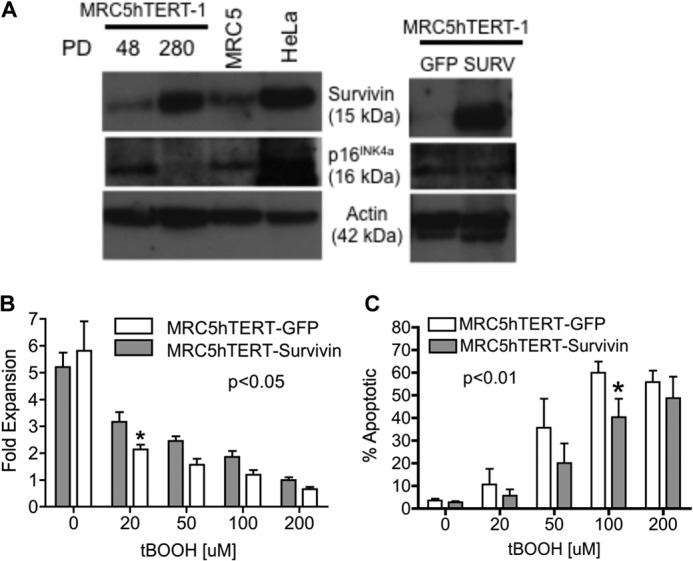
Up-regulation of survivin during immortalization confers resistance to tBOOH. A, immunoblot analysis of survivin and p16INK4a in parental MRC5 cells and MRC5hTERT-1 cells at the indicated PDs (left panel) as well as precrisis MRC5hTERT cells transduced with MIGR1-survivin (SURV) or MIGR1 control vector (GFP) (right panel). HeLa cells were included as a positive control for detection of survivin and p16INK4a. B, MRC5hTERT-1 cells transduced with SURV or GFP retroviral vectors were treated with tBOOH for 48 h. Cell expansion was determined by trypan blue exclusion. C, quantification of apoptotic cells by annexin V-APC/PI staining. Values in B and C are presented as means ± S.E. calculated from four to five independent experiments. p values indicate the overall effect of retroviral transduction as determined by two-way ANOVA. *, p < 0.05 in Bonferroni post-test comparisons with normal GFP cells.
To further confirm that survivin played a central role in the survival of cells subject to oxidative stress, immortal MRC5hTERT-1 cells that express high levels of endogenous survivin were transfected with siRNA targeting survivin or control siRNA (Sc) before being treated with tBOOH. A number of siRNAs targeting survivin were screened previously, and the siRNA that showed the greatest efficacy was utilized in this study (23). Transfection with survivin siRNA effectively ablated survivin protein expression and activated caspases in immortal MRC5hTERT-1 (Fig. 3, A and B). Annexin V/PI staining revealed that the survivin siRNA-transfected cells had a heightened sensitivity to tBOOH relative to Sc-transfected cells. Indeed, the survivin-depleted cells exhibited an ∼3-fold increase in apoptosis relative to control cells when treated with 50 μm tBOOH (p < 0.001, Bonferroni post-test) (Fig. 3C). Together these results demonstrate that an abundance of survivin in immortal MRC5hTERT cells is a crucial determinant of their vulnerability to oxidant-induced cell death.
FIGURE 3.

Down-regulation of survivin heightens sensitivity to oxidant-induced apoptosis via a p53-dependent pathway. A, immortal MRC5hTERT-1 cells were transfected with siRNA targeting survivin (siSurvivin) or a control oligonucleotide (Sc). Immunoblotting was performed to confirm depletion of survivin protein. The immunoblot analysis was subsequently hybridized to antibodies for detection of p53, p21cip1, and actin (loading control). B, caspase activity in immortal MRC5hTERT-1 cells transfected with siRNA targeting survivin or Sc. Caspase activity was measured using a caspase-profiling ELISA plate. Values are presented as means ± S.E. from four independent experiments, with each assay performed in duplicate. C, apoptotic cells were quantified by annexin V-APC/PI staining 48 h after tBOOH treatment. Values are presented as means ± S.E. calculated from four to five independent experiments. D, immunoblot analysis of p53 and p21 protein in immortal MRC5-hTERT-1 transduced with shRNA targeting GFP (control) or p53. The blot was incubated with actin antibody to control for protein loading. E, quantification of apoptotic cells by annexin V-APC/PI staining of immortal MRC5-hTERT-1 cells transduced with GFP or p53shRNA and then treated with tBOOH for 48 h. p values are from statistical comparisons made by two-way ANOVA and indicate the overall effect of siRNA (B and C) or shRNA (E). *, p < 0.05; **, p < 0.01; ***, p < 0.001 in Bonferroni post-test comparisons with control. F, precrisis MRC5hTERT-1 cells transduced with SURV or GFP retroviral vectors encoding survivin and GFP or GFP alone were maintained in long-term culture. The graphs show proliferation of cultures from two independent transductions (A and B).
Oxidant-induced Cell Death in Survivin-depleted Cells Is Mediated by p53
We have shown previously that immortal MRC5hTERT-1 cells retain p53 function (28). In this study, immunoblotting revealed that p53 and its transcriptional target, p21cip1, were elevated when survivin was depleted in MRC5hTERT-1 cells (Fig. 3A). To determine whether p53 was required for oxidant-induced death when cells were depleted of survivin, immortal MRC5hTERT-1 cells (321 PD) were stably transduced with retroviral vectors encoding shRNA that targeted either p53 or GFP (control) (29). After confirming that p53 and p21cip1 were effectively repressed in the p53shRNA-transduced cells (Fig. 3D), the cells were transfected with survivin siRNA or Sc and/or treated with 50 μm tBOOH. Staining with annexin V/PI demonstrated that apoptosis was reduced significantly in MRC5hTERT-p53shRNA cells compared with MRC5hTERT-GFPshRNA cells (Fig. 3E, p < 0.001, two-way ANOVA). The survival effect of p53 down-regulation was particularly evident with the combined treatment with survivin siRNA and tBOOH (p < 0.01, Bonferroni post-test). These results demonstrate that p53 is a critical mediator of oxidative stress-induced apoptotic death when survivin levels are low.
Up-regulation of Survivin Does Not Circumvent Crisis
To determine whether the up-regulation of survivin would rescue MRC5hTERT cells from crisis and facilitate immortalization, EP MRC5hTERT-1 cells transduced with SURV and GFP vectors at precrisis time points were maintained in culture for more than 400 days (Fig. 3F). In two independent experiments, overexpression of survivin had no effect on the rate of proliferation of EP MRC5hTERT-1 cells and did not appear to alter the overall expansion of the cultures during crisis. Thus, although a high level of survivin protects cells against tBOOH-induced apoptosis, it was not sufficient to overcome the stresses that induce crisis during immortalization.
Repression of p16INK4a Confers Resistance to Oxidative Stress and Circumvents Crisis
Because past studies suggest that the silencing of p16INK4a is required for overcoming “culture stress” associated with long-term exposure to 21% oxygen (18), we directly investigated whether repression of p16INK4a would protect EP MRC5hTERT-1 cells from oxidative stress. Precrisis MRC5hTERT cells were transduced with retroviral vectors encoding shRNA targeting p16INK4a (p16 shRNA) or a control shRNA (31). Cells transduced with p16 shRNA expressed p16INK4a mRNA at less than 20% of control cells (Fig. 4A). Following 48-hour exposure to increasing concentrations of tBOOH (20–200 μm), the shRNA-transduced MRC5hTERT cells were assessed for proliferation and death (Fig. 4, B and C). The results demonstrate that cells transduced with p16shRNA had improved survival and increased proliferation when subject to 48-hour treatment with tBOOH compared with control cells. The survival effect associated with p16INK4a repression was apparent when annexin V-positive/PI-negative (early apoptotic) cells or PI-positive cells were quantified (Fig. 4C and data not shown).
FIGURE 4.

Repression of p16INK4a enhances survival and proliferation of tBOOH-treated MRC5hTERT-1 cells. EP MRC5hTERT-1 cells were transduced with retroviral vectors carrying shRNA targeting p16INK4a (p16 shRNA) or an irrelevant gene (murine p63) as a control. A, qRT-PCR was performed to confirm down-regulation of p16INK4a mRNA in p16 shRNA-transduced cells relative to control vector-transduced cells. Values were calculated relative to p16INK4a expression in HeLa cells and are presented as means + S.E. from three assays, each with duplicate samples. B and C, transduced cells were treated for tBOOH for 48 h, counted using trypan blue (B), and assayed for apoptosis (C) by annexin V/PI staining. Values are expressed as means + S.E. calculated from four to five independent experiments. D, EP MRC5hTERT-1 cells transduced with p16 shRNA or control shRNA were maintained in replicate cultures and treated (or not treated) with tBOOH. On days 7 and 14, replicate cultures were stained with x-gal at pH 6.0 to detect senescence-associated β-galactosidase activity in senescent cells. Values are expressed as means + S.E. calculated from seven independent experiments. p values are from two-way ANOVA analysis of the effect of shRNA. *, p < 0.05; ***, p < 0.001 in Bonferroni post-test comparisons of p16 shRNA and control shRNA at the indicated tBOOH concentrations.
To examine the influence of p16INK4a expression on proliferation of cells under conditions of chronic hyperoxia, MRC5hTERT cells transduced with control or p16shRNA vectors were subject to low concentrations of tBOOH (10 μm and 20 μm) over an extended period (2 weeks). Replicate cultures were stained with X-Gal on days 7 and 14 for detection of senescent cells (Fig. 4D). Quantitation of the percentage of senescence-associated β-galactosidase-positive cells showed that a substantial portion of the control cells underwent a senescence-like growth arrest when exposed to 20 μm of tBOOH. Overall, the proportion of senescent cells was significantly less in cultures transduced with p16shRNA compared with control vector-transduced cultures (p < 0.001, two-way ANOVA).
To determine whether repression of 16INK4a would also enable MRC5hTERT cells to overcome stresses encountered at crisis, EP MRC5hTERT-1 cells expressing p16 shRNA or the control vector were maintained in long-term culture (more than 500 days). The cells were harvested at regular intervals to confirm stable suppression of p16INK4a mRNA and protein (Fig. 5, A and B). At early time points post-transduction, shRNA-mediated repression of p16INK4a had no apparent effect on the rate of proliferation (Fig. 5C). However, p16shRNA-transduced cells had a clear proliferative advantage during the crisis period. Together, the analyses of the p16 shRNA-transduced cells show that p16INK4a is a major determinant of the replicative potential of MRC5hTERT cells when cultured under stress conditions and that the down-regulation of p16INK4a facilitates proliferation and immortalization by mitigating both cell death and senescence.
FIGURE 5.

Repression of p16INK4a increases expression of survivin and promotes immortalization. MRC5hTERT-1 cells transduced with p16 shRNA or control shRNA were maintained in long-term culture and sampled intermittently to determine expression of p16INK4a and survivin. A, qRT-PCR analysis of p16INK4a (top panel) and survivin (bottom panel) gene expression in transduced cells. Population doublings counted from the initiation of the original culture are indicated beneath the bottom of the graph. The arrow highlights the spontaneous down-regulation of p16INK4a expression in the control cells. Values were normalized to expression in HeLa cells and presented as means + S.E. B, immunoblot analysis of p16INK4a and survivin in shRNA-transduced MRC5hTERT-1 cells. Actin was used as a loading control. C, graphical representation of the long-term proliferation of MRC5hTERT-1 cells transduced with p16 shRNA or control shRNA. D, qRT-PCR analysis of p16INK4a and survivin gene expression in early-passage and immortal hTERT-transduced WI-38 cells. Results are shown for two independently transduced and immortalized cell lines. Values were normalized to expression levels in HeLa cells and presented as means + S.E. from four assays, each performed with duplicates.
Up-regulation of Survivin Is Linked to Repression of p16INK4a and E2F1 Binding at the Promoter of the Gene Encoding Survivin (BIRC5)
Because the repression of p16INK4a is known to be involved in senescence but has not been directly linked previously to cell death pathways, we investigated the possibility that expression of p16INK4a and survivin may be linked. This possibility was brought to light by the observation that survivin gene expression increased in control vector-transduced cells around the same time that p16INK4a was spontaneously repressed in these cells (∼170 PD) (Fig. 5A). The detection of high levels of survivin mRNA and protein in p16 shRNA-transduced MRC5hTERT-1 cells provided further evidence of an intrinsic link between the repression of p16INK4a and up-regulation of survivin (Fig. 5, A and B). The inverse regulation of p16INK4a and survivin gene expression observed during immortalization of MRC5hTERT-1 cells was also evident in another fibroblast cell strain, WI-38, which was transduced with hTERT and immortalized in an independent laboratory (Fig. 5D) (21).
The functional significance of the link between p16INK4a repression and up-regulation of survivin was investigated by down-regulating survivin in cells transduced with shRNA targeting p16INK4a or control shRNA. The cells were then exposed to tBOOH for 48 h and assayed for apoptotic death using annexin/PI staining. Consistent with the data shown in Fig. 4C, repression of p16INK4a rendered MRC5hTERT cells resistant to oxidant-induced death (Fig. 6A, p < 0.05 at 50 μm tBOOH). However, this survival effect was completely abrogated by siRNA-mediated down-regulation of survivin. Taken together with the expression data (Fig. 5, A–C), these results identify survivin as a crucial mediator of cell survival associated with p16INK4a repression.
FIGURE 6.
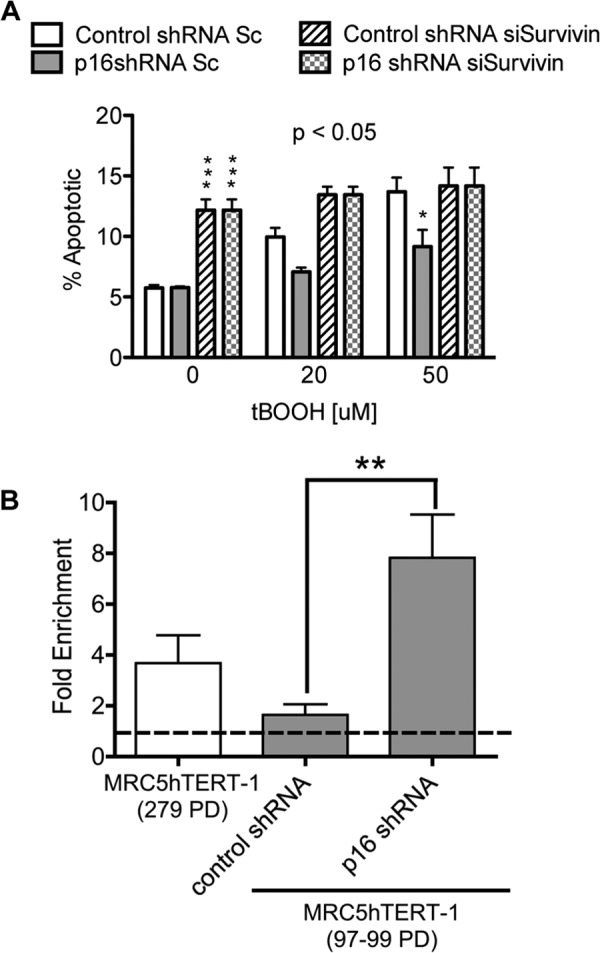
Repression of p16INK4a is intrinsically linked to survivin by E2F1 binding at the BIRC5 promoter and resistance to apoptotic cell death. A, EP MRC5hTERT-1 cells transduced with p16 shRNA or control shRNA were transfected with Sc or Survivin siRNA and then treated with tBOOH for 48 h. Apoptotic cells were quantified by annexin V-APC/PI staining. Values are presented as means ± S.E. from three independent experiments. The p value is from ANOVA analysis of the effect of shRNA. *, p < 0.05; ***, p < 0.001 in Bonferroni post-test comparisons of p16 shRNA and control shRNA at the indicated tBOOH concentrations. B, E2F1 and IgG (control) antibodies were used in ChIP assays of immortal MRC5hTERT-1 (279 PD) and precrisis MRC5hTERT-1 cells transduced with p16 shRNA or control shRNA. Values obtained with the E2F1 antibody are presented relative to IgG values. The results are means + S.E. from three assays, with each performed in duplicate. **, p < 0.01 in Student's t test comparisons of control and p16 shRNA-transduced cells.
A possible mechanism by which p16INK4a may influence gene expression is via its well described role in the regulation of pRB phosphorylation (39, 40). By inhibiting pRB phosphorylation, p16INK4a limits the availability of the E2F-1 transcription factor, which is sequestered into an inactive complex with hypophosphorylated pRB. Because it has been shown previously that E2F-1 directly binds and transactivates the promoter of the gene encoding survivin (BIRC5) (41), we employed the ChIP method to determine whether p16INK4a expression alters the interaction of E2F-1 with BIRC5 in MRC5hTERT-1 cells. These investigations confirmed that E2F-1 was bound to the BIRC5 promoter in immortal MRC5hTERT-1 cells (279 PD) that spontaneously repressed p16INK4a (Fig. 6B). Moreover, comparison of EP MRC5hTERT cells (98 PD) transduced with p16shRNA or control vector revealed a 7-fold enrichment of E2F1 protein at the BIRC5 promoter in the p16shRNA-transduced cells. These results indicate that p16INK4a is an important determinant of survivin expression and suggest that immortal cells acquire resistance to oxidative stress as a result of the spontaneous repression of p16INK4a and consequential up-regulation of survivin via E2F1 binding.
DISCUSSION
Past studies have established that an abundance of survivin contributes to drug resistance in advanced cancers and tumor-derived cell lines (27). However, prior to these investigations, the significance of high levels of survivin in premalignant cells was unclear (42, 43). Results from this study show that the up-regulation of survivin confers non-transformed immortal cell resistance to oxidative stress, a property that would potentially provide an advantage during the early stages of carcinogenesis and the transition to the high intracellular redox state induced by oncogenic activation (44–47).
It has been shown previously that the down-regulation of survivin activates TP53 and stimulates caspase activity in tumor cell lines (48, 49). In this study, repression of survivin in non-transformed immortal cells resulted in an accumulation of p53 and p21cip1 as well as the activation of the caspases 3, 8, and 9. This was accompanied by a moderate increase in basal apoptosis and heightened sensitivity to the organic peroxide tBOOH. Additional results from this study showing that repression of p53 rescued survivin-depleted cells from oxidant-induced cell death indicate that p53 plays a central role in mediating oxidative stress-induced cell death in survivin-deficient cells.
Although the up-regulation of survivin conferred resistance to oxidant-induced cell death, increased expression of survivin was not sufficient to rescue hTERT-transduced cells from a proliferative crisis that was mitigated by growth in low oxygen. These results suggest that survivin may protect against the specific type of oxidative damage elicited by tBOOH and/or that that crisis was the result of a combination of antiproliferative pathways. tBOOH has been used extensively in previous studies to model the effects of oxidative stress (38, 50–52). Its mechanism of action involves the generation of methyl and tert-butoxyl radicals that elicit cytotoxicity through lipid peroxidation, glutathione oxidation, and perturbation of intracellular Ca2+ (38, 52). These changes culminate in mitochondrial swelling, release of cytochrome c, and caspase activation. Mitochondrial apoptosis induced by tBOOH is overcome by diverse antioxidants or overexpression of Bcl-2 (53). These investigations are the first to demonstrate that the up-regulation of survivin also protects against tBOOH-induced apoptotic cell death.
The possibility that the proliferative impairment observed at crisis was the result of multiple pathways is supported by results from our previous study that showed evidence of both cell death and senescence-like growth arrest in cultures engaged in crisis (28). This is also consistent with the diverse effects of oxidative stress that have been documented (37). Results showing that p16INK4a repression circumvented crisis extend to prior studies that have shown that inactivation of p16INK4a increases the replicative lifespan of normal and premalignant human cells by overcoming telomere-independent stresses (6, 54, 55). In this study, it was shown that p16INK4a repression suppressed the senescence response to chronic exposure to low-dose tBOOH and mitigated cell death induced by acute oxidant exposure. The capacity of p16INK4a repression to avert both senescence and cell death presents a probable explanation for the highly efficient means by which the down-regulation of p16INK4a overcame crisis and promoted immortalization.
The function of p16INK4a in the regulation of G1-S phase cell cycle progression accounts for the means by which p16INK4a repression circumvents senescence (39, 56). In contrast to this well described paradigm, little is known about the interplay of p16INK4a and cell death pathways. Past studies that have drawn a link between p16INK4a expression and cell death include reports that showed enhanced survival of kidney epithelial cells in p16INK4a null mice, as well as a study that demonstrated resistance to mitochondrial- and death receptor-mediated apoptosis in human leukemia cell lines with deficient p16INK4a expression (57–59). In this study, spontaneous or shRNA-mediated down-regulation of p16INK4a rendered MRC5hTERT cells resistant to oxidative stress. siRNA-mediated down-regulation of survivin showed that the abundance of survivin was central to the relative resistance of p16INK4a-deficient immortal cells to oxidative stress. Collectively, our results demonstrate that loss of p16INK4a expression during immortalization promotes resistance to cell death and implicate survivin as an essential mediator of this effect. Because expression of survivin is tuned precisely to the cell cycle in both normal and immortal cells (23, 41), it will be of considerable interest for future studies to determine whether the survival effect of p16INK4a is cell cycle-dependent.
Our results linking the up-regulation of survivin to repression of p16INK4a add to past studies that demonstrated transcriptional activation of BIRC5 following the inactivation of tumor suppressors TP53, PTEN, and RB (41, 49, 60, 61). In our previous investigations, it was noted that the up-regulation of survivin in a panel of immortal MRC5hTERT clones did not always correspond to p16INK4a silencing (23). Those observations may be explained by defects in other tumor suppressors that regulate survivin. This study directly demonstrates that shRNA-mediated down-regulation of p16INK4a in TP53-competent cells increases the expression of survivin. This may be mediated (at least in part) by the E2F1 transcription factor, which has been shown previously to directly bind and transactivate the BIRC5 promoter (41). In the latter study, normal human fibroblasts (WI-38) were synchronized to ensure optimal E2F availability at the time of the ChIP assay. In contrast, our study employed unsynchronized hTERT-transduced cells with and without p16INK4a repression. Only a very low level of E2F-1 binding was detectable in unsynchronized, early-passage control cells that express p16INK4a. This is consistent with the low level of survivin mRNA detected in these cells. Results from this study further show that E2F1 binding at the BIRC5 promoter was increased when p16INK4a was either repressed spontaneously during immortalization or quenched by shRNA. These data strongly implicate E2F1 in the regulation of survivin gene expression during immortalization.
Collectively, these investigations show that repression of p16INK4a during immortalization of human myofibroblasts results in an up-regulation of survivin and consequential resistance to oxidative stress. Repression of p16INK4a overcame both senescence and cell death and thereby very effectively promoted immortalization of telomerase-transduced cells. In contrast, the up-regulation of survivin was not sufficient to circumvent the alternate cellular fates that impeded immortalization. Thus, the primary consequence of the up-regulation of survivin identified in these studies was resistance to stress-induced cell death, a property that would be favorable for cytoprotection against redox changes and other stresses associated with malignant transformation.
Acknowledgments
We thank Prof. John D. Crispino (North Western University) for retroviral MIGRSurvivin, Prof. Reuven Agami (Netherlands Cancer Institute, Amsterdam, The Netherlands) for pRetroSuper-p16shRNA, Prof. Varda Rotter (Weizmann Institute of Science, Israel) for pRetroSuper-mp63shRNA and hTERT-immortalized WI-38 cells, and Prof. Andrei Gudkov (Roswell Park Cancer Institute, Buffalo, NY) for the p53shRNA retroviral vector.
This work was supported by National Health and Medical Research Council (NHMRC) Career Development Award 510378 (to K. L. M.), by a senior research fellowship (to R. B. L.), by NHMRC Project Grant 568704, and by Cancer Institute New South Wales Career Development and Support Fellowship 07-CDF-1/23 (to K. L. M.).
- hTERT
- human telomerase reverse transcriptase
- tBOOH
- tert-butylhydroperoxide
- PD
- population doubling
- APC
- allophycocyanin
- PI
- propidium iodide
- qRT-PCR
- real-time reverse transcriptase PCR
- ANOVA
- analysis of variance
- SURV
- MIGR1-survivin.
REFERENCES
- 1. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer. The next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 2. Bryan T. M., Englezou A., Gupta J., Bacchetti S., Reddel R. R. (1995) Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 14, 4240–4248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meyerson M., Counter C. M., Eaton E. N., Ellisen L. W., Steiner P., Caddle S. D., Ziaugra L., Beijersbergen R. L., Davidoff M. J., Liu Q., Bacchetti S., Haber D. A., Weinberg R. A. (1997) hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 90, 785–795 [DOI] [PubMed] [Google Scholar]
- 4. Bodnar A. G., Ouellette M., Frolkis M., Holt S. E., Chiu C. P., Morin G. B., Harley C. B., Shay J. W., Lichtsteiner S., Wright W. E. (1998) Extension of life-span by introduction of telomerase into normal human cells. Science 279, 349–352 [DOI] [PubMed] [Google Scholar]
- 5. Whitaker N. J., Bryan T. M., Bonnefin P., Chang A. C., Musgrove E. A., Braithwaite A. W., Reddel R. R. (1995) Involvement of RB-1, p53, p16INK4 and telomerase in immortalisation of human cells. Oncogene 11, 971–976 [PubMed] [Google Scholar]
- 6. Kiyono T., Foster S. A., Koop J. I., McDougall J. K., Galloway D. A., Klingelhutz A. J. (1998) Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 396, 84–88 [DOI] [PubMed] [Google Scholar]
- 7. Huschtscha L. I., Noble J. R., Neumann A. A., Moy E. L., Barry P., Melki J. R., Clark S. J., Reddel R. R. (1998) Loss of p16INK4 expression by methylation is associated with lifespan extension of human mammary epithelial cells. Cancer Res. 58, 3508–3512 [PubMed] [Google Scholar]
- 8. Okamoto A., Demetrick D. J., Spillare E. A., Hagiwara K., Hussain S. P., Bennett W. P., Forrester K., Gerwin B., Serrano M., Beach D. H. (1994) Mutations and altered expression of p16INK4 in human cancer. Proc. Natl. Acad. Sci. U.S.A. 91, 11045–11049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shay J. W., Bacchetti S. (1997) A survey of telomerase activity in human cancer. Eur. J. Cancer 33, 787–791 [DOI] [PubMed] [Google Scholar]
- 10. Harley C. B., Futcher A. B., Greider C. W. (1990) Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460 [DOI] [PubMed] [Google Scholar]
- 11. d'Adda di Fagagna F., Reaper P. M., Clay-Farrace L., Fiegler H., Carr P., Von Zglinicki T., Saretzki G., Carter N. P., Jackson S. P. (2003) A DNA damage checkpoint response in telomere-initiated senescence. Nature 426, 194–198 [DOI] [PubMed] [Google Scholar]
- 12. Ksiazek K., Passos J. F., Olijslagers S., Saretzki G., Martin-Ruiz C., von Zglinicki T. (2007) Premature senescence of mesothelial cells is associated with non-telomeric DNA damage. Biochem. Biophys. Res. Commun. 362, 707–711 [DOI] [PubMed] [Google Scholar]
- 13. von Zglinicki T., Saretzki G., Döcke W., Lotze C. (1995) Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts. A model for senescence? Exp. Cell Res. 220, 186–193 [DOI] [PubMed] [Google Scholar]
- 14. Chen Q., Ames B. N. (1994) Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc. Natl. Acad. Sci. U.S.A. 91, 4130–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Passos J. F., Saretzki G., Ahmed S., Nelson G., Richter T., Peters H., Wappler I., Birket M. J., Harold G., Schaeuble K., Birch-Machin M. A., Kirkwood T. B., von Zglinicki T. (2007) Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 5, e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen Q., Fischer A., Reagan J. D., Yan L. J., Ames B. N. (1995) Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc. Natl. Acad. Sci. U.S.A. 92, 4337–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Napier C. E., Veas L. A., Kan C. Y., Taylor L. M., Yuan J., Wen V. W., James A., O'Brien T. A., Lock R. B., MacKenzie K. L. (2010) Mild hyperoxia limits hTR levels, telomerase activity, and telomere length maintenance in hTERT-transduced bone marrow endothelial cells. Biochim. Biophys. Acta 1803, 1142–1153 [DOI] [PubMed] [Google Scholar]
- 18. Forsyth N. R., Evans A. P., Shay J. W., Wright W. E. (2003) Developmental differences in the immortalization of lung fibroblasts by telomerase. Aging Cell 2, 235–243 [DOI] [PubMed] [Google Scholar]
- 19. Vaziri H., Benchimol S. (1998) Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr. Biol. 8, 279–282 [DOI] [PubMed] [Google Scholar]
- 20. MacKenzie K. L., Franco S., May C., Sadelain M., Moore M. A. (2000) Mass cultured human fibroblasts overexpressing hTERT encounter a growth crisis following an extended period of proliferation. Exp. Cell Res. 259, 336–350 [DOI] [PubMed] [Google Scholar]
- 21. Milyavsky M., Shats I., Erez N., Tang X., Senderovich S., Meerson A., Tabach Y., Goldfinger N., Ginsberg D., Harris C. C., Rotter V. (2003) Prolonged culture of telomerase-immortalized human fibroblasts leads to a premalignant phenotype. Cancer Res. 63, 7147–7157 [PubMed] [Google Scholar]
- 22. MacKenzie K. L., Franco S., Naiyer A. J., May C., Sadelain M., Rafii S., Moore M. A. (2002) Multiple stages of malignant transformation of human endothelial cells modelled by co-expression of telomerase reverse transcriptase, SV40 T antigen and oncogenic N-ras. Oncogene 21, 4200–4211 [DOI] [PubMed] [Google Scholar]
- 23. Yuan J., Yang B. M., Zhong Z. H., Shats I., Milyavsky M., Rotter V., Lock R. B., Reddel R. R., MacKenzie K. L. (2009) Up-regulation of survivin during immortalization of nontransformed human fibroblasts transduced with telomerase reverse transcriptase. Oncogene 28, 2678–2689 [DOI] [PubMed] [Google Scholar]
- 24. Ambrosini G., Adida C., Altieri D. C. (1997) A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 3, 917–921 [DOI] [PubMed] [Google Scholar]
- 25. Marusawa H., Matsuzawa S., Welsh K., Zou H., Armstrong R., Tamm I., Reed J. C. (2003) HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J. 22, 2729–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dohi T., Okada K., Xia F., Wilford C. E., Samuel T., Welsh K., Marusawa H., Zou H., Armstrong R., Matsuzawa S., Salvesen G. S., Reed J. C., Altieri D. C. (2004) An IAP-IAP complex inhibits apoptosis. J. Biol. Chem. 279, 34087–34090 [DOI] [PubMed] [Google Scholar]
- 27. Pennati M., Folini M., Zaffaroni N. (2007) Targeting survivin in cancer therapy. Fulfilled promises and open questions. Carcinogenesis 28, 1133–1139 [DOI] [PubMed] [Google Scholar]
- 28. Taylor L. M., James A., Schuller C. E., Brce J., Lock R. B., Mackenzie K. L. (2004) Inactivation of p16INK4a, with retention of pRB and p53/p21cip1 function, in human MRC5 fibroblasts that overcome a telomere-independent crisis during immortalization. J. Biol. Chem. 279, 43634–43645 [DOI] [PubMed] [Google Scholar]
- 29. Vaziri S. A., Hill J., Chikamori K., Grabowski D. R., Takigawa N., Chawla-Sarkar M., Rybicki L. R., Gudkov A. V., Mekhail T., Bukowski R. M., Ganapathi M. K., Ganapathi R. (2005) Sensitization of DNA damage-induced apoptosis by the proteasome inhibitor PS-341 is p53 dependent and involves target proteins 14-3-3sigma and survivin. Mol. Cancer Ther. 4, 1880–1890 [DOI] [PubMed] [Google Scholar]
- 30. Gurbuxani S., Xu Y., Keerthivasan G., Wickrema A., Crispino J. D. (2005) Differential requirements for survivin in hematopoietic cell development. Proc. Natl. Acad. Sci. U.S.A. 102, 11480–11485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Voorhoeve P. M., Agami R. (2003) The tumor-suppressive functions of the human INK4A locus. Cancer Cell 4, 311–319 [DOI] [PubMed] [Google Scholar]
- 32. Milyavsky M., Shats I., Cholostoy A., Brosh R., Buganim Y., Weisz L., Kogan I., Cohen M., Shatz M., Madar S., Kalo E., Goldfinger N., Yuan J., Ron S., MacKenzie K., Eden A., Rotter V. (2007) Inactivation of myocardin and p16 during malignant transformation contributes to a differentiation defect. Cancer Cell 11, 133–146 [DOI] [PubMed] [Google Scholar]
- 33. Dimri G. P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E. E., Linskens M., Rubelj I., Pereira-Smith O. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U.S.A. 92, 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kan C.-Y., Wen V. W., Pasquier E., Jankowski K., Chang M., Richards L. A., Kavallaris M., MacKenzie K. L. (2012) Endothelial cell dysfunction and cytoskeletal changes associated with repression of p16(INK4a) during immortalization. Oncogene 31, 4815–4827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu T., Tee A. E., Porro A., Smith S. A., Dwarte T., Liu P. Y., Iraci N., Sekyere E., Haber M., Norris M. D., Diolaiti D., Della Valle G., Perini G., Marshall G. M. (2007) Activation of tissue transglutaminase transcription by histone deacetylase inhibition as a therapeutic approach for Myc oncogenesis. Proc. Natl. Acad. Sci. U.S.A. 104, 18682–18687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Franco S., MacKenzie K. L., Dias S., Alvarez S., Rafii S., Moore M. A. (2001) Clonal variation in phenotype and life span of human embryonic fibroblasts (MRC-5) transduced with the catalytic component of telomerase (hTERT). Exp. Cell Res. 268, 14–25 [DOI] [PubMed] [Google Scholar]
- 37. Bladier C., Wolvetang E. J., Hutchinson P., de Haan J. B., Kola I. (1997) Cell Growth & Differ. 8, 589–598 [PubMed] [Google Scholar]
- 38. Rice-Evans C., Baysal E., Pashby D. P., Hochstein P. (1985) t-Butyl hydroperoxide-induced perturbations of human erythrocytes as a model for oxidant stress. Biochim. Biophys. Acta 815, 426–432 [DOI] [PubMed] [Google Scholar]
- 39. Serrano M., Hannon G. J., Beach D. (1993) A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 366, 704–707 [DOI] [PubMed] [Google Scholar]
- 40. Xiong Y., Zhang H., Beach D. (1993) Subunit rearrangement of the cyclin-dependent kinases is associated with cellular transformation. Genes Dev. 7, 1572–1583 [DOI] [PubMed] [Google Scholar]
- 41. Jiang Y., Saavedra H. I., Holloway M. P., Leone G., Altura R. A. (2004) Aberrant regulation of survivin by the RB/E2F family of proteins. J. Biol. Chem. 279, 40511–40520 [DOI] [PubMed] [Google Scholar]
- 42. Gianani R., Jarboe E., Orlicky D., Frost M., Bobak J., Lehner R., Shroyer K. R. (2001) Expression of survivin in normal, hyperplastic, and neoplastic colonic mucosa. Hum. Pathol. 32, 119–125 [DOI] [PubMed] [Google Scholar]
- 43. Frost M., Jarboe E. A., Orlicky D., Gianani R., Thompson L. C., Enomoto T., Shroyer K. R. (2002) Immunohistochemical localization of survivin in benign cervical mucosa, cervical dysplasia, and invasive squamous cell carcinoma. Am. J. Clin. Pathol. 117, 738–744 [DOI] [PubMed] [Google Scholar]
- 44. Szatrowski T. P., Nathan C. F. (1991) Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 51, 794–798 [PubMed] [Google Scholar]
- 45. Irani K., Xia Y., Zweier J. L., Sollott S. J., Der C. J., Fearon E. R., Sundaresan M., Finkel T., Goldschmidt-Clermont P. J. (1997) Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 275, 1649–1652 [DOI] [PubMed] [Google Scholar]
- 46. Lee A. C., Fenster B. E., Ito H., Takeda K., Bae N. S., Hirai T., Yu Z. X., Ferrans V. J., Howard B. H., Finkel T. (1999) Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 274, 7936–7940 [DOI] [PubMed] [Google Scholar]
- 47. Vafa O., Wade M., Kern S., Beeche M., Pandita T. K., Hampton G. M., Wahl G. M. (2002) c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function. A mechanism for oncogene-induced genetic instability. Mol. Cell 9, 1031–1044 [DOI] [PubMed] [Google Scholar]
- 48. Xu J. H., Wang A. X., Huang H. Z., Wang J. G., Pan C. B., Zhang B. (2010) Survivin shRNA induces caspase-3-dependent apoptosis and enhances cisplatin sensitivity in squamous cell carcinoma of the tongue. Oncol. Res. 18, 377–385 [DOI] [PubMed] [Google Scholar]
- 49. Mirza A., McGuirk M., Hockenberry T. N., Wu Q., Ashar H., Black S., Wen S. F., Wang L., Kirschmeier P., Bishop W. R., Nielsen L. L., Pickett C. B., Liu S. (2002) Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene 21, 2613–2622 [DOI] [PubMed] [Google Scholar]
- 50. Guidarelli A., Cattabeni F., Cantoni O. (1997) Alternative mechanisms for hydroperoxide-induced DNA single strand breakage. Free Radic. Res. 26, 537–547 [DOI] [PubMed] [Google Scholar]
- 51. Dumont P., Burton M., Chen Q. M., Gonos E. S., Frippiat C., Mazarati J. B., Eliaers F., Remacle J., Toussaint O. (2000) Induction of replicative senescence biomarkers by sublethal oxidative stresses in normal human fibroblast. Free Radic. Biol. Med. 28, 361–373 [DOI] [PubMed] [Google Scholar]
- 52. Guidarelli A., Brambilla L., Clementi E., Sciorati C., Cantoni O. (1997) Stimulation of oxygen consumption promotes mitochondrial calcium accumulation, a process associated with, and causally linked to, enhanced formation of tert-butylhydroperoxide-induced DNA single-strand breaks. Exp. Cell Res. 237, 176–185 [DOI] [PubMed] [Google Scholar]
- 53. Rincheval V., Bergeaud M., Mathieu L., Leroy J., Guillaume A., Mignotte B., Le Floch N., Vayssière J. L. (2012) Differential effects of Bcl-2 and caspases on mitochondrial permeabilization during endogenous or exogenous reactive oxygen species-induced cell death. A comparative study of HO, paraquat, t-BHP, etoposide and TNF-α-induced cell death. Cell Biol. Toxicol. 28, 239–253 [DOI] [PubMed] [Google Scholar]
- 54. Duan J., Zhang Z., Tong T. (2001) Senescence delay of human diploid fibroblast induced by anti-sense p16INK4a expression. J. Biol. Chem. 276, 48325–48331 [DOI] [PubMed] [Google Scholar]
- 55. Brenner A. J., Stampfer M. R., Aldaz C. M. (1998) Increased p16 expression with first senescence arrest in human mammary epithelial cells and extended growth capacity with p16 inactivation. Oncogene 17, 199–205 [DOI] [PubMed] [Google Scholar]
- 56. Medema R. H., Herrera R. E., Lam F., Weinberg R. A. (1995) Growth suppression by p16ink4 requires functional retinoblastoma protein. Proc. Natl. Acad. Sci. U.S.A. 92, 6289–6293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wolstein J. M., Lee D. H., Michaud J., Buot V., Stefanchik B., Plotkin M. D. (2010) INK4a knockout mice exhibit increased fibrosis under normal conditions and in response to unilateral ureteral obstruction. Am. J. Physiol. Renal Physiol. 299, F1486–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ausserlechner M. J., Obexer P., Wiegers G. J., Hartmann B. L., Geley S., Kofler R. (2001) J. Biol. Chem. 276, 10984–10989 [PubMed] [Google Scholar]
- 59. Obexer P., Hagenbuchner J., Rupp M., Salvador C., Holzner M., Deutsch M., Porto V., Kofler R., Unterkircher T., Ausserlechner M. J. (2009) p16INK4A sensitizes human leukemia cells to FAS- and glucocorticoid-induced apoptosis via induction of BBC3/Puma and repression of MCL1 and BCL2. J. Biol. Chem. 284, 30933–30940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Guha M., Plescia J., Leav I., Li J., Languino L. R., Altieri D. C. (2009) Endogenous tumor suppression mediated by PTEN involves survivin gene silencing. Cancer Res. 69, 4954–4958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hoffman W. H., Biade S., Zilfou J. T., Chen J., Murphy M. (2002) Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J. Biol. Chem. 277, 3247–3257 [DOI] [PubMed] [Google Scholar]