

Background: DDR1 is a receptor tyrosine kinase that signals in response to collagen and regulates cell-collagen interactions. MT-MMPs are membrane-anchored proteases that accomplish pericellular collagenolysis.
Results: MT-MMPs cleave DDR1 and regulate collagen-induced receptor phosphorylation.
Conclusion: MT-MMPs negatively regulate DDR1 activation by promoting receptor ectodomain shedding.
Significance: Cross-talk between membrane-anchored collagenases and RTKs integrates collagen-induced signaling and pericellular proteolysis.
Keywords: Cell Surface Receptor, Collagen, Matrix Metalloproteinase (MMP), Proteolytic Enzymes, Shedding, Discoidin Domain Receptors
Abstract
The discoidin domain receptors (DDRs) are receptor tyrosine kinases that upon binding to collagens undergo receptor phosphorylation, which in turn activates signal transduction pathways that regulate cell-collagen interactions. We report here that collagen-dependent DDR1 activation is partly regulated by the proteolytic activity of the membrane-anchored collagenases, MT1-, MT2-, and MT3-matrix metalloproteinase (MMP). These collagenases cleave DDR1 and attenuate collagen I- and IV-induced receptor phosphorylation. This effect is not due to ligand degradation, as it proceeds even when the receptor is stimulated with collagenase-resistant collagen I (r/r) or with a triple-helical peptide harboring the DDR recognition motif in collagens. Moreover, the secreted collagenases MMP-1 and MMP-13 and the glycosylphosphatidylinositol-anchored membrane-type MMPs (MT4- and MT6-MMP) have no effect on DDR1 cleavage or activation. N-terminal sequencing of the MT1-MMP-mediated cleaved products and mutational analyses show that cleavage of DDR1 takes place within the extracellular juxtamembrane region, generating a membrane-anchored C-terminal fragment. Metalloproteinase inhibitor studies show that constitutive shedding of endogenous DDR1 in breast cancer HCC1806 cells is partly mediated by MT1-MMP, which also regulates collagen-induced receptor activation. Taken together, these data suggest a role for the collagenase of membrane-type MMPs in regulation of DDR1 cleavage and activation at the cell-matrix interface.
Introduction
Tissue homeostasis is dictated in part by specific interactions between cells and their extracellular milieu. These interactions are essential to support normal cellular function and are usually disrupted during pathological processes. It is well established that cells respond to environmental cues through specific receptors that are activated and signal in response to extracellular matrix (ECM)2 proteins, in particular collagen, one of the major ECM components. Among the various proteins that can act as collagen receptors, the discoidin domain receptors (DDRs) are unique because they are receptor tyrosine kinases (RTKs) that are specifically activated in response to collagen (1–3). The DDR family includes two members, DDR1 and DDR2. DDR1 is activated by both fibrillar and nonfibrillar collagens, whereas DDR2 is only activated by fibrillar collagens. For instance, interstitial collagen I activates both receptors, but basement membrane collagen IV only activates DDR1 (4, 5). DDR2 preferentially binds collagen II (6) and collagen X (7). DDR1 can also bind collagen VIII (8), but it is not known whether DDR2 shares this property. Both DDRs only bind and signal in response to triple-helical collagen and thus do not recognize heat-denatured collagen (gelatin) or degraded collagen (4, 9). However, triple-helical peptides containing the collagen-binding motif of DDRs, GVMGFO (where O is hydroxyproline), which is present in fibrillar collagens I–III, are capable of inducing receptor activation (10, 11). In response to collagen, DDRs undergo tyrosine autophosphorylation within the intracellular juxtamembrane region and the kinase domain (KD), leading to the recruitment of adaptor proteins and activation of downstream effectors (3, 12). However, as opposed to most RTKs, the kinetics of DDR phosphorylation is slow and sustained (4, 5), indicative of a unique activation process that remains to be elucidated.
Structurally, the DDRs share a similar basic domain organization composed of an extracellular N-terminal discoidin domain (DS) followed by a predicted DS-like domain (DS-like) (13), a short extracellular juxtamembrane (EJXM) linker, a single-span transmembrane domain, and an intracellular juxtamembrane domain connected to a KD (1, 3, 14, 15). For a detailed description of DDR structure and domain organization, see Refs. 1–3. In the case of DDR1, alternative splicing generates five isoforms, which share a similar ectodomain organization. However, these isoforms differ in their intracellular juxtamembrane and KD. For instance, DDR1b and DDR1c contain an additional 37 residues in the intracellular juxtamembrane region (residues 505–541), whereas DDR1c possesses six additional residues in the KD. Thus, these DDR1 variants are fully functional RTKs that may activate different signaling pathways in response to collagen and elicit different cell functions (1–3). In contrast to DDR1, DDR2 is expressed as a single protein species.
A unique aspect of DDRs is the nature of their ligand, which includes several collagen types (1). The larger supramolecular structure of collagens does not appear to be critical for DDR activation, as triple-helical peptides bearing the DDR-binding motif are fully capable of inducing receptor phosphorylation. However, collagens are known to undergo multiple structural modifications that alter their mechanical properties, strength, and stability, which are caused in part by the action of membrane-anchored and secreted collagen-degrading proteases, specifically the members of the matrix metalloproteinase family of zinc-dependent endopeptidases (16). In particular, a triad of membrane-type MMPs (MT-MMPs), MT1-(MMP-14), MT2-(MMP-15), and MT3-(MMP-16), are known to be critical mediators of collagenolysis at the pericellular space (17–19). Because MT-MMPs and DDRs share a common substrate/ligand, we hypothesized that MT-MMPs can regulate collagen-induced activation of DDRs. In this study, we focused on DDR1 and examined the interaction of this receptor with membrane-anchored and secreted collagenases. Although we expected effects on DDR1 activation mediated by collagen degradation, we found that MT1-, MT2-, and MT3-MMP, but not the secreted collagenases, MMP-1 and MMP-13, inhibited DDR1 activation by promoting receptor cleavage at the EJXM region. Our results shed light on a novel interaction between surface proteases and RTKs that integrate collagen-induced signaling and pericellular proteolysis.
EXPERIMENTAL PROCEDURES
Cell Lines
Immortalized monkey kidney epithelial COS1 (CRL-1650) cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS), 2 mm l-glutamine, and antibiotics at 37 °C in an atmosphere of 95% air and 5% CO2. Human breast cancer T47D cells (HTB-133) were obtained from ATCC and cultured in RPMI 1640 medium supplemented with 10% FBS, insulin, and antibiotics. These cells were transfected to stably express human recombinant MT1-MMP, as described previously (20). Human breast cancer HCC1806 (CRL-2335) cells were obtained from ATCC and cultured in RPMI 1640 medium supplemented with 10% FBS and antibiotics.
Antibodies and Reagents
A rabbit polyclonal antibody against the C-terminal region of DDR1 (sc-532) and a mouse monoclonal antibody (mAb) to GAPDH (sc-47724) were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Goat polyclonal antibodies against the N-terminal region of DDR1 (AF2396) were from R&D Systems (Minneapolis, MN). mAbs against the catalytic domain of MT1-MMP (Lem2/15) were a kind gift from Dr. Alicia G Arroyo (Centro Nacional de Investigaciones Cardiovasulares, Madrid, Spain). Anti-phosphotyrosine mAb (clone 4G10®), referred to as anti-Tyr(P), and mAbs against human MT2-MMP (MAB3320), MMP-1 (MAB3307), and MMP-13 (MAB3321) were purchased from EMD Millipore (Billerica, MA). A polyclonal antibody against MT3-MMP (polyclonal antibody 318, directed against residues 318–335 of human MT3-MMP) was produced in our laboratory. Rabbit monoclonal antibodies to human MT4-MMP (EP1270Y) were purchased from Epitomics/Abcam (Burlingame, CA). A mouse mAb against Myc was a generous gift from Dr. Guri Tzivion (University of Mississippi Medical Center, Jackson, MS). Anti-FLAG M2 mAb (F1804) and anti-β-actin mAb (A5441) were both purchased from Sigma. Anti-TIMP-1 mAb (IM32) was purchased from Calbiochem, and anti-TIMP-2 mAb CA-101 was described previously (21). MIK-G2 (4-[4-(methanesulfonamido)phenoxy]phenylsulfonyl methylthiirane) was synthesized in the Mobashery laboratory, as described previously (22). Rat tail collagen type I (catalog no. 3440-100-01) and mouse collagen IV (catalog no. 3410-010-01) were purchased from Trevigen (Gaithersburg, MD). Collagenase-resistant (r/r) and wild type mouse collagen I were gifts from Dr. Stephen Weiss (University of Michigan, Ann Arbor, MI). The triple-helical collagen model peptide (GPO)5GPRGQOGVMGFO(GPO)5-NH2 (where O is 4-hydroxyproline) harboring the DDR recognition motif, GVMFO, was assembled using Fmoc solid-phase chemistry, as described previously (23). The peptide exhibited a triple-helix melting temperature (Tm) of 60.8 °C. Primers for RT-PCR were purchased from IDT (Coralville, IA), and the sequences are described in supplemental Table 1. Human recombinant TIMP-1 and TIMP-2 were purified to homogeneity from mammalian cells, as described previously (24).
Reagents for peptide synthesis and purification of crude products were purchased as follows: Fmoc-amino acids, 2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate, and N,N-dimethylformamide (AR grade) were from AGTC Bioproducts (Wilmington, MA); N-hydroxybenzotriazole·H2O was from Anaspec (Fremont, CA); thioanisole was from Fluka; trifluoroacetic acid (TFA) was from ThermoFisher Scientific; NovaPEG Rink amide resin (0.49 mmol/g) was from NovaBiochem; 1,2-ethanedithiol and piperazine were from Acros Organics; N-methylmorpholine, acetonitrile (HPLC grade), and water (HPLC grade) were from Sigma; and methyl tert-butyl ether was from Mallinckrodt Baker. Human recombinant pro-MT1-MMP ectodomain was purchased from EMD Millipore. Human recombinant pro-MMP-1 and p-aminophenylmercuric acetate were obtained from Calbiochem; trypsin-3 and 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride were from R&D Systems.
cDNA Constructs
All human DDR and MT-MMP cDNAs were cloned into the expression vector pcDNA3.1/myc-His(−) A (Invitrogen) using conventional cloning approaches. The human DDR1a and DDR1b cDNAs were kindly provided by Dr. Teizo Yoshimura (NCI, National Institutes of Health, Frederick, MD). In the case of the DDR1 isoforms, we constructed DDR1a or DDR1b cDNAs with or without a C-terminal Myc-His tag sequence inserted immediately after Val876 of DDR1a or Val913 of DDR1b to generate DDR1a/Myc and DDR1b/Myc. In addition, we also generated DDR1a/Myc bearing an N-terminal FLAG sequence (Asp-Tyr-Lys-Asp-Asp-Asp-Lys) inserted between His25 and Phe26, to generate FLAG/DDR1a/Myc. The Myc or Myc-FLAG-tagged DDR1 isoforms were phosphorylated in response to collagen I. To generate human DDR1a deletion mutants at the EJXM region, we deleted residues 377–399, 394–416, or 400–416 by PCR mutagenesis to generate DDR1aΔ377–399, DDR1aΔ394–416, and DDR1aΔ400–416, respectively, all harboring a Myc tag at the C-terminal end. The sequences of all mutant constructs were verified by DNA sequencing.
The cDNAs of human MT1-, MT2-, MT3-MT4, and MT6-MMP were all available in our laboratory. The human MT6-MMP cDNA construct included a FLAG tag inserted immediately after the furin motif. Also available in our laboratory were the following MT1-MMP mutants: catalytically inactive MT1-MMP bearing an Ala substitution at Glu240 (referred to as MT1-E/A); MT1-MMPΔCT, a deletion mutant lacking the cytosolic tail (Arg563 to Val582); and MT1-MMPΔTM/CT, a deletion mutant lacking both the transmembrane region and the cytosolic tail (Val542 to Val582), which were generated as described previously (25, 26). The human cDNAs of MMP-1 and MMP-13, harboring a proprotein convertase recognition motif (RXKR) located between the pro- and the catalytic domains of the proteases to allow activation of the zymogen prior to secretion (18, 27), referred to as MMP-1RXKR and MMP-13RXKR, and an MT1-MMP deletion mutant lacking the hemopexin-like domain (MT1-MMPΔHLD) were the generous gifts from Dr. Stephen Weiss. The presence of active MMP-1 and MMP13 in the media of the transfected cells was determined using specific peptide substrates.
Transient Transfections and DDR1 Stimulation
COS1 cells were split the day before transfection to 60–70% confluence in 60-mm dishes. The next day, equal amounts of the appropriate expression vectors were co-transfected using FuGENE 6 (Roche Applied Science) according to the manufacturer's instructions. For collagen I-induced DDR1 activation, COS1-transfected cells (4 h after transfection), T47D, or HCC1806 cells were preincubated (18 h, 37 °C) in serum-free media. Then, rat tail collagen type I (COS1, T47D, and HCC1806) or mouse collagen IV (COS1) was added to the serum-free media at the final concentration of 10 μg/ml for 2 h, and an equal volume of 20 mm acetic acid (collagen I) or 50 mm HCl (collagen IV) was added as vehicle control, as described previously (9). In the case of collagenase-resistant (r/r) mouse collagen I and GVMGFO triple-helical peptide, the DDR1-transfected COS1 cells were incubated (various times) with increasing concentrations of these ligands to identify optimal stimulation conditions (supplemental Figs. 4–6). After stimulation, the cells were washed twice with cold PBS, and the cells were lysed in RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS) supplemented with protease inhibitors (Roche Applied Science, complete, Mini, EDTA-free) and 10 mm NaF and 1 mm sodium orthovanadate. The cell lysates were cleared by centrifugation at 14,000 × g at 4 °C for 10 min; protein concentration was determined using the BCA kit (Pierce), and the lysates were frozen at −80 °C until used.
The relative level of MT1-MMP expressed in COS1 and HCC1806 cells was determined by immunoblot analyses using the Lem2/15 antibody and a recombinant catalytic domain of MT1-MMP (Millipore, catalog no. 475935) as a reference. The bands in the blot were quantified using UN-SCAN-IT gel 6.1(Silk Scientific, Orem, UT), and a standard curve was plotted using SigmaPlot 12.0.
Detection of DDR1 Phosphorylation
To detect DDR1 phosphorylation, the lysates of stimulated and unstimulated COS1 cells transfected with DDR1 cDNA were divided in two fractions, and equal amounts of protein (usually 40 μg per lane) from each treatment were resolved by reducing SDS-PAGE followed by immunoblot analyses into two duplicate blots. One blot was probed with anti-Tyr(P) mAb (clone 4G10®), as reported (9), and the other, depending on the experiment, with either anti-DDR1, anti-Myc, or anti-FLAG antibodies. The latter blot was also reprobed with the appropriate antibodies against various MMPs and/or GAPDH or β-actin, in some cases without stripping of the DDR1 antibody.
In the case of T47D and HCC1806 cells expressing endogenous DDR1, receptor phosphorylation was evaluated by immunoprecipitation with a DDR1 antibody (sc-532) to enrich the pool of DDR1 followed by immunoblot analyses with anti-Tyr(P) mAb. Briefly, serum-starved cells were treated with or without collagen I, as described earlier, and lysed with RIPA buffer. The lysates (∼400 μg) were incubated (1 h on ice) with 0.3–0.5 μg/ml of DDR1 antibody (sc-532), and the mixtures were incubated (overnight at 4 °C) with protein A-agarose beads (Pierce). The beads were then washed four times with RIPA buffer, and the captured immune complexes were released by boiling the samples in 1× reducing Laemmli SDS-sample buffer. After a brief centrifugation, the supernatants were resolved by reducing SDS-PAGE, followed by immunoblot analyses using anti-Tyr(P) mAb. Total (pulled down) DDR1 was determined by reprobing the blots with DDR1 antibody (sc-532).
Detection of Released DDR1 Ectodomain in the Media
Transfected COS1 or HCC1806 cells were washed twice with warm PBS, and the cell monolayers were incubated (37 °C) in serum-free media for an additional 24-h period. The conditioned media were then collected and clarified by a brief centrifugation at 4 °C to remove cell debris. The supernatants were supplemented with 2 mm EDTA, final concentration, before a high speed centrifugation (100,000 × g, 45 min, 4 °C) to remove cell membranes and vesicles. Two to four hundred μl of each media were then precipitated with trichloroacetic acid (TCA), and the resultant pellets were washed with acetone and then resuspended in 1× reducing Laemmli SDS-sample buffer. The samples were then resolved by reducing SDS-PAGE and subjected to immunoblot analyses using antibody AF2396, which is directed against the N-terminal region of DDR1.
Effects of Inhibitors on DDR1 Shedding and Activation
Subconfluent cultures of HCC1806 cells seeded in 6-well plates were washed twice with warm PBS and then incubated (22 h, 37 °C) in serum-free medium supplemented with either 100 nm TIMP-1 or TIMP-2 or 10 μm MIK-G2 diluted in PBS (TIMPs) or DMSO (MIK-G2, 0.1% final concentration). As a control, some wells received serum-free media supplemented with PBS or 0.1% DMSO. The media and cell lysates were then analyzed for DDR1 forms by immunoblot analyses, as described earlier. To evaluate the effects of the inhibitors on DDR1 phosphorylation, the cells were incubated with the inhibitors as described above, and 20-h later the cells were treated (2 h) with or without 20 μg/ml of rat tail collagen I. The cells were then lysed with RIPA buffer (200 μl per well), and equal amounts of protein lysates were immunoprecipitated with DDR1 antibody sc-532 followed by immunoblot analyses with anti-Tyr(P) mAb, as described above.
N-terminal Sequencing
To determine the N-terminal sequence of the C-terminal fragment of DDR1, Myc-tagged DDR1a was transiently expressed in COS1 cells with or without MT1-MMP, as described earlier. The cell lysates in RIPA buffer were then immunoprecipitated using Myc mAbs coupled to protein G-agarose beads (Pierce). After an overnight incubation (4 °C), the beads were washed thoroughly with RIPA buffer, and the bound proteins were eluted by adding 1× reducing Laemmli SDS-sample buffer, boiled for 5 min, and resolved by 7% SDS-PAGE followed by transfer to a PVDF membrane. After staining with Coomassie Blue, the putative bands were cut out of the membrane and sent to the Protein Chemistry Laboratory (Department of Biochemistry, Texas A&M University, College Station, TX) for Edman protein N-terminal sequencing.
Peptide Synthesis and Peptide Hydrolysis Assay
Peptides derived from the EJXM region of DDR1 (Pro-Thr-Asn-Phe-Ser-Ser-Leu-Glu-Leu-Glu-Pro-Arg-Gly-Gln-Gln-Pro-Val-Aln-Lys-Aln-Glu-Gly-NH2) or DDR2 (Ser-Glu-Aln-Leu-Pro-Thr-Ser-Pro-Met-Aln-Pro-Thr-Thr-Tyr-Asp-Pro-Met-Leu-Lys-Val-Asp-Asp-NH2) were synthesized by Fmoc solid-phase methodology using the Liberty Microwave Peptide Synthesizer (CEM Corp., Mathews, NC). The coupling of Fmoc-amino acids was performed with 5 eq of each amino acid, 4.9 eq of 2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate/N-hydroxybenzotriazole, and 10 eq of N-methylmorphine. The reaction was performed for 5 min at 50 °C and 25 watts with the exception of DDR1-EJXM Gln15, Pro16, Val17, Ala18, Lys19, and Ala20 and DDR2-EJXM Glu2, Met9, Ala10, Pro16, Met17, Leu18, Lys19, and Val20, where coupling was extended to 20 min. DDR1-EJXM residues Arg12, Pro16, Val17, and Ala18 and DDR2-EJXM residues Pro16, Met17, and Leu18 were double-coupled. The Fmoc group was removed with 10% piperazine in N,N-dimethylformamide. The initial deprotection was carried out for 30 s followed by second deprotection for 180 s. The maximum of 75 °C with microwave power of 35 watts was set for both deprotection steps. DDR1-EJXM peptide was cleaved from the resin and deprotected by treatment with TFA/H2O/thioanisole (92.5:5:2.5) for 3 h in an argon atmosphere. DDR2-EJXM peptide was cleaved from the resin and deprotected using TFA/H2O/1,2-ethanedithiol/thioanisole (90:5:2.5:2.5) for 3 h in an argon atmosphere. Crude peptides were precipitated with cold methyl tert-butyl ether and centrifuged, and the resulting pellets were dissolved in water and lyophilized. Peptides were purified using reverse phase-HPLC (Agilent 1260 Infiniti with multiwavelength detector) on a Vydac C18 column (15–20 μm, 300 Å, 250 × 22 mm) at a flow rate of 15 ml/min. The elution gradient was 15–40% B over 50 min (where A was 0.1% TFA in water and B was 0.1% TFA in acetonitrile), and detection was at λ = 220 nm. HPLC fractions were analyzed by MALDI-TOF MS (Voyager DEPro), and the desired fractions were combined and freeze-dried.
Pro-MT1-MMP was activated by treatment with trypsin-3 at 37 °C for 1 h followed by deactivation of trypsin with 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (20 mm) for 30 min. Pro-MMP-1 was activated with p-aminophenylmercuric acetate (2 mm) at 37 °C for 3 h. Peptides were dissolved in Tris buffer (50 mm Tris, 100 mm NaCl, 10 mm CaCl2, 0.05% Brij-35, 0.02% NaN3) to a final concentration of 50 μm and incubated with the individual MMPs (final enzyme concentration of 5 nm). Hydrolysis progress was monitored at various time points for 18 h by RP-HPLC (Agilent 1260 Infinity with DAD detector) with detection at λ = 220 nm on a Vydac C18 column (5 μm, 300 Å, 150 × 4.6 mm). The analytical gradient was 2–98% B over 20 min (where A and B are as described above) and a flow rate of 1 ml/min. All species separated by HPLC were collected and identified using MALDI-TOF MS.
Computational Modeling
Two peptides derived from the regions surrounding the two cleavage sites at the DDR1 EJXM region by MT1-MMP, Phe-Ser-Ser-Leu-Glu-Leu and Gln-Gln-Pro-Val-Ala-Lys (underlined residues flank the cleavage sites), were built and energy-minimized using SYBYL-X 1.3 (Tripos International, 2011, St. Louis, MO). The two peptides have 30 and 27, respectively, rotatable bonds. To sample the many conformational states of these peptides, the conformational sampling was enhanced by generating a conformer library for each using the ConfGen program in Schrödinger Suite 2012. Conformations that were within a heavy atom root mean square deviation of 1.0 Å and those that were higher in energy by more than 100 kJ/mol from the starting position were discarded. The final library contained 20 distinct conformations for Phe-Ser-Ser- Leu-Glu-Leu and 49 for Gln-Gln-Pro- Val-Ala-Lys. Docking of the two peptides was carried out using Glide 5.8 (Schrödinger LLC, 2012, New York). The MT1-MMP x-ray crystal structure (Protein Data Bank code 3MA2) coordinates were used as receptor. The structure was processed using the Protein Preparation Wizard protocol in Schrödinger Suite 2012. A constrained energy minimization using the OPLS2005 forcefield was carried out with a 0.30-Å root mean square deviation convergence threshold. The docking grid was calculated for a cubic box of 30 Å at the catalytic site. The pre-generated peptide conformers were docked to the catalytic site and scored using the GlideXP methodology. A total of 10 top-scored poses for each of the docked conformers were retained for analysis. Finally, a docking pose was selected based on the high docking score, the direction of the peptide, and the positioning of the groups at the cleavage site (28, 29).
RESULTS
MT1-MMP Cleaves DDR1 and Inhibits Receptor Activation
To examine the effects of MT1-MMP on DDR1 (DDR1a and DDR1b) collagen-dependent activation, COS1 cells were transiently transfected to express human DDR1 with or without active or catalytically inactive MT1-MMP. The cells were then exposed to collagen I and analyzed for DDR1 phosphorylation using an anti-Tyr(P) antibody. It should be noted that, under the experimental conditions (expression of recombinant receptor), detection of phosphorylated DDR1 could be accomplished directly in cell lysates using anti-Tyr(P) antibody, without the need of an immunoprecipitation step, in agreement with previous studies (9). Under these conditions, unstimulated COS1 cells exhibited background recombinant DDR1 phosphorylation, which was significantly enhanced by exposure to collagen. To examine the effects of MT1-MMP on endogenous DDR1, we utilized human breast cancer T47D cells, which express high levels of DDR1 (30). T47D cells, however, do not express detectable levels of MT1-MMP (20) and thus can be used to test the effects of ectopically expressed MT1-MMP on endogenous DDR1 activation and cleavage. To this end, the cells were stimulated with collagen I and lysed, and the lysates were immunoprecipitated with the sc-532 antibody followed by detection of phosphorylated receptor with the anti-Tyr(P) antibody.
As shown in Fig. 1A, collagen I treatment of COS1 cells significantly induced the phosphorylation of an ∼120-kDa protein in cells expressing DDR1a (Fig. 1A, lane 2) or DDR1b (data not shown). This species was not detected in the absence of collagen I (Fig. 1A, lane 1) or in mock-transfected COS1 cells (data not shown), indicating that the ∼120-kDa band corresponds to phosphorylated DDR1. Co-expression of DDR1a (Fig. 1A) or DDR1b (data not shown) with wild type MT1-MMP completely abrogated collagen I-induced receptor phosphorylation, as determined by the absence of the ∼120-kDa phosphorylated form (Fig. 1A, lane 4). In contrast, co-expression of MT1-E/A with DDR1 did not inhibit receptor activation (Fig. 1A, lane 6), suggesting that protease activity was required for the lack of DDR1 phosphorylation observed in the presence of MT1-MMP, and it could not be attributed to disruption of receptor conformation and/or trafficking. Examination of the profile of phosphorylated proteins revealed that the anti-Tyr(P) antibody also detected two phosphorylated proteins of ∼62–65 and ∼35 kDa (Fig. 1A). Although the 35-kDa protein appeared to be nonspecific, we surmised that the ∼65-kDa protein represents a phosphorylated fragment of DDR1. This ∼62–65-kDa phosphoprotein was detected independently of collagen stimulation, but its levels increased upon expression of MT1-MMP (Fig. 1A, lanes 3 and 4). Thus, detection of the phosphorylated ∼65-kDa protein suggests that a small fraction of the DDR1 receptor pool undergoes MT1-MMP- and collagen I-independent cleavage and phosphorylation, respectively, with the latter possibly due to recombinant receptor expression in COS1 cells.
FIGURE 1.
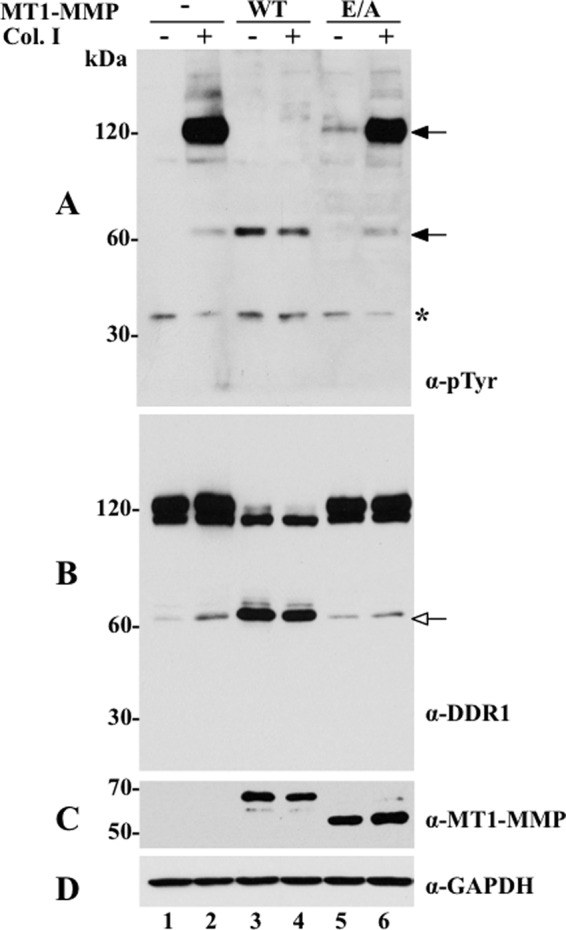
MT1-MMP cleaves DDR1 and inhibits collagen I-induced receptor activation. COS1 cells were transiently transfected to co-express DDR1a without (−) or with (+) wild type (WT) or catalytically inactive (E/A) MT1-MMP and serum-starved (18 h) before stimulation (2 h) with (+) 10 μg/ml of rat tail collagen I (Col. I) or vehicle control (−), as described under “Experimental Procedures.” After stimulation, the cells were lysed in RIPA buffer, and the lysates from each experimental condition were divided in two fractions. Equal amounts of the two fractions were then resolved by reducing 8% SDS-PAGE in two identical separate gels followed by immunoblot analyses. One blot was probed with Tyr(P) (α-pTyr) (4G10®) antibody (A) and the other with DDR1 antibody (sc-532) (B). The blot in B was then reprobed with antibodies to MT1-MMP (Lem2/15) (C) or GAPDH, as loading control (D). Black arrows in A indicate phosphorylated DDR1 forms, and white arrow in B indicates the CTF of DDR1. Asterisk indicates a nonspecific band.
To establish the nature of the DDR1 species detected in the absence or presence of MT1-MMP and/or with or without collagen I stimulation, the cell lysates were analyzed with antibody sc-532, directed against the C-terminal end of DDR1. Cells expressing only DDR1a displayed two major forms of ∼110 and ∼120 kDa and a minor species of ∼62-kDa (Fig. 1B, lanes 1 and 2). In the presence of MT1-MMP, the lysates displayed the ∼110-kDa form of DDR1a, although the ∼120-kDa species was no longer detected (Fig. 1B, lanes 3 and 4). In contrast, the levels of the ∼62-kDa form of DDR1 significantly increased, and this increment occurred regardless of collagen I treatment (Fig. 1B, lanes 3 and 4). Co-expression of DDR1a with MT1-E/A significantly reduced the levels of the ∼62-kDa species to its constitutive levels (Fig. 1B, lanes 5 and 6). The blot of Fig. 1C shows the presence of MT1-MMP forms in this experiment. The MT1/E/A protein (Fig. 1C, lanes 5 and 6) displays a somewhat faster electrophoretic mobility (and reduced molecular mass), a known characteristics of this protease mutant, which here is emphasized by the running conditions (reducing 8% SDS-PAGE).
Because the ∼62-kDa form was recognized with the antibody to the C terminus of DDR1, we surmised that this species represents a membrane-anchored C-terminal fragment (CTF) of DDR1a. Indeed, analyses of lysates of COS1 cells expressing a DDR1 tagged with a FLAG tag at the N terminus and a Myc tag at the C terminus (FLAG/DDR1a/Myc) with or without MT1-MMP revealed that the 62-kDa species was recognized by the anti-Myc (supplemental Fig. 1A) but not by the anti-FLAG M2 (supplemental Fig. 1B) antibody. The nature of the ∼110-kDa species of DDR1, detected after co-expression with MT1-MMP (Fig. 1B, lanes 3 and 4), is unclear. However, this species may represent a differentially glycosylated precursor form of DDR1 that is not targeted to the cell surface, and thus it remains unchanged in the presence of MT1-MMP.
To rule out a possible effect of MT1-MMP overexpression on DDR1 cleavage and activation, we transfected COS1 cells with a fixed amount of DDR1 cDNA and increasing amounts of the MT1-MMP-containing expression plasmid. We found that DDR1 cleavage and inhibition of collagen I-stimulated activation also occurred at very low levels of MT1-MMP expression (supplemental Fig. 2). In addition, quantification of the relative levels of recombinant MT1-MMP expressed in COS1 cells, under the conditions used here to detect DDR1 shedding, using a recombinant catalytic domain of MT1-MMP as a standard, revealed comparable levels of protease in COS1 cells and HCC1806 cells (data not shown), which express natural MT1-MMP (supplemental Fig. 10) and shed DDR1 (shown in Fig. 8). Thus, these data indicate that the effects of MT1-MMP on DDR1 shedding in the COS1 cell system cannot be attributed to protease overexpression.
FIGURE 8.
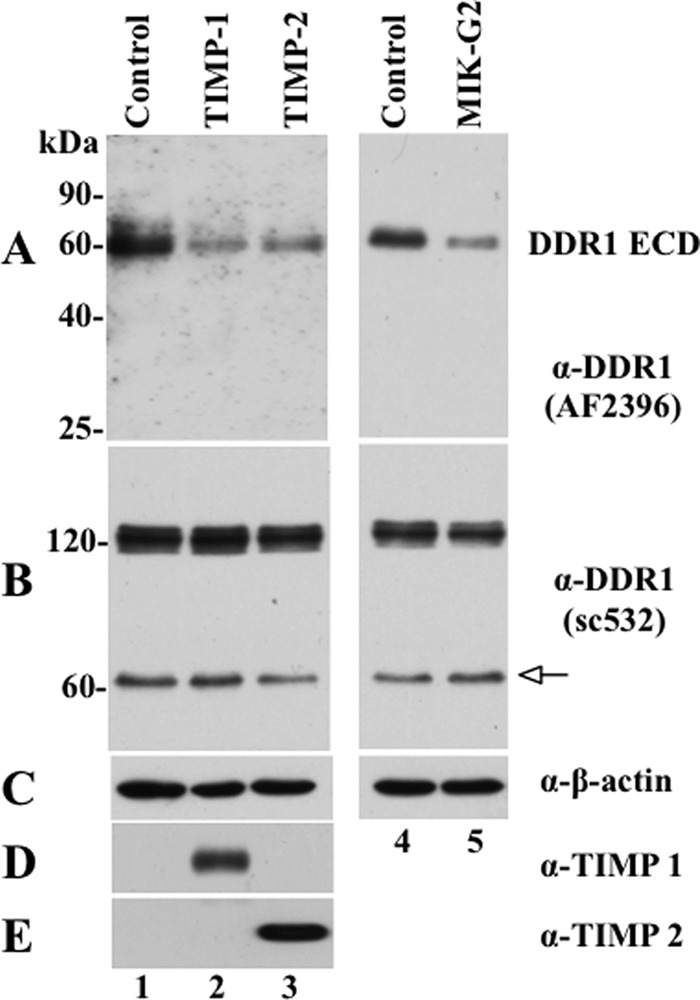
TIMPs and MIK-G2 inhibit DDR1 shedding in HCC1806 cells. HCC1806 cells seeded in 6-well plates were treated overnight without (Control) or with 100 nm of purified TIMP-1 or TIMP-2 or 10 μm MIK-G2. The serum-free media (400 μl/well) were then collected and TCA-precipitated, and the resultant pellets were resolved by reducing 12% SDS-polyacrylamide gel followed by immunoblot analyses. The blot was probed with N-terminal DDR1 antibody, AF2396 (A) and then reprobed with antibodies to TIMP-1 (D) and TIMP-2 (E). For total protein analysis, 30 μg of protein from each lysate were resolved by reducing 7.5% SDS-polyacrylamide gel followed by immunoblot analyses. The blot was probed with anti-DDR1 antibody (sc-532) (B), and then reprobed with antibodies to anti-β-actin antibody as loading control (C). White arrow indicates the CTF of DDR1.
We also examined whether the effects of MT1-MMP on DDR1 phosphorylation and cleavage were also observed upon stimulation with basement membrane collagen IV, another DDR1 ligand. These studies showed that although collagen IV induces a much weaker phosphorylation of the receptor in transfected COS1 cells, when compared with collagen I, the presence of MT1-MMP also results in DDR1a/b cleavage and inhibition of phosphorylation (data not shown).
Next, we investigated DDR1 activation and cleavage in T47D cells. As shown in supplemental Fig. 3A, collagen I induced the phosphorylation of DDR1 in control T47D cells for up to 18 h (supplemental Fig. 3A, lanes 3, 5, 7, and 9). However, expression of MT1-MMP completely blocked collagen I-induced DDR1 activation, as indicated by the lack of phosphorylated receptor (∼120 kDa; supplemental Fig. 3A, lanes 4, 6, 8, and 10). The total precipitated fraction demonstrated similar amounts of precipitated DDR1 from each lysates (supplemental Fig. 3B). Immunoblot analyses of the total cell lysates with the sc-532 antibody (supplemental Fig. 3C) revealed the presence of the ∼62-kDa CTF of DDR1 only in T47D cells expressing MT1-MMP (supplemental Fig. 3D, lanes 2, 4, 6, 8, and 10) but not in the control T47D cells (supplemental Fig. 3D , lanes 1, 3, 5, 7, and 9), similar to the results obtained in the COS1 cell system. Collectively, these data show that MT1-MMP efficiently cleaves DDR1 independently of collagen (I and IV) stimulation and negatively regulates its activation in cells expressing recombinant or natural DDR1.
MT1-MMP Enhances DDR1 Ectodomain Shedding
We next examined the conditioned media of COS1 cells for the presence of a soluble DDR1 fragment. As shown in Fig. 2A, serum-free conditioned media derived from COS1 cells co-expressing DDR1a without or with wild type or inactive mutant MT1-MMP, contained a major ∼62-kDa protein in all conditions, albeit at different levels, that was recognized by an antibody directed to the ectodomain of DDR1 (AF2396). In the presence of wild type MT1-MMP, the levels of the soluble DDR1a fragment significantly increased (Fig. 2A, lane 2), when compared with the levels detected in the media of cells expressing DDR1 alone or DDR1 with MT1-E/A (Fig. 2A, lanes 1 and 3, respectively). Fig. 2, B and C, shows the lysate of the same experiment, which demonstrates the cleavage of DDR1a only by wild type MT1-MMP (Fig. 2B, lane 2), and the corresponding expression of wild type and E/A MT1-MMP (Fig. 2C, lanes 2 and 3, respectively). These results demonstrate that in COS1 cells constitutive shedding of the DDR1a ectodomain is significantly enhanced by MT1-MMP, in a process that requires its catalytic activity.
FIGURE 2.
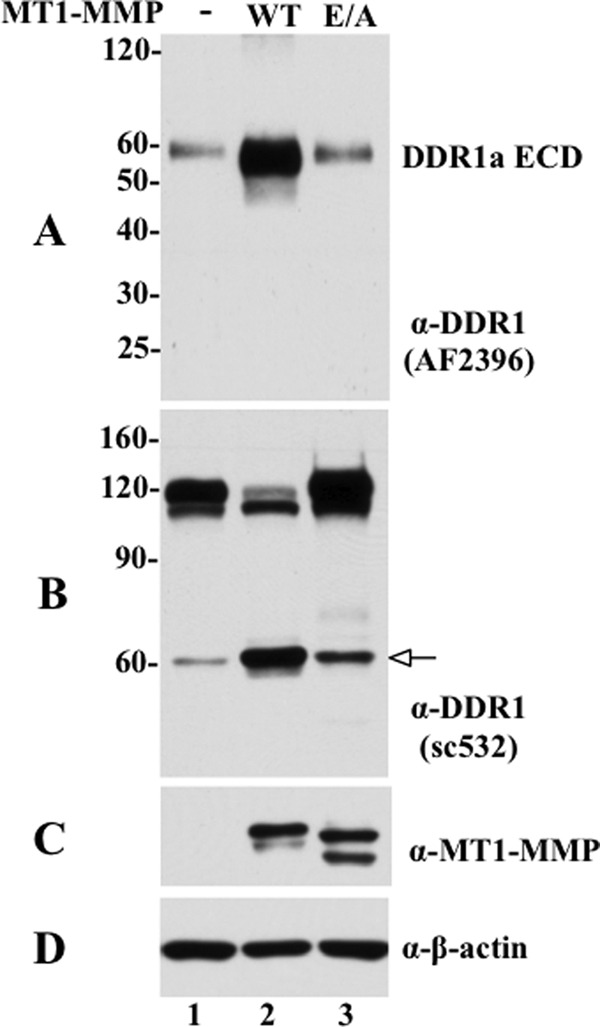
MT1-MMP enhances DDR1 ectodomain shedding. COS1 cells were transfected to co-express DDR1a without (−) or with wild type (WT) or catalytically inactive (E/A) MT1-MMP, and the conditioned media were collected, as described under “Experimental Procedures.” Equal volumes of the conditioned media were TCA-precipitated and resolved by reducing 12% SDS-PAGE followed by immunoblot analyses using a DDR1 antibody against the N-terminal ectodomain from R&D Systems (AF2396). A single immunoreactive fragments of ∼60 kDa was detected, indicated as soluble DDR1 (A). B and C, cells from the same experiment were lysed in RIPA buffer, and equal amounts of protein from each sample was resolved by reducing 7.5% SDS-PAGE followed by immunoblot analyses. The blot was probed with DDR1 antibody (sc-532) (B). The blot in B was then reprobed with antibodies to MT1-MMP (Lem2/15) (C) or β-actin, as loading control (D). White arrow in B indicates the CTF of DDR1.
Involvement of MT1-MMP Domains in DDR1 Cleavage
We investigated the structural features of MT1-MMP involved in DDR1 cleavage. To this end, various MT1-MMP deletion mutants, MT1-MMPΔCT, MT1-MMPΔHLD, and MT1-MMPΔTM/CT, were co-expressed with DDR1a in COS1 cells, and the cells were then examined for collagen I-induced DDR1 activation and receptor cleavage. Fig. 3 shows that DDR1a expressed alone was activated in response to collagen I (Fig. 3, A, lane 2, and E, lane 10). However, DDR1 activation was inhibited when the receptor was co-expressed with either wild type MT1-MMP (Fig. 3A, lane 4), MT1-MMPΔCT (Fig. 3A, lane 6), or MT1-MMPΔHLD (Fig. 3A, lane 8). In contrast, MT1-MMPΔTM/CT, which is not retained at the plasma membrane and is secreted, had no effect on receptor phosphorylation (Fig. 3E, lane 12). Analyses of the cell lysates revealed that wild type MT1-MMP, MT1-MMPΔCT, and MT1-MMPΔHLD all cleaved DDR1a generating the ∼62-kDa CTF, regardless of the presence of collagen I (Fig. 3B, lanes 3–8). In contrast, MT1-MMPΔTM/CT failed to cleave DDR1a (Fig. 3F, lanes 11 and 12), consistent with the lack of effect of this deletion mutant on receptor phosphorylation (Fig. 3E, lane 12). No cleavage products of DDR1a were detected in the lysates of cells expressing receptor alone (Fig. 3B, lanes 1 and 2, and E, lanes 9 and 10), as expected. All protease mutants were detected in the cell lysates (Fig. 3, D and H). Taken together, these results suggest that although neither the cytosolic tail nor the hemopexin-like domain of MT1-MMP is required for MT1-MMP-mediated DDR1 cleavage, this process requires a membrane-anchored catalytic domain.
FIGURE 3.

Structural requirements for MT1-MMP-mediated cleavage of DDR1. COS1 cells were transiently transfected to express DDR1a without (−) or with (+) wild type MT1-MMP (WT), MT1-MMPΔCT (ΔCT), MT1-MMPΔHLD (ΔHLD) (A–D), or MT1-MMPΔTM/CT (ΔTM/CT) (E–H). The cells were then serum-starved (18 h) before stimulation (2 h) with (+) 10 μg/ml of rat tail collagen I (Col. I) or vehicle control (−). After stimulation, the cells were lysed, and the lysates were examined for DDR1 phosphorylation (A and E) and cleavage (B and F), as described in the legend of Fig. 1. The blot in B was then reprobed with antibodies to MT1-MMP (Lem2/15) (D) or GAPDH, as loading control (C). The blot in F was reprobed with anti-GAPDH antibodies (G). The blot in H shows serum-free conditioned media collected from COS1 cells expressing secreted MT1-MMPΔTM/CT (lanes 11 and 12) or vector control (lanes 9 and 10). Black arrows in A and E indicate phosphorylated DDR1 and white arrow in B indicates the CTF of DDR1. α-pTyr, Tyr(P).
Differential Effects of MMPs on DDR1 Activation and Cleavage
We next examined the effects of MT2- and MT3-MMP, the close homologues of MT1-MMP, and the secreted collagenases MMP-1 and MMP-13 on DDR1 activation and cleavage. As shown in Fig. 4, MT2- and MT3-MMP, like MT1-MMP, inhibited collagen I-induced DDR1 activation and accomplished receptor cleavage, as determined by the absence of phosphorylated DDR1 (∼120 kDa) (Fig. 4A, lanes 6 and 8) and the appearance of the corresponding ∼65-kDa CTF (Fig. 4B, lanes 5–8), respectively. In contrast, co-expression of DDR1a (Fig. 4E) or DDR1b (data not shown) with either MMP-1RXKR or MMP-13RXKR had no effect on collagen I-induced receptor activation (Fig. 4E, lanes 12 and 14), when compared with cells expressing only DDR1 (Fig. 4E, lane 10). Moreover, neither MMP-1RXKR nor MMP-13RXKR generated a detectable CTF of DDR1 (Fig. 4F). Measurement of enzymatic activity in the media of COS1 cells expressing MMP-1RXKR or MMP-13RXKR using synthetic peptide substrates for these proteases demonstrated the presence of catalytic activity, indicating that these collagenases were activated (data not shown), as expected (18, 27). Thus, the inability of the secreted collagenases to cleave DDR1 could not be ascribed to a lack of activated proteases.
FIGURE 4.

Effects of membrane-anchored and secreted collagenases on DDR1 phosphorylation and cleavage. COS1 cells were transiently transfected to co-express DDR1a without (−) or with (+) MT1-MMP, MT2-MMP, MT3-MMP, MMP-1RXKR, or MMP-1RXKR. Serum-starved cells were then stimulated (2 h) with (+) 10 μg/ml rat tail collagen I (Col. I) or vehicle control (−). After stimulation, the cells were lysed, and the lysates were examined for DDR1 phosphorylation (A and E) and cleavage (B and F), as described in the legend of Fig. 1. The blots in B and F were then reprobed with antibodies to the respective MMPs (D and H) or GAPDH, as loading control (C and G). Black arrows in A and E indicate phosphorylated DDR1 and white arrow in B indicates the CTF of DDR1. α-pTyr, Tyr(P).
We also evaluated the ability of the glycosylphosphatidylinositol-anchored MT4- and MT6-MMP to cleave DDR1 and/or to affect collagen I-stimulated receptor activation in COS1 cells co-transfected to express DDR1 with or without these MT-MMPs. These studies showed that neither MT4- nor MT6-MMP was able to cleave DDR1 or to alter receptor activation (data not shown). Taken together, these results demonstrate that, among the members of the MMP family tested here, the transmembrane MT-MMPs specifically promote the shedding of DDR1 isoforms (a and b) and regulate collagen-evoked receptor activation.
Role of Collagenolysis on DDR1 Regulation by MT1-MMP
Proteolytic degradation or denaturation of collagen I has been reported to diminish DDR1 activation (4). Therefore, because MT1-MMP is a collagenase (17, 18), we evaluated the contribution of collagenolysis, over receptor proteolysis, on the inhibition of DDR1 activation in the presence of MT1-MMP using a collagenase-resistant collagen I and a synthetic triple-helical collagen peptide spanning the DDR1-binding site. The collagenase-resistant collagen I was isolated from homozygous r/r mice (31), which harbor a Pro substitution at both Gln774 and Ala777 in the α1(I) chain. Thus, the resulting collagen I heterotrimer is less susceptible to cleavage by collagenases (31), including MT1-MMP (18). The synthetic triple-helical collagen I peptide harbors the sequence GVMGFO, which has been shown to be present in collagens I, II, and III, and to constitute a major binding site for DDRs on these fibrillar collagens (1, 11). However, this peptide is not a substrate of MT1-MMP collagenase activity.3 Preliminary time course and dose-dependent experiments showed that mouse r/r collagen I stimulated DDR1 phosphorylation albeit less efficiently than mouse wild type collagen I (supplemental Figs. 4 and 5). Moreover, although wild type collagen I elicited a sustained DDR1 phosphorylation over an 18-h time period (supplemental Fig. 5A), the r/r collagen I-evoked DDR1 phosphorylation was short lived, and after 4 h it was significantly diminished (supplemental Fig. 5C). The triple-helical GVMGFO peptide also induced DDR1 phosphorylation. However, relatively high concentrations of peptide were required (supplemental Fig. 6) (11). The reason(s) for the relatively weaker phosphorylation of DDR1 in response to these ligands when compared with wild type collagen I is unknown and remains to be further explored. Based on these results, we used the optimal conditions to test the effects of MT1-MMP on DDR1 activation in response to r/r collagen I and the GVMGFO-containing triple-helical peptide. These analyses showed that the collagenase-resistant ligands did not rescue DDR1 phosphorylation in the presence of MT1-MMP (Fig. 5, A and C). Moreover, analyses of the cell lysates revealed the presence of the CTF of DDR1, when the receptor was co-expressed with MT1-MMP (Fig. 5, B and D), regardless of the ligand type. These results suggest that, under the experimental conditions, receptor cleavage and not ligand degradation is the main cause for the reduction in DDR1 collagen I-induced phosphorylation in the presence of MT1-MMP.
FIGURE 5.

MT1-MMP dampens DDR1a phosphorylation in response to r/r collagen I and triple-helical GVMGFO peptide. COS1 cells were transiently transfected to co-express DDR1a without (−) or with (+) wild type (WT) or catalytically inactive (E/A) MT1-MMP, and serum-starved (18 h) before stimulation (2 h) with (+) either 20 μg/ml wild type (WT) or r/r (r/r) mouse tail type I collagen (A and C) or with 200 μg/ml triple-helical GVMGFO peptide. After stimulation, the cells were lysed, and the lysates were examined for DDR1 phosphorylation (A and B) and cleavage (C and D), as described in the legend of Fig. 1. The blots in C and D were also probed with anti-GAPDH antibodies, without stripping. Black arrows in A and C indicate phosphorylated DDR1 and white arrow in B and D indicate the CTF of DDR1. α-pTyr, Tyr(P).
DDR1 Is Cleaved at the Extracellular Juxtamembrane Region
To identify the cleavage site(s) of DDR1 catalyzed by MT1-MMP, Myc-tagged DDR1a CTF was purified from lysates of COS1 cells co-expressing DDR1a with MT1-MMP, as described under “Experimental Procedures.” The purified proteins were resolved by SDS-PAGE and transferred to a PVDF membrane. Two bands within the range of ∼62–65 kDa were excised from the membrane and subjected to N-terminal sequencing by Edman degradation (supplemental Fig. 7A). These analyses were consistent with the upper protein displaying the N-terminal sequence Leu398-Gln-Leu-Gln-Pro-Arg, whereas the lower protein, Val408-Ala-Lys-Ala-Glu-Gly-Ser-Pro, which corresponds to two DDR1a fragments, one starting at Leu398 and the other at Val408, respectively. These results are consistent with the presence of two cleavage sites at the Ser397–Leu398 and Pro407–Val408 peptide bonds located within the EJXM region of DDR1 (depicted in the schematic of Fig. 6A and in supplemental Fig. 7B).
FIGURE 6.

A, alignment of the EJXM region of DDR1 and DDR2. B, close-up stereo view of the docked peptide FSSLEL (as capped sticks; nitrogen in blue, oxygen in red, carbon in gray; hydrogens not shown) into the active site of MT1-MMP. The protein is depicted as a translucent green Connolly surface. The Zn2+ ion is shown as a gray sphere, and the histidines coordinated to it are shown as capped sticks. C, close-up stereo view of the docked peptide QQPVAK shown in the same representation as in B.
The ability of MT1-MMP to cleave within the EJXM region was further investigated using a synthetic peptide from the EJXM region of DDR1 (Pro392-Thr-Asn-Phe-Ser-Ser-Leu-Glu-Leu-Glu-Pro-Arg-Gly-Gln-Gln-Pro-Val-Aln-Lys-Aln-Glu-Gly-NH2) comprising the two cleavage sites identified by Edman-based sequencing (underlined) (see supplemental Fig. 7). The peptide was incubated with activated MT1-MMP, and the cleaved products were identified by MALDI-TOF MS. These analyses showed that MT1-MMP cleaved the DDR1 peptide at the Ser397–Leu398 peptide bond, as observed with the full-length receptor. The reason for the lack of cleavage at the Pro407–Val408 site in the DDR1 peptide by MT1-MMP is unclear. However, it is possible that access to this site is conformationally dependent. In fact, as described later, a DDR1aΔ377–399 deletion mutant, which lacks the Ser397–Leu398 peptide bond but preserves the Pro407–Val408 site, generates a CTF only in the presence of MT1-MMP (Fig. 7D, lanes 7 and 8). In addition, we found that treatment of the DDR1-EJXM peptide with active MMP-1 resulted in no peptide hydrolysis, in agreement with our cell data (Fig. 4F).
FIGURE 7.

Effects of deletions at the EJXM region of DDR1 on receptor activation and cleavage. A, schematic depicting the structural domain organization of DDR1 and the sequence of the deletions generated at the EJXM region of DDR1. B–G, COS1 cells were transiently transfected to express wild type or EJXM deletion mutants without (−) or with (+) wild type MT1-MMP (B–E) or with wild type (WT) or catalytically inactive (E/A) MT1-MMP (F and G). Serum-starved cells were then stimulated (2 h) with (+) 10 μg/ml rat tail collagen I (Col. I) or vehicle control (−). After stimulation, the cells were lysed, and the lysates were examined for DDR1 cleavage (B, D, and F) and phosphorylation (C, E and G), with antibodies to DDR1 (sc-532) or Tyr(P) (α-pTyr) (4G10®). The blots in B, D, and F were also reprobed with anti-GAPDH antibodies without stripping. White arrows in B, D, and F indicate the CTF of DDR1, and black arrows in C, E, and G indicate phosphorylated DDR1.
Computational Analysis of the DDR1 Cleavage Sites
A recent x-ray structure of human DDR1 provides insights into the structure of the DS and DS-like domains (residues 30–367) for the first time (13). However, our experiments indicate that DDR1 undergoes proteolytic cleavage within the EJXM region, which is outside of the crystallized portion of the structure. An inspection of the EJXM sequence reveals the presence of many glycine and proline residues, which are hallmarks of flexible mobile structural elements in proteins. Such flexible structural elements are usually prone to proteolysis, as is documented in this study by the existence of MT1-MMP cleavage sites within this region. Notwithstanding this observation, our attempts to identify any known three-dimensional structure for similar sequences from other proteins did not bear fruit. Therefore, to understand how the recognition by MT1-MMP takes place within this region, it was necessary to sample all significant conformational states for the peptides in the two cleavage sites, Ser397–Leu398 and Pro407–Val408. We used the program ConfGen to allow the peptides comprising the cleavage sites, Phe-Ser-Ser-Leu-Glu-Leu and Gln-Gln-Pro-Val-Ala-Lys, to sample many permissible conformational states, which our selection criteria narrowed down to 20 and 49 energetically distinct and significant conformers for the respective peptides. These conformations were docked into the active site of MT1-MMP. Suitable poses that could account for the requisite hydrolytic reaction subsequent to appropriate coordination of the amide carbonyl of the scissile bond with the zinc ion emerged from analysis (Fig. 6, B and C). Ser397 and Pro407 backbone carbonyls interact with the Zn2+ ion, and for both interactions the distance between Zn2+ and the oxygen of the carbonyl is 2.2 Å. Ser397 and Pro407 side chains in these peptides are ensconced in the S1 pocket, and the Leu398 and Val408 side chains are in the S1′ pocket. These pockets are documented peptide-like substrate recognition sites (32). Additionally, the Phe-Ser-Ser-Leu-Glu-Leu peptide forms five hydrogen bonds with the protein atoms; one is between the Lys396 side chain and the backbone, and the rest are among the backbone atoms. The Gln-Gln-Pro-Val-Ala-Lys peptide also forms five hydrogen bonds; four are among the backbone atoms, and one is between Lys410 Nζ and a backbone carbonyl of the protein. Thus, our analyses reveal that both of the sites identified can readily be accommodated within the active site of MT1-MMP, en route to the hydrolytic turnover.
The EJXM Region of DDR1 Is Critical for MT1-MMP-mediated Receptor Cleavage
Because inhibition of phosphorylation was a consequence of receptor cleavage, as suggested by the data with the collagenase-resistant ligands (Fig. 5), we hypothesized that an MT1-MMP-resistant DDR1 receptor will maintain collagen I-induced activation in the presence of MT1-MMP. To test this hypothesis, we generated DDR1a mutants harboring deletions at the EJXM linker. This approach was chosen because the sequence of the EJXM is considered a conformationally flexible one, and thus deletion of specific residues represents a more advantageous approach than would be generation of point mutations. Indeed, we found that double (S397T/L398H and P407V/V408H, each) or quadruple (S397T/L398H + P407V/V408H, each) substitutions at the two cleavage sites, chosen based on previous analyses of nonconcurring amino acids in natural MT1-MMP substrates (33), failed to prevent DDR1a cleavage by MT1-MMP (data not shown). Therefore, we generated three EJXM deletion mutants of DDR1a (Fig. 7A). DDR1Δ400–416 lacks the Pro407–Val408 site; DDR1aΔ377–399 lacks the Ser397–Leu398 site; and DDR1aΔ394–416 lacks both sites (Fig. 7A). As shown in Fig. 7B, lysates of cells expressing DDR1aΔ400–416, which lacks the Pro407–Val408 site, did not display generation of a CTF in the presence of MT1-MMP, suggesting that this mutant is MT1-MMP-resistant. However, in spite of no evidence of cleavage, DDR1aΔ400–416 was not phosphorylated upon stimulation with collagen I (Fig. 7C, lane 6), when compared with wild type DDR1a (Fig. 7C, lane 2). This result suggested that the structure and/or trafficking of the DDR1aΔ400–416 deletion mutant was compromised. Indeed, a surface biotinylation study confirmed that the DDR1aΔ400–416 mutant is not detected at the cell surface (supplemental Fig. 8A, lane 4). Thus, the importance of the Pro407–Val408 site for MT1-MMP cleavage could not be determined with this deletion mutant. Lysates of cells expressing DDR1aΔ377–399 contained a CTF (Fig. 7D, lanes 7 and 8) similar to that detected with wild type DDR1a (Fig. 7D, lanes 3 and 4). The CTF was detected only in the presence of MT1-MMP, suggesting cleavage at the Pro407–Val408 site by MT1-MMP. The DDR1aΔ377–399 mutant was also phosphorylated in response to collagen I (Fig. 7E, lane 6), albeit to a lower extent than the wild type receptor (Fig. 7E, lane 2), and, in agreement, it could be detected at the cell surface (supplemental Fig. 8A , lane 6). Moreover, the activation of the DDR1aΔ377–399 mutant was also inhibited in the presence of MT1-MMP (Fig. 7E, lane 8), consistent with receptor cleavage. Thus, under these conditions, MT1-MMP-dependent ectodomain cleavage of DDR1a can take place in the absence of the Ser397–Leu398 site, possibly through the hydrolysis of the Pro407–Val408 peptide bond.
Deletion of residues 394–416 in DDR1aΔ394–416, which removes both cleavage sites, significantly reduced MT1-MMP-dependent cleavage, as indicated by the lack of a detectable CTF (Fig. 7F, lanes 3 and 4). Because the control (wild type DDR1 with MT1-MMP) shown in the blot of Fig. 7F (lane 8) exhibits an unusual lower level of CTF, an additional similar experiment is shown in supplemental Fig. 9B, which clearly demonstrates that the DDR1aΔ394–416 mutant cannot be cleaved by MT1-MMP to generate a CTF, when compared with wild type DDR1 (supplemental Fig. 9B, lanes 4 and 8, respectively) under equal loading and exposure conditions.
Interestingly, stimulation with collagen I revealed that DDR1aΔ394–416, in contrast to wild type DDR1a, retained receptor phosphorylation in the presence of MT1-MMP (Fig. 7G, lanes 4 and 8, respectively, and supplemental Fig. 9A , lanes 4 and 8, respectively), consistent with its resistance to MT1-MMP-mediated cleavage. However, under similar conditions of collagen I stimulation, the extent of DDR1aΔ394–416 phosphorylation was consistently lower than that displayed by wild type DDR1a (Fig. 7G and supplemental Fig. 9A). Although we cannot yet explain the reason(s) for the relatively lower activation rate of the DDR1aΔ394–416 mutant when compared with wild type DDR1, co-expression of this mutant with MT1-E/A recovered receptor phosphorylation (Fig, 7G, lane 6, and supplemental Fig. 9A , lane 6) to the extent of receptor expressed alone (Fig. 7G, lane 2, and supplemental Fig. 9A, lane 7). This suggests that DDR1aΔ394–416 remains partially sensitive to MT1-MMP proteolysis. However, a detectable cleavage product could not be detected under these conditions. It is also possible that the reduced phosphorylation of the DDR1aΔ394–416 mutant is partly due to the deletion of Asn394, which constitutes a putative N-glycosylation site of DDR1 within the EJXM region. However, DDR1aΔ394–416 was clearly detected at the cell surface (supplemental Fig. 8, lane 8).
Nonetheless, these studies demonstrate that DDR1aΔ394–416, in contrast to the wild type receptor, is able to undergo collagen I-induced phosphorylation in the presence of MT1-MMP. Collectively, these results provide further support to the notion that cleavage of DDR1 occurs at the EJXM region and that this proteolytic event is a major cause for the inhibition of collagen I-induced receptor activation by MT1-MMP.
Effects of Inhibitors on DDR1 Shedding and Activation
Next, we sought to investigate the relative contribution of MT1-MMP to DDR1 shedding in breast cancer HCC1806 cells, which we found to naturally express both MT1-MMP and DDR1 (supplemental Fig. 10). PCR analyses showed that HCC1086 cells express the mRNA of both DDR1a and DDR1b isoforms but not of DDR2 (supplemental Fig. 10A). HCC1806 cells also express the mRNA of several secreted and membrane-anchored collagenases, including MT1-MMP, MT2-MMP, MMP-1, and MMP-13. In addition, we also examined the expression of ADAM (a disintegrin and metalloproteinase) 10 and 17 mRNAs, two well studied members of the ADAM subfamily of metalloproteinases with established sheddase activity (34), which were also implicated in collagen I-induced shedding of DDR1 in human embryonic kidney 293 cells transfected to express recombinant DDR1 (35). We found that HCC1806 cells express the mRNA of these ADAMs (supplemental Fig. 10A). The expression of DDR1 and MT1-MMP protein was confirmed by immunoblot analyses of cell lysates. Two DDR1 immunoreactive proteins of ∼110–120 and ∼60 kDa were detected, consistent with these forms representing the full-length and the CTF of DDR1 based on the recognition of antibody sc-532, which binds to the C terminus of the receptor (supplemental Fig. 10B). In addition, the lysates displayed the active form of MT1-MMP (∼57 kDa) (supplemental Fig. 10C). Analyses of the serum-free media revealed a protein of ∼62 kDa, which was recognized by antibody AF2396 to the N-terminal region of DDR1, indicating that HCC1806 cells display constitutive ectodomain shedding of DDR1 (supplemental Fig. 10D). However, we also found that DDR1 shedding in HCC1806 cells can be stimulated by exposure to either collagen I, phorbol ester, or concanavalin A (data not shown), suggesting a multifactorial process. Here, we have focused on constitutive DDR1 shedding to address our findings with the co-expression of DDR1 and MT-MMPs in unstimulated COS1 cells.
Next, we examined the contribution of MT1-MMP to the constitutive shedding of DDR1 in HCC1806 cells. To this end, we used several approaches, including RNA interference and inhibitors. Interestingly, under the conditions tested, knockdown of MT1-MMP mRNA expression by siRNA had no effect on DDR1 shedding despite an ∼70% reduction in MT1-MMP protein (data not shown). We interpret this finding as a result of DDR1 being cleaved by an alternate protease(s) in the absence of MT1-MMP (discussed later) and/or an indication of the relatively high efficiency of MT1-MMP-mediated cleavage of DDR1, which may proceed even at reduced protease levels (supplemental Fig. 2). Therefore, we examined the effects of two natural and one synthetic inhibitor of metalloproteinases as follows: TIMP-2, TIMP-1, and MIK-G2 on DDR1 shedding in HCC1806 cells. Whereas TIMP-2 inhibits all MMPs, TIMP-1 is a poor inhibitor of the transmembrane MT-MMPs and MMP-19. In contrast, both TIMP-2 and TIMP-1 do not inhibit most members of the ADAM family with the exception of ADAM10 and ADAM12, which are sensitive to TIMP-1 and TIMP-2, respectively (36). MIK-G2 (referred to as compound 3 in the original publication and as SB-3CT pMS in commercial sources) is a mechanism-based and selective inhibitor of the gelatinases and MT1-MMP (22), and it does not inhibit ADAM-mediated shedding of N-cadherin (37). As depicted in Fig. 8, treatment (20 h) of HCC1806 cells with 100 nm of either TIMP-1 (Fig. 8A, lane 2) or TIMP-2 (Fig. 8A, lane 3) inhibited (48 and 39%, respectively, over untreated cells) the release of the DDR1 ectodomain, as determined by the reduction in the ∼62-kDa soluble form of DDR1 in the media, when compared with media of untreated cells (Fig. 8A, lane 1). Likewise, addition of MIK-G2 (10 μm for 20 h) inhibited DDR1 shedding by ∼50% (Fig. 8A, lane 5). Analyses of the lysates revealed the full-length and the CTF of DDR1 in all conditions (Fig. 8B, lanes 1–5). However, the relative levels of these forms did not match the extent of inhibition of DDR1 shedding observed in the media. This is an agreement with a previous study in which treatment with either the metalloproteinase inhibitor TAPI or TIMP-3 in T47D and DDR1-transfected 293 cells, respectively, had no effect on the levels of full-length or CTF of DDR1, despite a significant inhibition of collagen I-induced DDR1 shedding by these inhibitors (35). In our cell system, we ascribed this paradoxical finding with the TIMPs and MIK-G2 to differences in antibody affinity (sc-532 versus AF2396), technique sensitivity (immunoblot analyses only able to detect drastic shifts in receptor pool), and/or issues of receptor turnover (compensatory mechanisms), all of which may highlight the complex relationship between receptor shedding and generation of degradation fragments (35).
Because of this limitation, we sought to evaluate the effects of TIMP/MIK-G2-mediated inhibition of DDR1 shedding on receptor phosphorylation in response to collagen I. To this end, HCC1806 cells were treated with TIMPs or MIK-G2 in the presence or absence of collagen I, and then examined for receptor activation by immunoprecipitation followed by detection of activated receptor with anti-Tyr(P) antibody, as described under “Experimental Procedures.” As shown in Fig. 9A, collagen I induces robust DDR1 phosphorylation in serum-starved HCC1806 cells (Fig. 9A, lanes 1 versus lane 2), as expected. Treatment with TIMP-1, TIMP-2, or MIK-G2 in the absence of collagen I had no effect on DDR1 phosphorylation (Fig. 9A, lanes 3, 5, and 9) indicating that protease inhibition per se does not affect DDR1 activation. In contrast, inhibitor treatment in the presence of collagen I increased the relative levels of receptor activation in response to collagen I, when compared with the untreated cells (Fig. 9A, lanes 4, 6, and 10 versus lanes 2 and 8, respectively), in two independent experiments. Collectively, the data in the COS1 and T47D cell systems strongly indicate a potential role for MT1-MMP in DDR1 ectodomain shedding and regulation of collagen-induced receptor activation, whereas the data with the HCC1806 cell system suggests that MT1-MMP might be only one of a number of specific DDR1 sheddases and regulators.
FIGURE 9.
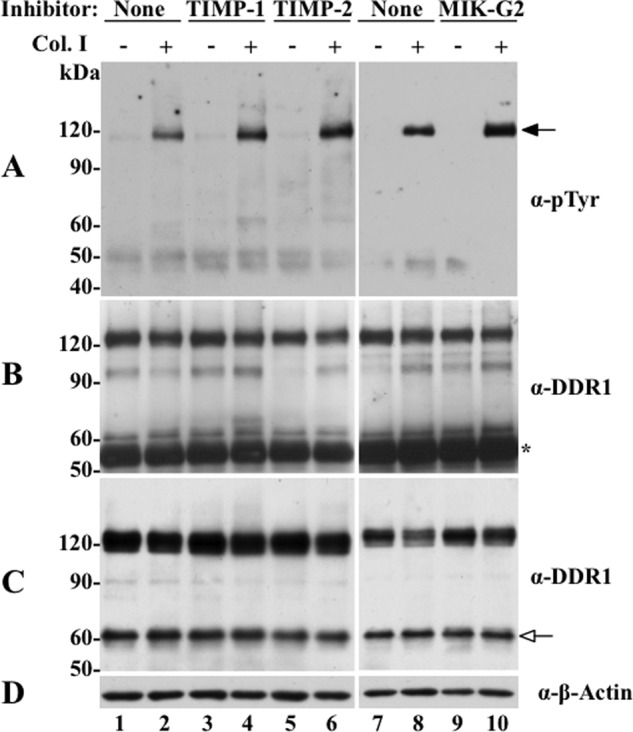
Effects of TIMPs and MIK-G2 on collagen I-induced DDR1 activation in HCC1806 cells. HCC1806 cells were incubated with the inhibitors as described in the legend of Fig. 8, and 20 h later the cells were treated (2 h) with or without 20 μg/ml of rat tail collagen I. The cells were then lysed with RIPA buffer (200 μl per well), and equal amounts of protein lysates were immunoprecipitated with DDR1 antibody sc-532. The immunoprecipitates were resolved by reducing 7.5% SDS-PAGE followed by immunoblot analyses with Tyr(P) (α-pTyr) (4G10®) antibody (A). The membrane was then stripped and probed with DDR1 (sc-532) for total DDR1 immunoprecipitated (B). Another fraction of the lysates was subjected to immunoblot analyses for total DDR1 expression level using DDR1 antibody (sc-532) antibody (C). The blot in C was then reprobed with antibodies to β-actin, as loading control (D). Black arrow in A indicates phosphorylated DDR1, and white arrow in C indicates the CTF of DDR1. Asterisk in B indicates the IgG fraction.
DISCUSSION
Many RTKs undergo proteolytic processing at their ectodomain in an event referred to as ectodomain shedding (38, 39). The sheddases cleave the ectodomain, generally at the stalk/linker region, and thus regulate the pool of active receptor available at the cell surface for ligand-induced signaling. In some surface receptors, the released N-terminal fragment elicits biological activity of its own in the pericellular space, whereas the remnant CTF undergoes intramembranous cleavage followed by translocation to the nucleus where it regulates transcriptional activity (40). Previous studies have shown that DDR1 is shed from the cell surface in both constitutive (41) and collagen-induced manners (35, 42), leading to the formation of two C-terminal fragments of 58 and 62 kDa (42). Based on sensitivity to protease inhibitors, it was postulated that DDR1 cleavage is mediated by the action of a metalloproteinase(s) (35). However, the nature of the metalloproteinase(s) was not identified. The data presented here show for the first time that membrane-anchored but not secreted collagenases cleave DDR1 releasing a soluble ectodomain and, consequently, negatively regulate collagen-dependent receptor phosphorylation. This effect of the membrane-anchored collagenases on DDR1 could not be reproduced by either MT4- or MT6-MMP further demonstrating the specificity of DDR1 as a substrate for the transmembrane MT-MMPs. The involvement of these MT-MMPs in shedding of DDR1 was also demonstrated by the effects of inhibitors, in particular TIMP-2 and MIK-G2, which are known to target transmembrane MT-MMPs, in HCC1806 cells. These findings establish MT-MMPs as one of the metalloproteinases that contribute to DDR1 shedding under physiological conditions. It should be noted, however, that our inhibitor and mutational data also point to a complex process of DDR1 shedding involving several proteases, including MMPs and ADAMs, which may collaborate and/or compensate each other to accomplish ectodomain cleavage. For instance, the partial inhibition caused by TIMP-1 in HCC1086 cells (Fig. 8A) may be explained by an effect on ADAM10 activity, which is expressed in HCC1806 cells and has been previously suggested to be a potential DDR1 sheddase (35). Thus, we cannot rule out members of the ADAM family of metalloproteinases as additional and important DDR1 sheddases. Further studies are required to identify and characterize the full spectrum of metalloproteases that cleave DDR1 under constitutive and stimulated conditions, in various cellular contexts.
Because the transmembrane MT-MMPs (MT1-, MT2-, and MT3-MMP) are known to exhibit collagenase activity, we thought it was important to determine whether abrogation of collagen I-evoked DDR1 phosphorylation observed in the presence of MT-MMPs was due to receptor cleavage and/or ligand degradation. Based on multiple lines of evidence, we conclude that the inhibitory action of MT-MMPs on collagen-induced DDR1 activation, under the experimental conditions, could only be ascribed to ectodomain shedding and not to collagenolysis. This is supported by the following: (i) inhibition of receptor activation by MT1-MMP was also observed in the presence of two DDR1 ligands, namely r/r collagen I and the GVMGFO-containing triple-helical peptide, which are not MT1-MMP substrates; (ii) receptor cleavage occurred independently of the collagen presence or collagen I-induced activation; (iii) DDR1 activation was not inhibited by MMP-1RXKR or MMP-13RXKR; and (iv) receptor phosphorylation was retained in an MT1-MMP-resistant DDR1 deletion mutant, DDR1Δ394–416. Although these conclusions are based on studies conducted with MT1-MMP, we surmise that these findings also apply to MT2- and MT3-MMP because these proteases also accomplish cleavage of DDR1 and are close homologues of MT1-MMP. Moreover, our data indicate that both DDR1a and DDR1b are equally sensitive to these MT-MMPs, both being unable to undergo receptor phosphorylation when co-expressed with these proteases. At present, the expression and activation of DDR1 isoforms in different cells/tissues as well as in different physiological and pathological conditions are unknown. Likewise, where and when they are co-expressed with MT-MMPs within a distinct subcellular locale also remains to be established. Nevertheless, our data suggest that these DDR1 isoforms, which may activate different downstream effector (2), are equally targeted by MT-MMPs, establishing these membrane-tethered collagenases as general DDR1 regulators.
As indicated earlier, the data presented here highlight the importance of receptor cleavage as a critical step in the regulation of DDR1 activation by MT-MMPs. Cleavage of DDR1 was independent of receptor activation, and it occurred in the absence of ligand. Thus, MT-MMPs may act to clear DDR1 receptors from the cell surface independently of collagen stimulation. However, the detection of phosphorylated CTF suggests the possibility that MT-MMPs may also cleave DDR1 after activation and thus alter DDR1-initiated signaling. Considering that DDRs exhibit slow activation kinetics (4), removal of phosphorylated DDR1 by MT-MMPs may significantly impact the nature of the signaling networks activated in response to DDR1 stimulation. Moreover, inhibition of metalloprotease activity under conditions of excess inhibitor secretion at the pericellular space may also enhance collagen-evoked DDR1 phosphorylation (Fig. 9) by increasing the pool of receptor available for ligand binding. However, the outcome of increased receptor phosphorylation in regulation of cell-collagen interactions needs to be defined, as the consequences of DDR1 signaling are only partially understood. The impact of collagenase activity should also be taken into account when evaluating effects of MT-MMPs on DDR1 signaling. Indeed, previous evidence demonstrated that degradation of collagen attenuates DDR1 phosphorylation (4). Thus, in vivo, collagenolysis would also compromise receptor activation by impairing ligand integrity, in particular during processes of intense remodeling of the pericellular ECM. However, limited collagen proteolysis may preserve DDR1 activation if the MMPs were to generate collagen fragments comprising an intact and functional DDR binding motif (1, 43), such as the GVMGFO-containing triple-helical peptide, as shown here. Thus, in vivo, the consequences of MT-MMP activity on DDR1 signaling are likely to be complex, involving both direct and indirect effects on receptor and ligand integrity.
We found that membrane anchoring of the catalytic domain was essential for MT1-MMP-mediated cleavage of DDR1. This suggests that DDR1 cleavage is strictly dependent on an entropically favorable close interaction between the protease and the receptor within the confines of the plasma membrane. Consistently, a secreted variant of MT1-MMP was unable to cleave DDR1, and consequently, receptor activation remained unchanged. This raises the possibility that the differential effects between membrane-anchored and secreted collagenases on DDR1 stem in part from their distinct tissue localization and may be independent of their substrate profile. We compared the structures of the catalytic sites of MMP-1, MMP-13, and MT1-MMP to explain the selectivity of MT1-MMP for DDR1 as a substrate. We observed that all three enzymes are almost identical in the vicinity of the catalytic Zn2+ and differences start to appear beyond. The importance of potential exosite interactions for substrate recognition of MMPs has been reported previously (44, 45). Thus, besides cellular localization, it is possible that the inability of MMP-1RXKR or MMP-13RXKR to cleave DDR1 has to do with these potential exosite interactions, which abrogate the ability of DDR1 to serve as substrate for these enzymes. However, as discussed earlier, an effect on DDR1 activation by these collagenases caused by hydrolysis of collagen cannot be ruled out and may be relevant under certain physiological and pathological conditions involving deregulated collagen degradation.
Our data highlight the EJXM linker of DDR1 as the major proteolytic target of MT1-MMP via cleavage at the Ser397–Leu398 and Pro407–Val408 peptide bonds. This was established by N-terminal sequencing of two degradation products of DDR1 and by the data with the DDR1aΔ394–416 mutant, which lacks the two cleavage sites. Although MT1-MMP cleavage site motifs possessing Ser/Pro∼Leu bonds have been reported previously based on analysis of phage display libraries (45, 46), DDRs have not been identified as MT1-MMP substrates or binding partners in proteomics studies (47–49). Nevertheless, structurally, the computational analyses supported the notion that the active site of MT1-MMP can accommodate the two sites within the EJXM of DDR1 and serve as targets for MT1-MMP proteolysis. However, our analyses could not predict which of the two sites would experience hydrolysis before the other. The disrupted trafficking of the DDR1aΔ400–416 deletion mutant also precluded us to evaluate the sequence of proteolytic steps that lead to DDR1 cleavage and whether both sites are indeed hydrolyzed by MT1-MMP alone or can be targeted by both MT-MMPs and ADAMs cooperatively and/or independently. We surmise that the inability to completely block DDR1 shedding using various inhibitors reflects the flexible nature of the EJXM, which might have evolved to confer DDR1 with the ability to undergo ectodomain shedding, among other functions, by various metalloproteinases (39, 50). However, the effects of these proteases on DDR1 may be differentially regulated. For instance, although early findings showed collagen-dependent shedding of DDR1, which was ascribed to ADAM activity (35, 42), we showed that cleavage of DDR1 by MT-MMPs proceeded independently of collagen exposure. In addition, although these proteases may overlap during promotion of receptor cleavage, MMPs may provide an extra level of receptor regulation by altering ligand integrity. Although the fate and function of the soluble DDR1 ectodomain released by MT-MMPs remains to be established, previous evidence showed that the isolated DDR1 ectodomain maintains collagen binding activity (9) and biological function (51, 52), including blocking the activity of membrane-anchored receptor (53). The role of the CTF of DDR1 is also unknown. However, as reported for other surface receptors (54), the CTF of DDR1 may elicit biological activity via regulation of DDR1 downstream effectors (2, 12) and/or possibly by translocation to the nucleus. These possibilities are currently under investigation in our laboratory.
The fact that DDR receptors exhibit structural and ligand binding differences begs the question as to whether they are differentially regulated by MT-MMP proteolysis. Alignment of the EJXM regions of DDR1 (residues 368–416) and DDR2 (residues 368–398) reveals a significant lack of sequence similarity in this region (Fig. 6A). Moreover, the EJXM of DDR2 lacks the Ser397–Leu398 site, whereas Val408 of DDR1 is replaced with a Met390 residue (Fig. 6A). Ongoing studies in our laboratory indicate that DDR2, in contrast to DDR1, is not cleaved at the EJXM region by MT1-MMP when co-expressed in COS1 cells, and the receptor maintains collagen I-induced phosphorylation.4 Moreover, a DDR2-EJXM synthetic peptide was not hydrolyzed by MT1-MMP (data not shown). Collectively, this evidence suggests that the EJXM region of DDRs evolved to impart these RTKs with a differential sensitivity to MT-MMP-mediated proteolysis, which may have profound effects on receptor turnover and signaling. At present, the consequences of MT-MMP activity on DDR1-mediated cell behavior are unknown and remain to be elucidated. However, this is not a trivial task considering the diverse and opposing biological activities of DDR1 isoforms in different cellular backgrounds and environments (1–3). This challenge will abate as we learn more about the intricate biological actions of these unique receptors. Nevertheless, the data presented here identify the first set of metalloproteinases involved in shedding and regulation of DDR1, a unique RTK activated by collagen.
Acknowledgments
We are indebted to Dr. Stephen Weiss for providing essential reagents for this study and helpful comments. We thank Dr. Lawrence Dangott (Texas A&M University) for the great help and service with the Edman degradation analyses
This work was supported, in whole or in part, by National Institutes of Health Grants CA-61986 (to R. F.) and CA-98799 (to G. B. F.).

This article contains supplemental Figs. 1–10 and Table 1.
M. Bhowmick and G. B. Fields, unpublished data.
H.-L. Fu, A. Sohail, and R. Fridman, unpublished data.
- ECM
- extracellular matrix
- DDR
- discoidin domain receptor
- MMP
- matrix metalloproteinase
- MT
- membrane type
- CTF
- C-terminal fragment
- DS
- discoidin
- EJXM
- extracellular juxtamembrane
- KD
- kinase domain
- RTK
- receptor tyrosine kinase
- TIMP
- tissue inhibitor of metalloproteinase
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl.
REFERENCES
- 1. Leitinger B. (2011) Transmembrane collagen receptors. Annu. Rev. Cell Dev. Biol. 27, 265–290 [DOI] [PubMed] [Google Scholar]
- 2. Valiathan R. R., Marco M., Leitinger B., Kleer C. G., Fridman R. (2012) Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. 31, 295–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vogel W. F., Abdulhussein R., Ford C. E. (2006) Sensing extracellular matrix: an update on discoidin domain receptor function. Cell. Signal. 18, 1108–1116 [DOI] [PubMed] [Google Scholar]
- 4. Vogel W., Gish G. D., Alves F., Pawson T. (1997) The discoidin domain receptor tyrosine kinases are activated by collagen. Mol. Cell 1, 13–23 [DOI] [PubMed] [Google Scholar]
- 5. Shrivastava A., Radziejewski C., Campbell E., Kovac L., McGlynn M., Ryan T. E., Davis S., Goldfarb M. P., Glass D. J., Lemke G., Yancopoulos G. D. (1997) An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol. Cell 1, 25–34 [DOI] [PubMed] [Google Scholar]
- 6. Leitinger B., Steplewski A., Fertala A. (2004) The D2 period of collagen II contains a specific binding site for the human discoidin domain receptor, DDR2. J. Mol. Biol. 344, 993–1003 [DOI] [PubMed] [Google Scholar]
- 7. Leitinger B., Kwan A. P. (2006) The discoidin domain receptor DDR2 is a receptor for type X collagen. Matrix Biol. 25, 355–364 [DOI] [PubMed] [Google Scholar]
- 8. Hou G., Vogel W., Bendeck M. P. (2001) The discoidin domain receptor tyrosine kinase DDR1 in arterial wound repair. J. Clin. Invest. 107, 727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leitinger B. (2003) Molecular analysis of collagen binding by the human discoidin domain receptors, DDR1 and DDR2. Identification of collagen-binding sites in DDR2. J. Biol. Chem. 278, 16761–16769 [DOI] [PubMed] [Google Scholar]
- 10. Konitsiotis A. D., Raynal N., Bihan D., Hohenester E., Farndale R. W., Leitinger B. (2008) Characterization of high affinity binding motifs for the discoidin domain receptor DDR2 in collagen. J. Biol. Chem. 283, 6861–6868 [DOI] [PubMed] [Google Scholar]
- 11. Xu H., Raynal N., Stathopoulos S., Myllyharju J., Farndale R. W., Leitinger B. (2011) Collagen binding specificity of the discoidin domain receptors: binding sites on collagens II and III and molecular determinants for collagen IV recognition by DDR1. Matrix Biol. 30, 16–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lemeer S., Bluwstein A., Wu Z., Leberfinger J., Müller K., Kramer K., Kuster B. (2012) Phosphotyrosine-mediated protein interactions of the discoidin domain receptor 1. J. Proteomics 75, 3465–3477 [DOI] [PubMed] [Google Scholar]
- 13. Carafoli F., Mayer M. C., Shiraishi K., Pecheva M. A., Chan L. Y., Nan R., Leitinger B., Hohenester E. (2012) Structure of the discoidin domain receptor 1 extracellular region bound to an inhibitory Fab fragment reveals features important for signaling. Structure 20, 688–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abdulhussein R., McFadden C., Fuentes-Prior P., Vogel W. F. (2004) Exploring the collagen-binding site of the DDR1 tyrosine kinase receptor. J. Biol. Chem. 279, 31462–31470 [DOI] [PubMed] [Google Scholar]
- 15. Abdulhussein R., Koo D. H., Vogel W. F. (2008) Identification of disulfide-linked dimers of the receptor tyrosine kinase DDR1. J. Biol. Chem. 283, 12026–12033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lauer-Fields J. L., Juska D., Fields G. B. (2002) Matrix metalloproteinases and collagen catabolism. Biopolymers 66, 19–32 [DOI] [PubMed] [Google Scholar]
- 17. Holmbeck K., Bianco P., Yamada S., Birkedal-Hansen H. (2004) MT1-MMP: a tethered collagenase. J. Cell. Physiol. 200, 11–19 [DOI] [PubMed] [Google Scholar]
- 18. Hotary K. B., Allen E. D., Brooks P. C., Datta N. S., Long M. W., Weiss S. J. (2003) Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell 114, 33–45 [DOI] [PubMed] [Google Scholar]
- 19. Rowe R. G., Weiss S. J. (2009) Navigating ECM barriers at the invasive front: the cancer cell-stroma interface. Annu. Rev. Cell Dev. Biol. 25, 567–595 [DOI] [PubMed] [Google Scholar]
- 20. Cho J. A., Osenkowski P., Zhao H., Kim S., Toth M., Cole K., Aboukameel A., Saliganan A., Schuger L., Bonfil R. D., Fridman R. (2008) The inactive 44-kDa processed form of membrane type 1 matrix metalloproteinase (MT1-MMP) enhances proteolytic activity via regulation of endocytosis of active MT1-MMP. J. Biol. Chem. 283, 17391–17405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hernandez-Barrantes S., Toth M., Bernardo M. M., Yurkova M., Gervasi D. C., Raz Y., Sang Q. A., Fridman R. (2000) Binding of active (57 kDa) membrane type 1-matrix metalloproteinase (MT1-MMP) to tissue inhibitor of metalloproteinase (TIMP)-2 regulates MT1-MMP processing and pro-MMP-2 activation. J. Biol. Chem. 275, 12080–12089 [DOI] [PubMed] [Google Scholar]
- 22. Ikejiri M., Bernardo M. M., Bonfil R. D., Toth M., Chang M., Fridman R., Mobashery S. (2005) Potent mechanism-based inhibitors for matrix metalloproteinases. J. Biol. Chem. 280, 33992–34002 [DOI] [PubMed] [Google Scholar]
- 23. Lauer-Fields J. L., Tuzinski K. A., Shimokawa Ki, Nagase H., Fields G. B. (2000) Hydrolysis of triple-helical collagen peptide models by matrix metalloproteinases. J. Biol. Chem. 275, 13282–13290 [DOI] [PubMed] [Google Scholar]
- 24. Olson M. W., Gervasi D. C., Mobashery S., Fridman R. (1997) Kinetic analysis of the binding of human matrix metalloproteinase-2 and -9 to tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2. J. Biol. Chem. 272, 29975–29983 [DOI] [PubMed] [Google Scholar]
- 25. Toth M., Osenkowski P., Hesek D., Brown S., Meroueh S., Sakr W., Mobashery S., Fridman R. (2005) Cleavage at the stem region releases an active ectodomain of the membrane type 1 matrix metalloproteinase. Biochem. J. 387, 497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Toth M., Hernandez-Barrantes S., Osenkowski P., Bernardo M. M., Gervasi D. C., Shimura Y., Meroueh O., Kotra L. P., Gálvez B. G., Arroyo A. G., Mobashery S., Fridman R. (2002) Complex pattern of membrane type 1 matrix metalloproteinase shedding. Regulation by autocatalytic cells surface inactivation of active enzyme. J. Biol. Chem. 277, 26340–26350 [DOI] [PubMed] [Google Scholar]
- 27. Hotary K., Li X. Y., Allen E., Stevens S. L., Weiss S. J. (2006) A cancer cell metalloprotease triad regulates the basement membrane transmigration program. Genes Dev. 20, 2673–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Babine R. E., Bender S. L. (1997) Molecular recognition of protein-ligand complexes: applications to drug design. Chem. Rev. 97, 1359–1472 [DOI] [PubMed] [Google Scholar]
- 29. Tallant C., Marrero A., Gomis-Rüth F. X. (2010) Matrix metalloproteinases: fold and function of their catalytic domains. Biochim. Biophys. Acta 1803, 20–28 [DOI] [PubMed] [Google Scholar]
- 30. L'hôte C. G., Thomas P. H., Ganesan T. S. (2002) Functional analysis of discoidin domain receptor 1: effect of adhesion on DDR1 phosphorylation. FASEB J. 16, 234–236 [DOI] [PubMed] [Google Scholar]
- 31. Wu H., Byrne M. H., Stacey A., Goldring M. B., Birkhead J. R., Jaenisch R., Krane S. M. (1990) Generation of collagenase-resistant collagen by site-directed mutagenesis of murine pro-α1(I) collagen gene. Proc. Natl. Acad. Sci. U.S.A. 87, 5888–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grams F., Crimmin M., Hinnes L., Huxley P., Pieper M., Tschesche H., Bode W. (1995) Structure determination and analysis of human neutrophil collagenase complexed with a hydroxamate inhibitor. Biochemistry 34, 14012–14020 [DOI] [PubMed] [Google Scholar]
- 33. Hawinkels L. J., Kuiper P., Wiercinska E., Verspaget H. W., Liu Z., Pardali E., Sier C. F., ten Dijke P. (2010) Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 70, 4141–4150 [DOI] [PubMed] [Google Scholar]
- 34. Reiss K., Saftig P. (2009) The “a disintegrin and metalloprotease” (ADAM) family of sheddases: physiological and cellular functions. Semin. Cell Dev. Biol. 20, 126–137 [DOI] [PubMed] [Google Scholar]
- 35. Slack B. E., Siniaia M. S., Blusztajn J. K. (2006) Collagen type I selectively activates ectodomain shedding of the discoidin domain receptor 1: involvement of Src tyrosine kinase. J. Cell. Biochem. 98, 672–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Murphy G. (2011) Tissue inhibitors of metalloproteinases. Genome Biol. 12, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kohutek Z. A., diPierro C. G., Redpath G. T., Hussaini I. M. (2009) ADAM-10-mediated N-cadherin cleavage is protein kinase C-alpha dependent and promotes glioblastoma cell migration. J. Neurosci. 29, 4605–4615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Arribas J., Borroto A. (2002) Protein ectodomain shedding. Chem. Rev. 102, 4627–4638 [DOI] [PubMed] [Google Scholar]
- 39. van Kilsdonk J. W., van Kempen L. C., van Muijen G. N., Ruiter D. J., Swart G. W. (2010) Soluble adhesion molecules in human cancers: sources and fates. Eur. J. Cell Biol. 89, 415–427 [DOI] [PubMed] [Google Scholar]
- 40. Higashiyama S., Nanba D., Nakayama H., Inoue H., Fukuda S. (2011) Ectodomain shedding and remnant peptide signalling of EGFRs and their ligands. J. Biochem. 150, 15–22 [DOI] [PubMed] [Google Scholar]
- 41. Alves F., Vogel W., Mossie K., Millauer B., Höfler H., Ullrich A. (1995) Distinct structural characteristics of discoidin I subfamily receptor tyrosine kinases and complementary expression in human cancer. Oncogene 10, 609–618 [PubMed] [Google Scholar]
- 42. Vogel W. F. (2002) Ligand-induced shedding of discoidin domain receptor 1. FEBS Lett. 514, 175–180 [DOI] [PubMed] [Google Scholar]
- 43. Schenk S., Quaranta V. (2003) Tales from the crypt[ic] sites of the extracellular matrix. Trends Cell Biol. 13, 366–375 [DOI] [PubMed] [Google Scholar]
- 44. Robichaud T. K., Steffensen B., Fields G. B. (2011) Exosite interactions impact matrix metalloproteinase collagen specificities. J. Biol. Chem. 286, 37535–37542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kridel S. J., Sawai H., Ratnikov B. I., Chen E. I., Li W., Godzik A., Strongin A. Y., Smith J. W. (2002) A unique substrate binding mode discriminates membrane type-1 matrix metalloproteinase from other matrix metalloproteinases. J. Biol. Chem. 277, 23788–23793 [DOI] [PubMed] [Google Scholar]
- 46. Ohkubo S., Miyadera K., Sugimoto Y., Matsuo K., Wierzba K., Yamada Y. (1999) Identification of substrate sequences for membrane type-1 matrix metalloproteinase using bacteriophage peptide display library. Biochem. Biophys. Res. Commun. 266, 308–313 [DOI] [PubMed] [Google Scholar]
- 47. Butler G. S., Dean R. A., Tam E. M., Overall C. M. (2008) Pharmacoproteomics of a metalloproteinase hydroxamate inhibitor in breast cancer cells: dynamics of membrane type 1 matrix metalloproteinase-mediated membrane protein shedding. Mol. Cell. Biol. 28, 4896–4914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tam E. M., Morrison C. J., Wu Y. I., Stack M. S., Overall C. M. (2004) Membrane protease proteomics: Isotope-coded affinity tag MS identification of undescribed MT1-matrix metalloproteinase substrates. Proc. Natl. Acad. Sci. U.S.A. 101, 6917–6922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tomari T., Koshikawa N., Uematsu T., Shinkawa T., Hoshino D., Egawa N., Isobe T., Seiki M. (2009) High throughput analysis of proteins associating with a proinvasive MT1-MMP in human malignant melanoma A375 cells. Cancer Sci. 100, 1284–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hayashida K., Bartlett A. H., Chen Y., Park P. W. (2010) Molecular and cellular mechanisms of ectodomain shedding. Anat. Rec. 293, 925–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Agarwal G., Mihai C., Iscru D. F. (2007) Interaction of discoidin domain receptor 1 with collagen type 1. J. Mol. Biol. 367, 443–455 [DOI] [PubMed] [Google Scholar]
- 52. Flynn L. A., Blissett A. R., Calomeni E. P., Agarwal G. (2010) Inhibition of collagen fibrillogenesis by cells expressing soluble extracellular domains of DDR1 and DDR2. J. Mol. Biol. 395, 533–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hachehouche L. N., Chetoui N., Aoudjit F. (2010) Implication of discoidin domain receptor 1 in T cell migration in three-dimensional collagen. Mol. Immunol. 47, 1866–1869 [DOI] [PubMed] [Google Scholar]
- 54. Ancot F., Foveau B., Lefebvre J., Leroy C., Tulasne D. (2009) Proteolytic cleavages give receptor tyrosine kinases the gift of ubiquity. Oncogene 28, 2185–2195 [DOI] [PubMed] [Google Scholar]