

Background: Pax6, Pax6(5a), and Pax2 are derived from a common Pax2-like ancestral gene.
Results: Pax6(5a) and Pax2 can partially substitute for Pax6 in neural development.
Conclusion: The specificities of Pax6(5a) and Pax2 paired domains correspond to the extent of the rescues in forebrain but not in eye development.
Significance: The unique function of Pax6 in eye development requires the combined activities of paired domain and homeodomain.
Keywords: Development, Homeobox, Neurodevelopment, Retina, Transcription Factors
Abstract
The advent of the ocular and nervous system in metazoan evolution coincides with the diversification of a single ancestral paired box (Pax) gene into Pax6, Pax6(5a), and Pax2. To investigate the role of these Pax genes in neural development, we have generated an allelic series of knock-in models at the Pax6 locus. We showed that although Pax6(5a) and Pax2 could not replace Pax6 for its autoregulation in lens induction or for neural differentiation in retina, Pax6(5a) was sufficient for corneal-lenticular detachment. In brain development, cell proliferation in the cerebral cortex and dorsoventral patterning of the telencephalon and neural tube were partially rescued in either knock-in mutant. Contrary to the previous belief, our genetic studies showed that the Pax6 isoform Pax6(5a) could potentially play a role in neuronal differentiation in brain development. Importantly, Pax2 showed greater rescue efficiency than Pax6(5a) in the telencephalon even though the latter was identical to Pax6 outside the paired domain. In studying Ngn2, a Pax6 direct target gene in telencephalon, we showed that the level of Ngn2 expression correlated with the in vitro binding of Pax2, Pax6, and Pax6(5a) paired domain on its enhancer. Our results show that Pax6 is uniquely required for eye development, but in brain development, Pax6 can be functionally substituted by related Pax family genes that share a similar paired domain binding specificity.
Introduction
The Pax2 family of transcription factors is a metazoan innovation that can be traced back evolutionarily to a single Pax2-like gene in sponge, which has no eye or nervous system (for a review, see Ref. 1). The emergence of the visual system in bilateria coincides with the separation of Pax2 and Pax6 genes, whereas further gene duplications in Drosophila resulted in two canonical Pax6 paralogues (ey and toy) and two Pax6(5a)-like genes (eyg and toe). The importance of Pax6, Pax6(5a), and Pax2 in Drosophila eye development has been clearly demonstrated by the ey and the eyg mutants, which have no eyes, and by the Pax2 mutants, which have abnormal cone cells (2–4). It was further shown that Drosophila Pax6, Pax6(5a), and Pax2 have separate functions with ey controlling retinal specification, eyg regulating cell growth, and Pax2 involved in cone and pigment cell development (2, 5). However, the Pax2 and Pax6 paired domains share remarkably similar consensus DNA binding sites, and at least in Drosophila, the paired domain (PD) but not the homeodomain (HD) of Pax6 was required for eye development (6). Furthermore, Pax6(5a), but not Pax6, was shown to be able to induce ectopic retina in chick (7). These results raise the question whether Pax6, Pax6(5a), and Pax2 are functionally exchangeable in mammalian eye development.
Pax6 plays multiple roles in neural development. Humans heterozygous for PAX6 develop blindness, aniridia, colobomas, and cataracts, whereas Pax6-null mice (Pax6Sey/Sey; small eye) fail to form any mature eye structures (8–11). This is because Pax6 expression, which appears in the head surface ectoderm prior to lens placode formation, is crucial for lens induction as well as differentiation (12, 13). Although the remaining Pax6-null retinal primordia initially up-regulate retina-specific markers, such as Crx for photoreceptor cells and VC1.1 for amacrine cells, the neurogenic program is eventually aborted (14–16). During neural tube development, Pax6 controls ventral patterning through its antagonistic interaction with Nkx2.2, establishing distinct populations of progenitor cells (17). In contrast, Pax6 expression in forebrain is primarily restricted to the dorsal telencephalon where Pax6 activates the dorsal telencephalic transcription factor Ngn2 expression to prevent the expansion of ventral transcription factor Mash1 while maintaining the boundary structures to restrict cell migration (18, 19). In addition to these neural patterning defects, the Pax6-null progenitors in the dorsal telencephalon also present with cell cycle and migratory abnormalities, resulting in a thinner cortical plate (20–23).
Underlying its complex biological functions, Pax6 protein has three distinct domains, the PD, the HD, and the transactivation domain. The PD contains two helix-turn-helix motifs, referred to as the “PAI” and the “RED” motif regions, respectively, that are necessary for DNA recognition and binding (24). The PD may also cooperate with the DNA-binding HD to promote transcription via the transactivation domain, a proline-, serine-, threonine-rich linker domain at the C terminus (25). Pax6(5a), an alternatively spliced isoform of Pax6, contains a 14-amino acid insert within the PD that considerably changes its DNA binding specificity: the canonical PD binds to DNA via its N-terminal PAI domain, whereas the Pax6(5a) PD targets DNA with the C-terminal RED domain (24). Although the Pax6(5a) isoform only constitutes up to 20% of total Pax6 transcripts, loss of Pax6(5a) in human and mouse results in distinct ocular phenotypes (24, 26, 27). In addition, overexpression of Pax6(5a) in cerebral cortex progenitors was observed to inhibit cell proliferation without affecting cell fate, whereas overexpression of the canonical Pax6 inhibited cell proliferation but also potently increased neurogenesis (26, 28). These results led to the conclusion that Pax(5a) only controls cell proliferation, whereas Pax6 canonical isoform regulates both cell growth and fate determination.
A homologous PD is also the defining feature of the larger Pax gene family, which otherwise harbors considerable sequence variations within its nine family members. Unlike Pax6, for example, Pax2 retains a partial one-helix HD and an eight-amino acid octapeptide motif, which is functionally important for transcriptional inhibition (for a review, see Ref. 29). Using a PCR-based seletion method, Epstein et al. (30) showed that the Pax6 PD and Pax2 PD share strikingly similar consensus binding sequences. Nevertheless, although ectopic expression of Pax6 can sometimes induce eye formation outside the ocular region, Pax2 is known for its distinct control of urogenital development. Only during early eye development when Pax2 and Pax6 are initially co-expressed in the optic vesicle do they play redundant roles in retinal pigmented epithelium specification (31, 32). But even in the eye, Pax2 and Pax6 expressions quickly diverge through mutual repression to control optic stalk and neural retina development, respectively (33). It remains unclear how these two transcriptional regulators that share an almost identical consensus PD binding sequence can have sometimes redundant but often divergent functions in embryogenesis.
In our present study, we sought to examine the functional specificity of the Pax genes in neural development by replacing the Pax6 coding region with either Pax2 or Pax6(5a) cDNA, which led to induction of Pax2 and Pax6(5a) in the endogenous Pax6 expression domains. Although neither ectopically expressed transcription factor could rescue eye development, the ventral patterning of the neural tube was partially restored by the Pax6(5a) and Pax2 knock-in. In contrast to previous reports that Pax6(5a) only affects cell proliferation but not cell fate in the telencephalon, we showed that ectopic Pax6(5a) reversed both dorsoventral patterning and progenitor proliferation defects, although boundary formation was not recovered. Interestingly, ectopic Pax2 achieved stronger rescue of Pax6 telencephalon patterning and neurogenesis than Pax6(5a) despite the fact that Pax2 diverges from Pax6 and Pax6(5a) outside the PD domain. Using a Pax6 direct downstream target, the proneural gene Ngn2, as an example, we showed that the extent of rescue closely correlated with the binding affinities of Pax2 and Pax6(5a) PDs on the known Ngn2 enhancer. Therefore, whereas the entire Pax6 protein is necessary for eye development, the binding specificity of the PD is largely sufficient to determine the functional specificity of Pax2, Pax6, and Pax6(5a) in forebrain and neural tube development.
EXPERIMENTAL PROCEDURES
Pax6 Targeting Vector Construction
The Pax6 targeting vector was generated using the recombineering method (34, 35). Briefly, a minitargeting vector containing a Neo selection cassette and a Pax6(5a) full-length cDNA (IMAGE clone number 4008490) was used to replace the Pax6 genomic sequence from exons 4 to 13 contained in a 129S6/SvEvTac Bac clone (BACPAC Resources Center at Children's Hospital Oakland Research Institute, catalogue number RP22-55A14). Through homologous recombination, the translation start site of Pax6(5a) cDNA was fused exactly at the original ATG codon within Pax6 exon 4. The excision of the Neo cassette by Cre recombinase left behind a de novo NcoI site and a single loxP site in front of the Pax6(5a) cDNA. Using the gap repair method, the Pax6(5a) cDNA and the flanked 4.2- and 6-kb Pax6 genomic sequences were cloned into pAY253, a low copy number MC1TK-containing plasmid that can accommodate large DNA inserts. A second round of minitargeting then placed behind the Pax6(5a) cDNA another Neo cassette, a stop cassette (five copies of poly(A) sequence), a single loxP site, and a full-length cDNA for Pax2 followed by a new BamHI site (Fig. 1A). The resulting Pax6 targeting vector was verified by direct sequencing.
FIGURE 1.

Cloning strategy for Pax65a and Pax6Pax2 knock-ins. A, the coding region of Pax6 (exons 4–13) was replaced by a cDNA for Pax6(5a) followed by a neomycin (Neo) cassette, a stop cassette (five copies of poly(A) sequence), and a cDNA for Pax2. Frt and LoxP sites are represented by open arrows and solid triangles, respectively. Nc, NcoI; B, BamHI. B, for Southern blot confirmation, genomic DNA extracted from targeted ES cells digested with either NcoI or BamHI was hybridized with the 5′ or the 3′ external probes, respectively. Predicted fragment sizes were obtained for the correctly targeted clones. C, genotyping for Pax65a and Pax6Pax2 alleles. D–G, using a Pax6 3′-UTR probe (common to both Pax6 and Pax6(5a)) and a Pax2 probe, we showed that Pax6(5a) and Pax2 RNAs were expressed ectopically within the endogenous Pax6 expression domains at embryonic day 10.5 (arrows). tc, telecephalon; ey, eye; nt, neural tube. H–K, in Pax6Pax2/+ eyes, there was a decrease in Pax6 transcripts but a corresponding expansion of Pax2 expression from the optic disc to the whole retina (arrowhead) and the lens (arrow). L, quantitative real time PCR was performed on E14.5 brain tissues with primers that recognize the domain shared by Pax6 and Pax6(5a). After normalization using Gapdh expression, the combined expression of Pax6 and Pax6(5a) was shown to be the same in Pax6+/+ and Pax65a/+ embryos, demonstrating that Pax6(5a) was transcribed as efficiently as endogenous Pax6. M, the numbers of Pax6 and Pax2 transcripts in Pax6Pax2/+ embryos were determined by quantitative real time PCR using standard curves generated with Pax6 and Pax2 cDNA plasmids and normalized using Gapdh expression. After subtracting endogenous expression of Pax2 determined from wild type samples, the expression level of Pax2 knock-in allele was found to be the same as that for endogenous Pax6 in Pax6Pax2/+ embryos. n = 3 for each genotype. N.S., not significant. Scale bar, 100 μm. Error bars represent S.E.
Generation of Mouse Lines
ES cells (129S6/SvEvTac) electroporated with the linearized Pax6 targeting vector were screened for successful homologous recombination by Southern blot using both 5′ (NcoI) and 3′ (BamHI) external probes (Fig. 1B). The positive ES cells were used to generate Pax65a chimeric mice by pronuclear injection into C57BL/6 mouse blastocysts. Tail biopsies were collected, and genotype PCR was performed to confirm the Pax65a allele (primers: Pax65a F, 5′-GATGCAAAAGTCCAGGTGCT-3′; Pax65a R, 5′-TTCCCAAGCAAAGATGGAAG-3′) (Fig. 1C). The Pax65a/+ mice were crossed to the Ella-Cre mice (The Jackson Laboratory, stock number 003724) to remove the Pax6(5a) cDNA and the Neo and the stop cassettes in the germ line. The resulting Pax6Pax2/+ mice were confirmed by genotype PCR (primers: Pax6Pax2 F, 5′-AAAGTGGTGGACAAGATTGC-3′; Pax6Pax2 R, 5′-TTAGGGACAGAGCCCTCAGA-3′). Pax6 small eye mutant embryos (Pax6Sey-Neu/Sey-Neu) were kindly provided by James Li (University of Connecticut Health Center, Farmington, CT) (10, 36).
The presence of a vaginal plug was considered 0.5 days postcoitum or E0.5. All experimental procedures involving mice were humanely performed in accordance with the Laboratory Animal Research Center at Indiana University.
Immunohistochemistry and Histology
Fluorescent immunohistochemistry of cryosections and paraffin sections were performed as described previously (37–39). The following antibodies were used: mouse anti-Islet1 (1:10), anti-Pax6 (1:10), and anti-Nkx2.2 (1:10) (all from Developmental Studies Hybridoma Bank, Iowa City, IA); mouse anti-Mitf (1:50) (Thermo Scientific, Fremont, CA); rabbit anti-Sox2 (1:800) (Chemicon International, Billerica, MA); rabbit anti-Pax2 (1:100) and rabbit anti-Pax6 (1:250) (both from Covance, Berkeley, CA); rabbit anti-phosphohistone H3 (Ser-10) (1:500) (Upstate, Temecula, CA); and rabbit anti-Ptf1a (1:100) (kindly provided by Dr. Jane E. Johnson, University of Texas Southwestern Medical Center, Dallas, TX). Secondary antibodies for all experiments were either Alexa Fluor 488- (1:250) or Alexa Fluor 555 (1:500)-conjugated anti-mouse and anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA). For histology of E14.5 paraffin sections of cerebral cortex, hematoxylin and eosin staining was performed as described previously (37).
RNA in Situ Hybridization
RNA in situ hybridization experiments for whole-mount embryos and slide sections were performed as described previously (37–39). Antisense probes were generated from cDNAs for Crx (from Valerie Wallace, Ottawa Health Research Institute, Ottawa, Ontario, Canada), Foxe3 (from Dr. Milan Jamrich, Baylor College of Medicine, Houston, TX), Pax2 and Pax6 (both generously provided by Richard Maas, Brigham and Women's Hospital, Harvard Medical School, Boston, MA), Ngn2 (kindly provided by Lin Gan, University of Rochester, Rochester, NY), Mash1 (kindly provided by Alexandra Joyner, Memorial Sloan-Kettering Cancer Center New York, NY), Math5 (from Dr. Tom Glaser, University of California, Davis, CA), and Sfrp2 (a generous gift from Andrew McMahon, Harvard University, Boston, MA).
Quantitative Real Time RT-PCR
To compare the expression levels of knock-in Pax6(5a) and Pax2 in embryonic brain at E14.5, they were individually correlated with the endogenous Pax6 expression level in Pax65a/+ and Pax6Pax2/+ mutants by quantitative real time RT-PCR as described previously (34). PCR primers used were as follows: Pax6(5a)/Pax6: F, GCGCAGACGGCATGTATGATA; R, GGGTTGCCCTGGTACTGAAG; Pax2: F, AAGCCCGGAGTGATTGGTG; R, CAGGCGAACATAGTCGGGTT (acquired from Harvard PrimerBank). To determine any differences in expression levels of Pax6 and Pax6(5a), the combined expression levels of Pax6 and Pax6(5a) were compared in wild type and Pax65a/+ embryos. In another experiment, Pax2 knock-in and Pax6 expression levels were compared within Pax6Pax2/+ mutants. The knock-in Pax2 expression levels were calculated by subtracting endogenous Pax2 levels in wild type littermates. To determine the absolute transcript levels, plasmids encoding Pax6 and Pax2 cDNA were used to calibrate Ct values. Gapdh was used as an endogenous control to normalize the Ct values. Three embryos were used for each genotype, and all experiments were run in triplicates. The statistical significance was calculated by Student's t test.
Data Analysis
Quantification of cortical plate and phosphorylated histone H3-positive (pHH3+) cells was performed as described previously (26). Briefly, the quantification of cortical plate was determined in hematoxylin- and eosin-stained E14.5 cerebral cortex sections throughout rostral, intermediate, and caudal levels (40, 41). The length of a line, measured by the ImageJ program, from the ventricular surface to the pial surface served as the total cortical thickness. The length of a second line drawn from the apical and basal side of the cortical plate served as the thickness of the cortical plate. The relative width of the cortical plate was expressed as the proportion of the overall thickness of the cerebral cortex. Quantification of pHH3+ cells at subventricular zone (SVZ) cells was performed by placing a 150-μm-wide square covering the entire cortical thickness parallel to the ventricular surface and counting all pHH3+ cells five or more cell diameters away from the ventricular surface (26). Quantification of the Nkx2.2-positive domain in the developing neural tube was performed by counting the number of cell rows in the neural tube positive for Nkx2.2 immunofluorescence and expressing it as the proportion of the total number of neural tube cell rows in each E10.5 C1-R7 level section (17). The statistical significance was calculated by one-way analysis of variance and Tukey's post hoc tests.
Paired Domain-Glutathione S-Transferase Fusion Protein Preparations
The pGEX-Pax6 and pGEX-Pax6(5a) GST-paired domain expression plasmids were generously provided by Richard Maas (Brigham and Women's Hospital, Harvard Medical School, Boston MA) (24, 30). For pGEX-Pax2 GST, a full-length cDNA clone for murine Pax2, pPax2-CMV (also kindly provided by Richard Maas, Brigham and Women's Hospital, Harvard Medical School, Boston MA) was used as a template in a PCR with primers (F, 5′-CTCGGATCCATGGATATGCACTGCAAAGCAGACC-3′; R, 5′-ATCGAATTCGAACTTTGGTCCGGATGATCCTGTT-3′) to amplify the sequence corresponding to amino acids 1–128 for Pax2. This PCR product was then cloned into the BamHI and EcoRI sites of pGEX4T1, and the resulting clone was verified by direct sequencing. Large scale purification of GST fusion proteins was performed in BL21 Escherichia coli.
Electrophoretic Mobility Shift Assay (EMSA)
EMSA reactions were carried out as described previously (42, 43). The following oligonucleotide probes were used: P6CON, 5′-GATCAGGAAAAATTTTCACGCTTGAGTTCACAG-3′ (24); 5aCON, 5′-GATCCAATGTTCATTGACTCTCGAG-3′ (24); E1.1, 5′-TCATTCACGCCTAGAAGCAG-3′ (44); mtE.1, 5′-TCACTAGTAACGAGAAGCAG-3′ (44); E1.5a, 5′-ATCTCAATCAACAATCCATTAGAACTCA-3′; and mtE1.5a, 5′-TCACTAGTAACGCCATTAGAACTCA. These oligonucleotides were end-radiolabeled by T4 kinase with [γ-32P]ATP prior to annealing oligonucleotides of complimentary sequence.
RESULTS
Generation of Pax65a and Pax6Pax2 Alleles
To investigate the functional specificity of Pax6, we took a cDNA knock-in approach to determine whether Pax6 can be functionally replaced by either Pax2, which shares a similar DNA-binding PD, or Pax6(5a), which shares the same homeodomain and the transactivation domain. The Pax65a/+ allele was generated by fusing a Pax6(5a) cDNA in-frame to the original Pax6 start codon, whereas the Pax6Pax2/+ allele was subsequently derived by Cre-mediated excision of the Pax6(5a) and a stop cassette from the Pax65a/+ allele, allowing the remaining Pax2 cDNA to be expressed (Fig. 1, A–C). In homozygous mutants, Pax6(5a) and Pax2 RNAs were expressed in the endogenous Pax6 spatiotemporal pattern in the eye, telencephalon, and neural tube, whereas in heterozygous eyes (Pax6Pax2/+), ectopic Pax2 expression arose at the expense of the endogenous Pax6 transcripts (Fig. 1, D–K). By quantitative real time PCR, we confirmed that Pax6(5a) and Pax2 were both expressed at a level similar to that of the endogenous Pax6 in Pax65a/+ and Pax6Pax2/+ embryos, respectively (Fig. 1, L and M). Furthermore, no read-through transcription or translation of Pax2 was detected in Pax65a/5a mutants (see Figs. 5, C and G, and 6C). These results supported the correct gene targeting and preservation of major transcriptional regulatory elements in the Pax65a and Pax6Pax2 alleles.
FIGURE 5.

Correction of Nkx2.2 expression by Pax2 and Pax6(5a) in neural tube. A–H, Pax6(5a) and Pax2 were expressed within the endogenous Pax6 neural tube domain in the E10.5 Pax65a/5a and Pax6Pax2/Pax2 mutants, respectively. I–L, the suppression of Nkx2.2 by Pax6 was disrupted in the Pax6-null (Pax6Sey-Neu/Sey-Neu) mutants, allowing for Nkx2.2 expression to expand dorsally (arrows). A significant decrease in Nkx2.2 expansion was observed in the Pax65a/5a and Pax6Pax2/Pax2 mutants. M, measurement of the relative length of Nkx2.2-positive neural tube (*, p < 0.05; n = 3). Scale bar, 100 μm. Error bars represent S.E.
FIGURE 6.

Pax2 and Pax6(5a) partially rescued cell proliferation defects in telencephalon. A–D, an increase in cortical progenitor cell proliferation as indicated by pHH3 staining was evident in the SVZ (arrow) of the Pax6Sey-Neu/Sey-Neu mutants (B). A statistically significant decrease in SVZ cell proliferation was observed in both knock-in mutants. E–H, hematoxylin and eosin staining of E14.5 telencephalon showed a partial rescue of cortical plate (CP) thickness in the Pax6Pax2/Pax2 telencephalon. I, measurements of SVZ proliferation and cortical plate thickness (*, p < 0.05; n = 3). VZ, ventricular zone; CTX, cortex; MZ, marginal zone. Scale bar, 100 μm. Error bars represent S.E.
Pax6(5a), but Not Pax2, Prevents Persistent Lens Stalk in Heterozygous Animals
We first examined Pax65a/+ and Pax6Pax2/+ embryos to determine whether they resembled Pax6 heterozygous nulls. At E14.5, Pax6 staining revealed clear separations between lens and cornea in both wild type and Pax65a/+ embryos (Fig. 2, A and B, arrows). In Pax6Pax2/+ embryos, however, there still existed a persistent corneal-lenticular stalk (Fig. 2C, arrow), a known ocular defect in Pax6Seu/+ mutants. Consistent with this, Pax6 downstream gene Foxe3 was only modestly reduced in Pax65a/+ embryos but was nearly absent in Pax6Pax2/+ lens (Fig. 2, D–F, arrows) (45). In contrast, the budding of lacrimal gland and retinal expression of Math5, two features also known to be sensitive to Pax6 gene dosage, were both disrupted in Pax65a/+ and Pax6Pax2/+ embryos (Fig. 2, G–L, arrows) (34, 46, 47). Therefore, Pax6(5a), but not Pax2, partially substituted for Pax6 function in lens development.
FIGURE 2.

Partial rescue of Pax6 heterozygous lens phenotype by Pax6(5a) but not by Pax2. A–C, persistent corneal-lenticular stalk (arrow in C) was present only in E14.5 Pax6Pax2/+ mutants but not in wild type and Pax65a/+ eyes. D–F, Foxe3 expression (arrows) was reduced more significantly in Pax6Pax2/+ mutants than in Pax65a/+ eyes. G–L, both Pax6Pax2/+ and Pax65a/+ mutants displayed smaller lacrimal gland buds (outlined in white dashed lines) and reduced expression of retinal differentiation gene Math5 (arrows). Scale bar, 100 μm.
Failure of Lens Induction in the Pax65a/5a and Pax6Pax2/Pax2 Embryos
The Pax65a/5a and Pax6Pax2/Pax2 embryos were recovered at the expected Mendelian ratio at E15.5, but they lacked any obvious eye structures (Fig. 3, A–C, arrows). This prompted us to examine lens induction, the pivotal morphogenetic event in early eye development. The murine embryonic eye develops in response to signaling interactions between two Pax6-expressing embryonic tissues, the head surface ectoderm and optic vesicle. After coming in physical contact with the optic vesicle at E9.5, the wild type surface ectoderm thickened to form the presumptive lens placode where Pax6 up-regulated its own expression (Fig. 3D, arrow). In contrast, no Pax6-positive lens placode was observed in the Pax65a/5a and Pax6Pax2/Pax2 mutants, indicating a failure of the Pax6 autoregulatory loop (Fig. 3, E–F, arrows). As a consequence, the expression of Sox2, a Pax6 downstream transcription factor in lens placode development, was also extinguished (Fig. 3, G–I, arrows). At E10.5 when the wild type lens placode invaginated to form a lens vesicle, the Pax65a/5a and Pax6Pax2/Pax2 mutants failed to form any lens structures, and Pax6, Pax2, and Sox2 were completely absent on the remaining surface ectoderm (Fig. 3, J–R, arrows). It is important to note that Pax2 expression was still detected in Pax6Pax2/+ lens (Fig. 1K, arrow), indicating that our knock-in alleles did not lose any essential lens enhancer. Taken together, these results demonstrated that Pax6(5a) and Pax2 were unable to activate the Pax6 autoregulatory mechanism in the presumptive lens placode, which led to the loss of lens development.
FIGURE 3.

Lens induction failure in Pax65a/5a and Pax6Pax2/Pax2 knock-in mutants. A–C, no obvious eye structure was observed in the E15.5 Pax65a/5a and Pax6Pax2/Pax2 mutants (arrows). D–F, using an antibody that recognized both Pax6 and Pax6(5a) protein, we detected Pax6 in the wild type lens placode (thickening of lens surface ectoderm) but not Pax6(5a) in the Pax65a/5a mutant surface ectoderm (compare D and E, arrows). No lens placode was formed in either E9.5 Pax65a/5a or Pax6Pax2/Pax2 mutants. ov, optic vesicle; lp, lens placode; le, lens ectoderm. G–I, Sox2 expression was absent from lens surface ectoderm (arrows) but present in developing optic vesicles of both knock-in mutants at E9.5 (arrowheads). J–R, in both knock-in mutants at E10.5, lens vesicle and optic cup failed to form, and Pax6, Pax6(5a), Pax2, and Sox2 expressions were absent from lens surface ectoderm (arrows). lv, lens vesicle; oc, optic cup. Scale bar, 50 μm.
Retinal Development Is Disrupted in the Pax65a/5a and Pax6Pax2/Pax2 Mutants
Previous studies have shown that retinal differentiation was initially accelerated in the Pax6-null mutants, but the neurogenesis was eventually aborted (15, 16). Thus, we examined whether retinogenesis still occurred in the Pax65a/5a and Pax6Pax2/Pax2 mutants. In the E14.5 wild type embryos, Pax6 was detectable throughout the neural retina, whereas Pax2 was restricted to the optic stalk and the optic disc (Fig. 4, A and B, arrow and arrowhead). In contrast, the Pax65a/5a optic vesicle expressed both Pax6(5a) and Pax2 (Fig. 4, A′ and B′), whereas the Pax6Pax2/Pax2 optic vesicle only expressed Pax2 (Fig. 4, A″ and B″). Furthermore, both mutant optic vesicles were positive for Sox2, here a retinal progenitor marker, but negative for Mitf, a retinal pigmented epithelium marker (Fig. 4, C–D″). These results indicated that the patterning of optic vesicle into retinal pigmented epithelium, optic disc, and neural retina was abolished in the Pax65a/5a and Pax6Pax2/Pax2 mutants. We also provide evidence that neurogenesis was disrupted in the mutants. Ptf1a is a transcription factor important for retinal amacrine and horizontal cells. Its expression was completely lost in the Pax65a/5a and Pax6Pax2/Pax2 mutant optic vesicles (Fig. 4, E–E″). Brn3a and NF165, markers for newly differentiated ganglion cells, were similarly abrogated (Fig. 4, F–G″). Islet1, which marks differentiating retinal ganglion and amacrine cells, was also absent in the mutants (Fig. 4, H–H″). It has been shown previously that the Pax6-null retina still expresses the photoreceptor determination gene Crx and amacrine cell differentiation marker VC1.1 (15). Interestingly, we did not detect any VC1.1 expression in our mutants, but Crx expression was indeed expanded to most of the residual optic vesicles (Fig. 4, I–J″, arrows). Taken together, these ocular defects demonstrate the strict requirement of canonical Pax6 for lens and retinal development.
FIGURE 4.

Defective retinal development in Pax65a/5a and Pax6Pax2/Pax2 mutants. A–B″, Pax6(5a) and Pax2 were expressed in the residual Pax65a/5a and Pax6Pax2/Pax2optic vesicles (arrowhead). nr, neural retina; le, lens; os, optic stalk; od, optic disc. C–D″, entire mutant optic vesicles (dashed lines) expressed neural retinal marker Sox2 but not retinal pigmented epithelium marker Mitf. E–J″, the retinal differentiation factors Ptf1a, Brn3a, NF165, Islet1, and VC1.1 were lost in the mutant optic vesicles (dashed lines), but photoreceptor cell differentiation gene Crx was ectopically expressed (arrows). Scale bar, 100 μm.
Pax6(5a) and Pax2 Partially Substitute for Pax6 in Suppressing Ectopic Nkx2.2 Expansion in the Neural Tube
Considering that Pax6 is required for patterning the ventral neural tube, we next asked whether Pax6(5a) or Pax2 could recapitulate the role of Pax6 as a repressor of homeodomain transcription factor Nkx2.2 expression (17, 48). By E10.5, wild type Pax6 expression was detected in the ventral neural tube, whereas Pax2 was expressed in the neural tube bilaterally flanking the dorsoventral midline (Fig. 5, A and E). This was in contrast to the ectopic expression of Pax6(5a) and Pax2 within the neural tube-Pax6 expression domains in the corresponding knock-in mutants (Fig. 5, C, D, and F–H). As expected, Nkx2.2 expression expanded dorsally in the Pax6-null (Pax6Sey-Neu/Sey-Neu) mutants, confirming the antagonistic interaction between Pax6 and Nkx2.2 (Fig. 5, I and J, arrows). In both Pax65a/5a and Pax6Pax2/Pax2 mutants, however, the dorsal expansion of Nkx2.2 appeared to be partially reduced compared with the Pax6-null mutants (Fig. 5, K and L, arrows). By calculating the proportion of Nkx2.2-positive cells per total number of neural tube cells, we confirmed that the suppression of ectopic Nkx2.2 expression was statistically significant for Pax65a/5a and Pax6Pax2/Pax2 mutants (Fig. 5M).
Partial Rescue of Telencephalic Cell Proliferation and Neurogenesis by Pax2
Transgenic overexpression studies suggest that Pax6 and Pax6(5a) both have antiproliferative roles in the embryonic cortex (26, 28). To assess any changes in cortex cell proliferation in our mutants, mitotic cells in E14.5 coronal cortex sections were immunolabeled with anti-pHH3. Consistent with previous studies, E14.5 Pax6Sey-Neu/Sey-Neu mutants exhibited a significant increase in cell proliferation within the SVZ compared with the wild type cortex, whereas the ventricular zone mitotic index remained the same (Fig. 6, A and B, arrows) (26). However, there was a statistically significant reduction in the number of pHH3-positive subventricular zone cells in both Pax65a/5a and Pax6Pax2/Pax2 mutants compared with the Pax6-null mutants (Pax6Sey-Neu/Sey-Neu), indicating a partial rescue of telencephalic progenitor cell inhibition (Fig. 6, C, D, and I). After terminal cell division, newborn neurons migrate to beneath the pial surface to form a conspicuous band called the cortical plate (Fig. 6, E–H, telencephalic area flanked by dotted lines). Relative to the total cortical thickness in the lateral cortex, the cortical plates in the Pax6Sey-Neu/Sey-Neu and the Pax65a/5a mutants were both thinner than that of the wild type embryos. The Pax6Pax2/Pax2 mutants, however, exhibited a significant increase in cortical thickness, suggesting that, unlike Pax(5a), Pax2 could partially rescue neurogenesis in the Pax6-deficient cortex (Fig. 6I).
Stronger Rescue of Telencephalon Patterning in Pax6Pax2/Pax2 Mutants than in Pax65a/5a Mutants
Given the vital role of Pax6 in the dorsoventral specification of telencephalic progenitors, we next examined the expression patterns of three well characterized Pax6 downstream targets within the developing telencephalon at E14, namely Ngn2, Mash1, and Sfrp2. Pax6 expression was normally confined to the dorsal E14.5 telencephalon in the wild type embryos as was the expression of Pax6(5a) and Pax2 in the Pax65a/5a and the Pax6Pax2/Pax2 mutants, respectively (Fig. 7, A–D, arrows). Consistent with previous reports, we observed in the Pax6Sey-Neu/Sey-Neu mutants a complete abrogation of telencephalic Ngn2 expression in addition to an expanded expression of the ventral telencephalon-specific transcription factor Mash1 into the dorsal telencephalon (Fig. 7, F and J, arrows) (26). In the Pax65a/5a mutants, however, there was a modest recovery of Ngn2 expression in the dorsal telencephalon, but Mash1 was still ectopically expressed (Fig. 7, G and K, arrows). Remarkably, Mash1 expression in the Pax6Pax2/Pax2 mutant was correctly confined to the ventral telencephalon despite the lack of endogenous Pax6 expression, whereas Ngn2 was strongly expressed in the dorsal telencephalon (Fig. 7, H and L, arrow and arrowhead). Of note, Sfrp2, a boundary marker between the dorsal cortex and ventral ganglionic eminence, was still absent in the Pax65a/5a and Pax6Pax2/Pax2 mutants, suggesting an incomplete rescue of telencephalic patterning (Fig. 7, M–P, arrowheads). Nevertheless, it is clear that Pax2 and to a lesser extent Pax6(5a) can at least substitute for some of the canonical Pax6 functions in the regionalization of the forebrain telencephalon.
FIGURE 7.

Differential rescue of dorsoventral patterning in Pax65a/5a and Pax6Pax2/Pax2 mutant telencephalon. A–D, Pax2 and Pax6 immunohistochemistry confirmed endogenous and ectopic protein expression within the dorsal telencephalon at E15.5 (arrows). E–H, Pax6-proneural gene target Ngn2 RNA expression was lost in the Pax6Sey-Neu/Sey-Neu mutant dorsal telencephalon but partially recovered in the Pax65a/5a mutant and fully rescued in the Pax6Pax2/Pax2 mutant (arrows). I–L, Pax6Sey-Neu/Sey-Neu and Pax65a/5a mutants showed misexpression of proneural gene Mash1 in the dorsal telencephalon (arrows), whereas Pax6Pax2/Pax2 displayed normal Mash1 expression in ventral telencephalon (arrowheads). M–P, Wnt inhibitor Sfrp2, normally expressed at telencephalon dorsal-ventral boundary (arrowheads), was not detected in any of the mutants. All sections were coronal. Scale bar, 100 μm.
Differential Binding of Pax6(5a) and Pax2 Paired Domains on Ngn2 E1 Enhancer
To understand the mechanism of differential rescue in the Pax65a/5a and Pax6Pax2/Pax2 mutants, we next investigated the regulation of Ngn2, one of the best characterized Pax6 downstream targets in neural development. Previous studies have shown that the canonical Pax6 isoform through its PD binds to a low affinity site on the Ngn2 E1 enhancer, which promotes Ngn2 expression only in the telencephalic domains of high Pax6 expression (44). To assess whether there could exist a direct physical interaction between the Ngn2 E1 enhancer and Pax6(5a) or Pax2 PD, we performed DNA EMSAs using oligonucleotide probes containing sequences corresponding to the Pax6 binding sites. As expected, the P6CON and 5aCON control probes, which contained previously identified consensus binding site sequences, strongly bound GST-Pax6-PD and GST-Pax6(5a)-PD respectively (Fig. 8A) (24, 30). In addition, we confirmed the previous finding that GST-Pax6-PD weakly bound the Ngn2 E1 enhancer element probe E1.1 but not the mutated E1 enhancer probe mtE1.1 (Fig. 8A) (44). Although GST-Pax6-PD could also recognize the 5aCON control probe, GST-Pax6(5a)-PD was unable to bind the P6CON site effectively (Fig. 8A and data not shown). Consistent with this, we did not observe any binding between GST-Pax6(5a)-PD and the E1.1 element, confirming that the E1.1 sequence was indeed a canonical Pax6 PD binding site. Nevertheless, in the same Ngn2 E1 enhancer, we identified a putative Pax6(5a)-specific binding sequence, named E1.1(5a), that is evolutionally conserved from human to frog (Xenopus tropicalis) (Fig. 8B). By EMSA, we observed that GST-Pax6(5a)-PD could bind to probe E1.1(5a) but not to the mutated negative control probe mtE1.1(5a), although the overall binding was weaker than that of GST-Pax6-PD on probe E1.1 (Fig. 8A). This suggested that Pax6(5a) could potentially utilize the E1.1(5a) site, but the interaction was likely weaker than that of canonical Pax6 on the Ngn2 E1 enhancer. Finally, we showed that the GST-Pax2-PD bound to P6CON and 5aCON sequences with affinities similar to that of GST-Pax6-PD, whereas control GST protein failed to bind any of these probes, demonstrating that Pax6 and Pax2 PD indeed share the same DNA binding specificity (Fig. 8A). Importantly, the binding of GST-Pax2-PD on E1.1 was also significantly stronger than that of GST-Pax6(5a)-PD on E1.1(5a). Taken together, these results showed that Pax6 and Pax2 PDs formed a stronger binding complex on the Ngn2 E1 enhancer than did Pax6(5a) PD, which correlated well with the differential level of Ngn2 expression in the Pax65a/5a and Pax6Pax2/Pax2 mutant telencephalon.
FIGURE 8.
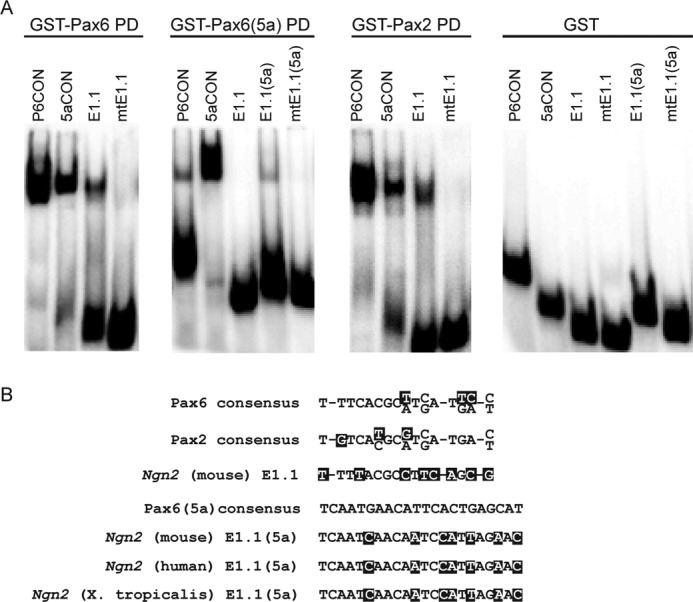
Pax6(5a) and Pax2 paired domains bound the Ngn2 telencephalic enhancer. A, electrophoretic mobility shift assays performed with the recombinant Pax6, Pax6(5a), and Pax2 PD-GST fusion proteins. Pax6 and Pax2 paired domains bound the Pax6 and Pax6(5a) consensus binding sites (P6CON and 5aCON) and with a weaker affinity the Ngn2 E1.1 enhancer element. Pax6(5a) paired domain preferentially bound 5aCON and the Ngn2 E1.1(5a) site. The Pax6(5a)-E1.1(5a) binding was weaker than that of Pax2-E1.1. No binding complexes were observed with GST and the individual oligonucleotide probes. B, Pax2 and Pax6 paired domain consensus binding sites as well as the corresponding Ngn2 enhancer E1.1 target sequence. The Pax6(5a) consensus binding site and its phylogenetically conserved binding sequence within the Ngn2 E1.1 enhancer are shown.
DISCUSSION
In this study, we performed gene replacement experiments to rigorously test the functional specificity of Pax6, Pax6(5a), and Pax2 in vivo. In both the Pax65a/5a and Pax6Pax2/Pax2 mutants, lens development was abolished because lens induction never occurred. Further analysis showed that Pax2 and Pax6(5a) failed to be expressed in the lens placode, demonstrating that the lens-specific enhancer activity in the Pax6 locus required canonical Pax6. The known Pax6 lens enhancer, also termed the ectoderm enhancer, contains binding sites for Meis/Prep, Sox2, Oct1, and Pax6 (34, 43, 49–51). Because the recombinant Pax2 PD bound as strongly as Pax6 PD on this lens enhancer in EMSA (data not shown), the lack of lens enhancer activity in the Pax6Pax2/Pax2 mutants suggests that Pax2 may be unable to synergistically interact with the other cofactors on this enhancer. Another possibility was that the Pax6 HD was also required for its autoregulation in lens development, which was supported by the previous finding that mutations in the Pax6 HD disrupted eye but not brain development (52). In support of this idea, we observed persistent lens stalk in Pax6Pax2/+, but not in Pax65a/+, embryos, suggesting that Pax6(5a) could partially substitute for canonical Pax6 in lens development. However, it should be noted that the Pax6 lens enhancer remained inactive in the Pax65a/5a mutants despite the intact HD in Pax6(5a). Therefore, it is likely that both the PD and HD of Pax6 were necessary for its optimum function in lens induction.
In retinal development, Pax2 and Pax6 have been shown to cooperate initially in retinal pigmented epithelium specification, but later on, they play antagonistic roles in optic stalk and optic cup development, respectively (31–33). In the Pax6Pax2/Pax2 mutants, however, the residual optic vesicle expressed the neural retina markers Sox2 and Crx but not the retinal pigmented epithelium marker Mitf. This suggested that, without Pax6, Pax2 alone could not maintain retinal pigmented epithelium or optic stalk fate. On the other hand, although ectopic Pax6(5a) could induce well differentiated retina in chick, replacement of Pax6 by Pax6(5a) in the Pax65a/5a mutants failed to correct the retinal differentiation observed in the Pax6-null mutant. Together with the lens development failure in our mutants, these results showed that canonical Pax6 was uniquely required for mammalian eye development.
Although Pax2 and Pax6(5a) failed to reverse Pax6 homozygous null ocular defects, our studies showed that they could partially replace Pax6 for telencephalon and neural tube development. In the Pax65a/5a and Pax6Pax2/Pax2 mutants, we showed that Ngn2 expression was reactivated in dorsal telencephalon, and Nkx2.2 was indeed suppressed in ventral neural tube. Thus, contrary to the previous gain-of-function studies that indicated that Pax6(5a) only affected cell proliferation in telencephalon, our genetic knock-in experiments supported that Pax6(5a) could complement Pax6 in neural patterning and differentiation. In brain development, Pax6, Pax6(5a), and Pax2 likely share a common set of downstream neurogenesis genes recognizable by their PDs. This is because even Pax2, which lacks a functional HD, could rescue Ngn2 and Nkx2.2 expression in the Pax6Pax2/Pax2 mutants. On the other hand, Pax6(5a) and Pax2 were still unable to activate Sfrp2 at the dorsoventral boundary, consistent with the previous reports that Sfrp2 regulation required both the PD and the HD of Pax6 (26, 53). Furthermore, we were able to identify an evolutionarily conserved binding site for the Pax6(5a) PD in the Ngn2 telencephalic enhancer and showed that the in vitro binding affinities of Pax6(5a) and Pax2 PD correlate well with the Ngn2 expression level. Therefore, at least for Ngn2, the strength of the PD binding dictates the functional activity of Pax6, Pax6(5a), and Pax2 in vivo.
The diversification of the ancestral Pax gene into Pax6, Pax6(5a), and Pax2 accompanied the arise of sophisticated vision systems in both invertebrates and vertebrates. We have shown that although Pax6, Pax6(5a), and Pax2 are partially interchangeable in brain development where their HDs are dispensable, Pax6(5a) and Pax2 cannot substitute for Pax6 in eye development where PD and HD are both required. This is in contrast with a previous knock-in study of the Pax2/5/8 subfamily of Pax genes that demonstrated the complete functional equivalence between Pax2 and Pax5 (54). Most of the previous efforts in determining Pax6 downstream targets have focused on its well characterized PD binding site. Our gene replacement studies suggest that it is essential to study the combinatorial activities of PD and HD to understand the unique function of Pax6 in neural development.
Acknowledgments
We thank Dr. James Li for the Pax6Sey-Neu/Sey-Neu mice and Drs. Lin Gan, Tom Glaser, Jane E. Johnson, Alexandra Joyner, Richard Maas, Andrew McMahon, Milan Jamrich, and Valerie Wallace for reagents.
This work was supported, in whole or in part, by National Institutes of Health Grants EY017061 and EY018868 (to X. Z.).
- Pax
- paired box
- PD
- paired domain
- HD
- homeodomain
- SVZ
- subventricular zone
- R
- reverse
- F
- forward
- pHH3
- phosphorylated histone H3
- E
- embryonic day.
REFERENCES
- 1. Kozmik Z. (2005) Pax genes in eye development and evolution. Curr. Opin. Genet. Dev. 15, 430–438 [DOI] [PubMed] [Google Scholar]
- 2. Fu W., Noll M. (1997) The Pax2 homolog sparkling is required for development of cone and pigment cells in the Drosophila eye. Genes Dev. 11, 2066–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Quiring R., Walldorf U., Kloter U., Gehring W. J. (1994) Homology of the eyeless gene of Drosophila to the Small eye gene in mice and Aniridia in humans. Science 265, 785–789 [DOI] [PubMed] [Google Scholar]
- 4. Jang C. C., Chao J. L., Jones N., Yao L. C., Bessarab D. A., Kuo Y. M., Jun S., Desplan C., Beckendorf S. K., Sun Y. H. (2003) Two Pax genes, eye gone and eyeless, act cooperatively in promoting Drosophila eye development. Development 130, 2939–2951 [DOI] [PubMed] [Google Scholar]
- 5. Dominguez M., Ferres-Marco D., Gutierrez-Aviño F. J., Speicher S. A., Beneyto M. (2004) Growth and specification of the eye are controlled independently by Eyegone and Eyeless in Drosophila melanogaster. Nat. Genet. 36, 31–39 [DOI] [PubMed] [Google Scholar]
- 6. Punzo C., Kurata S., Gehring W. J. (2001) The eyeless homeodomain is dispensable for eye development in Drosophila. Genes Dev. 15, 1716–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Azuma N., Tadokoro K., Asaka A., Yamada M., Yamaguchi Y., Handa H., Matsushima S., Watanabe T., Kohsaka S., Kida Y., Shiraishi T., Ogura T., Shimamura K., Nakafuku M. (2005) The Pax6 isoform bearing an alternative spliced exon promotes the development of the neural retinal structure. Hum. Mol. Genet. 14, 735–745 [DOI] [PubMed] [Google Scholar]
- 8. Glaser T., Walton D. S., Maas R. L. (1992) Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat. Genet. 2, 232–239 [DOI] [PubMed] [Google Scholar]
- 9. Grindley J. C., Davidson D. R., Hill R. E. (1995) The role of Pax-6 in eye and nasal development. Development 121, 1433–1442 [DOI] [PubMed] [Google Scholar]
- 10. Hill R. E., Favor J., Hogan B. L., Ton C. C., Saunders G. F., Hanson I. M., Prosser J., Jordan T., Hastie N. D., van Heyningen V. (1991) Mouse small eye results from mutations in a paired-like homeobox-containing gene. Nature 354, 522–525 [DOI] [PubMed] [Google Scholar]
- 11. Jordan T., Hanson I., Zaletayev D., Hodgson S., Prosser J., Seawright A., Hastie N., van Heyningen V. (1992) The human PAX6 gene is mutated in two patients with aniridia. Nat. Genet. 1, 328–332 [DOI] [PubMed] [Google Scholar]
- 12. Ashery-Padan R., Marquardt T., Zhou X., Gruss P. (2000) Pax6 activity in the lens primordium is required for lens formation and for correct placement of a single retina in the eye. Genes Dev. 14, 2701–2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shaham O., Smith A. N., Robinson M. L., Taketo M. M., Lang R. A., Ashery-Padan R. (2009) Pax6 is essential for lens fiber cell differentiation. Development 136, 2567–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marquardt T., Ashery-Padan R., Andrejewski N., Scardigli R., Guillemot F., Gruss P. (2001) Pax6 is required for the multipotent state of retinal progenitor cells. Cell 105, 43–55 [DOI] [PubMed] [Google Scholar]
- 15. Oron-Karni V., Farhy C., Elgart M., Marquardt T., Remizova L., Yaron O., Xie Q., Cvekl A., Ashery-Padan R. (2008) Dual requirement for Pax6 in retinal progenitor cells. Development 135, 4037–4047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Philips G. T., Stair C. N., Young Lee H., Wroblewski E., Berberoglu M. A., Brown N. L., Mastick G. S. (2005) Precocious retinal neurons: Pax6 controls timing of differentiation and determination of cell type. Dev. Biol. 279, 308–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ericson J., Rashbass P., Schedl A., Brenner-Morton S., Kawakami A., van Heyningen V., Jessell T. M., Briscoe J. (1997) Pax6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell 90, 169–180 [DOI] [PubMed] [Google Scholar]
- 18. Stoykova A., Fritsch R., Walther C., Gruss P. (1996) Forebrain patterning defects in Small eye mutant mice. Development 122, 3453–3465 [DOI] [PubMed] [Google Scholar]
- 19. Chapouton P., Gärtner A., Götz M. (1999) The role of Pax6 in restricting cell migration between developing cortex and basal ganglia. Development 126, 5569–5579 [DOI] [PubMed] [Google Scholar]
- 20. Nomura T., Osumi N. (2004) Misrouting of mitral cell progenitors in the Pax6/small eye rat telencephalon. Development 131, 787–796 [DOI] [PubMed] [Google Scholar]
- 21. Carić D., Gooday D., Hill R. E., McConnell S. K., Price D. J. (1997) Determination of the migratory capacity of embryonic cortical cells lacking the transcription factor Pax-6. Development 124, 5087–5096 [DOI] [PubMed] [Google Scholar]
- 22. Estivill-Torrus G., Pearson H., van Heyningen V., Price D. J., Rashbass P. (2002) Pax6 is required to regulate the cell cycle and the rate of progression from symmetrical to asymmetrical division in mammalian cortical progenitors. Development 129, 455–466 [DOI] [PubMed] [Google Scholar]
- 23. Götz M., Stoykova A., Gruss P. (1998) Pax6 controls radial glia differentiation in the cerebral cortex. Neuron 21, 1031–1044 [DOI] [PubMed] [Google Scholar]
- 24. Epstein J. A., Glaser T., Cai J., Jepeal L., Walton D. S., Maas R. L. (1994) Two independent and interactive DNA-binding subdomains of the Pax6 paired domain are regulated by alternative splicing. Genes Dev. 8, 2022–2034 [DOI] [PubMed] [Google Scholar]
- 25. Mikkola I., Bruun J. A., Holm T., Johansen T. (2001) Superactivation of Pax6-mediated transactivation from paired domain-binding sites by DNA-independent recruitment of different homeodomain proteins. J. Biol. Chem. 276, 4109–4118 [DOI] [PubMed] [Google Scholar]
- 26. Haubst N., Berger J., Radjendirane V., Graw J., Favor J., Saunders G. F., Stoykova A., Götz M. (2004) Molecular dissection of Pax6 function: the specific roles of the paired domain and homeodomain in brain development. Development 131, 6131–6140 [DOI] [PubMed] [Google Scholar]
- 27. Singh S., Mishra R., Arango N. A., Deng J. M., Behringer R. R., Saunders G. F. (2002) Iris hypoplasia in mice that lack the alternatively spliced Pax6(5a) isoform. Proc. Natl. Acad. Sci. U.S.A. 99, 6812–6815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Berger J., Berger S., Tuoc T. C., D'Amelio M., Cecconi F., Gorski J. A., Jones K. R., Gruss P., Stoykova A. (2007) Conditional activation of Pax6 in the developing cortex of transgenic mice causes progenitor apoptosis. Development 134, 1311–1322 [DOI] [PubMed] [Google Scholar]
- 29. Lang D., Powell S. K., Plummer R. S., Young K. P., Ruggeri B. A. (2007) PAX genes: roles in development, pathophysiology, and cancer. Biochem. Pharmacol. 73, 1–14 [DOI] [PubMed] [Google Scholar]
- 30. Epstein J., Cai J., Glaser T., Jepeal L., Maas R. (1994) Identification of a Pax paired domain recognition sequence and evidence for DNA-dependent conformational changes. J. Biol. Chem. 269, 8355–8361 [PubMed] [Google Scholar]
- 31. Bäumer N., Marquardt T., Stoykova A., Spieler D., Treichel D., Ashery-Padan R., Gruss P. (2003) Retinal pigmented epithelium determination requires the redundant activities of Pax2 and Pax6. Development 130, 2903–2915 [DOI] [PubMed] [Google Scholar]
- 32. Bharti K., Gasper M., Ou J., Brucato M., Clore-Gronenborn K., Pickel J., Arnheiter H. (2012) A regulatory loop involving PAX6, MITF, and WNT signaling controls retinal pigment epithelium development. PLoS Genet. 8, e1002757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schwarz M., Cecconi F., Bernier G., Andrejewski N., Kammandel B., Wagner M., Gruss P. (2000) Spatial specification of mammalian eye territories by reciprocal transcriptional repression of Pax2 and Pax6. Development 127, 4325–4334 [DOI] [PubMed] [Google Scholar]
- 34. Carbe C., Hertzler-Schaefer K., Zhang X. (2012) The functional role of the Meis/Prep-binding elements in Pax6 locus during pancreas and eye development. Dev. Biol. 363, 320–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu P., Jenkins N. A., Copeland N. G. (2003) A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 13, 476–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Glaser T., Jepeal L., Edwards J. G., Young S. R., Favor J., Maas R. L. (1994) PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat. Genet. 7, 463–471 [DOI] [PubMed] [Google Scholar]
- 37. Pan Y., Carbe C., Powers A., Feng G. S., Zhang X. (2010) Sprouty2-modulated Kras signaling rescues Shp2 deficiency during lens and lacrimal gland development. Development 137, 1085–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pan Y., Carbe C., Powers A., Zhang E. E., Esko J. D., Grobe K., Feng G. S., Zhang X. (2008) Bud specific N-sulfation of heparan sulfate regulates Shp2-dependent FGF signaling during lacrimal gland induction. Development 135, 301–310 [DOI] [PubMed] [Google Scholar]
- 39. Pan Y., Woodbury A., Esko J. D., Grobe K., Zhang X. (2006) Heparan sulfate biosynthetic gene Ndst1 is required for FGF signaling in early lens development. Development 133, 4933–4944 [DOI] [PubMed] [Google Scholar]
- 40. Schmahl W., Knoedlseder M., Favor J., Davidson D. (1993) Defects of neuronal migration and the pathogenesis of cortical malformations are associated with Small eye (Sey) in the mouse, a point mutation at the Pax-6-locus. Acta Neuropathol. 86, 126–135 [DOI] [PubMed] [Google Scholar]
- 41. Guénette S., Chang Y., Hiesberger T., Richardson J. A., Eckman C. B., Eckman E. A., Hammer R. E., Herz J. (2006) Essential roles for the FE65 amyloid precursor protein-interacting proteins in brain development. EMBO J. 25, 420–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang X., Rowan S., Yue Y., Heaney S., Pan Y., Brendolan A., Selleri L., Maas R. L. (2006) Pax6 is regulated by Meis and Pbx homeoproteins during pancreatic development. Dev. Biol. 300, 748–757 [DOI] [PubMed] [Google Scholar]
- 43. Zhang X., Friedman A., Heaney S., Purcell P., Maas R. L. (2002) Meis homeoproteins directly regulate Pax6 during vertebrate lens morphogenesis. Genes Dev. 16, 2097–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scardigli R., Bäumer N., Gruss P., Guillemot F., Le Roux I. (2003) Direct and concentration-dependent regulation of the proneural gene Neurogenin2 by Pax6. Development 130, 3269–3281 [DOI] [PubMed] [Google Scholar]
- 45. Brownell I., Dirksen M., Jamrich M. (2000) Forkhead Foxe3 maps to the dysgenetic lens locus and is critical in lens development and differentiation. Genesis 27, 81–93 [DOI] [PubMed] [Google Scholar]
- 46. Makarenkova H. P., Ito M., Govindarajan V., Faber S. C., Sun L., McMahon G., Overbeek P. A., Lang R. A. (2000) FGF10 is an inducer and Pax6 a competence factor for lacrimal gland development. Development 127, 2563–2572 [DOI] [PubMed] [Google Scholar]
- 47. Brown N. L., Kanekar S., Vetter M. L., Tucker P. K., Gemza D. L., Glaser T. (1998) Math5 encodes a murine basic helix-loop-helix transcription factor expressed during early stages of retinal neurogenesis. Development 125, 4821–4833 [DOI] [PubMed] [Google Scholar]
- 48. Briscoe J., Sussel L., Serup P., Hartigan-O'Connor D., Jessell T. M., Rubenstein J. L., Ericson J. (1999) Homeobox gene Nkx2.2 and specification of neuronal identity by graded Sonic hedgehog signalling. Nature 398, 622–627 [DOI] [PubMed] [Google Scholar]
- 49. Aota S., Nakajima N., Sakamoto R., Watanabe S., Ibaraki N., Okazaki K. (2003) Pax6 autoregulation mediated by direct interaction of Pax6 protein with the head surface ectoderm-specific enhancer of the mouse Pax6 gene. Dev. Biol. 257, 1–13 [DOI] [PubMed] [Google Scholar]
- 50. Donner A. L., Ko F., Episkopou V., Maas R. L. (2007) Pax6 is misexpressed in Sox1 null lens fiber cells. Gene Expr. Patterns 7, 606–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rowan S., Siggers T., Lachke S. A., Yue Y., Bulyk M. L., Maas R. L. (2010) Precise temporal control of the eye regulatory gene Pax6 via enhancer-binding site affinity. Genes Dev. 24, 980–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Favor J., Peters H., Hermann T., Schmahl W., Chatterjee B., Neuhäuser-Klaus A., Sandulache R. (2001) Molecular characterization of Pax6(2Neu) through Pax6(10Neu): an extension of the Pax6 allelic series and the identification of two possible hypomorph alleles in the mouse Mus musculus. Genetics 159, 1689–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim A. S., Anderson S. A., Rubenstein J. L., Lowenstein D. H., Pleasure S. J. (2001) Pax-6 regulates expression of SFRP-2 and Wnt-7b in the developing CNS. J. Neurosci. 21, RC132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bouchard M., Pfeffer P., Busslinger M. (2000) Functional equivalence of the transcription factors Pax2 and Pax5 in mouse development. Development 127, 3703–3713 [DOI] [PubMed] [Google Scholar]