

Background: Although antagonism of LINGO-1 is known to improve neuronal survival after injury, the mechanism is unknown.
Results: LINGO-1 interacts with and inhibits WNK3 kinase activity, thereby facilitating neuronal apoptosis.
Conclusion: LINGO-1 potentiates neuronal apoptosis, likely by inhibiting WNK3 kinase activity.
Significance: Learning how LINGO-1/WNK3 signaling regulates neuronal survival is crucial for understanding the pathology of neurodegenerative diseases and injury.
Keywords: Neurobiology, Neurodegeneration, Neurodevelopment, Neurons, Neuroprotection, LINGO-1, WNK3, Apoptosis, Interaction
Abstract
LINGO-1 is a functional component of the Nogo receptor 1·p75NTR·LINGO-1 and Nogo receptor 1·TAJ (TNFRSF19/TROY)·LINGO-1 signaling complexes. It has recently been shown that LINGO-1 antagonists significantly improve neuronal survival after neural injury. However, the mechanism by which LINGO-1 signaling influences susceptibility to apoptosis remains unknown. In an effort to better understand how LINGO-1 regulates these signaling pathways, we used an established model of serum deprivation (SD) to induce neuronal apoptosis. We demonstrate that treatment either with a construct containing the intracellular domain of LINGO-1 or with Nogo66, a LINGO-1 receptor complex agonist, resulted in an enhanced rate of apoptosis in primary cultured cortical neurons under SD. Reducing the expression levels of the serine/threonine kinase WNK3 using shRNA or inhibiting its kinase activity had similar effects on the survival of serum-deprived neurons. Consistent with these observations, we found that LINGO-1 and WNK3 co-localized and co-precipitated in cultured cortical neurons and brain tissue. Significantly, this co-association was enhanced by Nogo66 treatment. Binding of WNK3 to the intracellular domain of LINGO-1 led to a reduction in WNK3 kinase activity, as did Nogo66 stimulation. Moreover, in vitro and in vivo evidence indicates that endogenous WNK3 suppresses SD-induced neuronal apoptosis in a kinase-dependent manner, as the expression of either a WNK3 RNAi construct or a kinase-dead N-terminal fragment of WNK3 led to increased apoptosis. Taken together, our results show that LINGO-1 potentiates neuronal apoptosis, likely by inhibiting WNK3 kinase activity.
Introduction
After injury to the central nervous system, myelin-associated inhibitory factors inhibit cellular and axonal regeneration, resulting in permanent disability. The three major myelin-associated inhibitory factors, Nogo, oligodendrocyte myelin glycoprotein, and myelin-associated glycoprotein, share a trimolecular receptor complex composed of Nogo receptor 1 and its co-receptors p75NTR (neurotrophin receptor p75) or TROY and LINGO-1 (leucine-rich repeat and Ig domain-containing 1). As a glycosylphosphatidylinositol-anchored receptor, Nogo receptor 1 relies on its co-receptors for transmembrane signaling (1, 2).
LINGO-1 is a transmembrane glycoprotein with 12 leucine-rich repeat motifs flanked by N- and C-terminal capping domains, one Ig domain, one transmembrane domain, and a short cytoplasmic tail (1). The extracellular domain of LINGO-1 associates with itself to form ring-like tetramers that are thought to serve as a scaffold to facilitate the assembly of the receptor complex (3). The intracellular tail contains a canonical EGF receptor (EGFR)3-like tyrosine phosphorylation site (residue 591) that is critical for intracellular signaling (4, 5). LINGO-1 is expressed in neurons, oligodendrocytes, and a subset of reactive hypertrophic astrocytes (6–8). In neurons, it is involved in mediating growth cone collapse and inhibiting neurite extension (1, 2, 5). LINGO-1 also plays an important role in the inhibition of oligodendrocyte differentiation and myelination (6, 7, 9). A number of studies in diverse animal CNS disease models have shown that targeted inhibition of LINGO-1 can benefit neuronal and oligodendrocyte survival after injury (10–13). LINGO-1-Fc (a recombinant protein of human LINGO-1 residues 1–532 fused to the hinge and Fc regions of human IgG1) treatment significantly increased oligodendrocyte and neuronal survival after either rubrospinal or corticospinal tract transection (10). In LINGO-1 knock-out mice, dopaminergic neuronal survival was increased and behavioral abnormalities were reduced compared with wild-type animals. LINGO-1 antagonists were shown to provide similar neuroprotective effects to midbrain dopaminergic neurons both in vitro and in vivo. These neuroprotective effects were shown to result from activation of the EGFR/Akt signaling pathway through direct inhibition of LINGO-1 binding to the EGFR (12). In a model of retinal ganglion cell (RGC) injury, exogenously added soluble LINGO-1-Fc and anti-LINGO-1 antibody 1A7 significantly reduced RGC loss after ocular hypertension and also promoted RGC survival after optic nerve transection (11). Pretreatment with LINGO-1-Fc was also shown to inhibit low potassium-induced cerebellar granular neuron apoptosis by suppression of GSK3β activation (13). Recent evidence indicates that LINGO-1 also functions as a potent regulator of neuronal apoptosis in neural stem cell maturation, as the number of cells going through apoptosis during the early phase of differentiation was significantly decreased in cultures treated with anti-LINGO-1 antibodies (14). Although evidence supports a role for LINGO-1 antagonists in neuronal survival, the underlying mechanism remains elusive.
We have previously demonstrated that LINGO-1 interacts with WNK1 (with no lysine (K) kinase-1) to regulate Nogo-induced inhibition of neurite extension (5), suggesting the involvement of WNK proteins in the regulation of LINGO-1 signaling. WNK proteins are a subfamily of serine/threonine kinases composed of four members, WNK1, WNK2, WNK3, and WNK4 (15). WNK3 is the isoform that is most abundant in the brain and is best known for its role in regulating the Cl− transporters NKCC1 (Na+-K+-2Cl− cotransporter 1) and KCC2 (K+-Cl− cotransporter 2) in GABAergic neurons (16). Interestingly, WNK3 has also been shown to exert an anti-apoptotic effect by restraining procaspase-3 activation in HeLa cells (17). However, little is known about the role of WNK3 in regulation of neuronal apoptosis. In this study, we demonstrate that LINGO-1 can interact with WNK3 to promote neuronal apoptosis by inhibition of WNK3 kinase activity.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
Human WNK3(49–436), generated from pcDNA3-myc-hWNK3 (kindly provided by Dr. Peter Jordan, Centro de Genética Humana, Instituto Nacional de Saúde, Lisbon, Portugal), was subcloned into the pEGFP-N1 vector to generate pEGFP-WNK3(49–436). The mutant plasmid pEGFP-WNK3(49–436)K159M was generated using the QuikChange site-directed mutagenesis kit (Stratagene). All constructs were validated by DNA sequencing.
Generation of GST-Nogo66 and TAT-LINGO-1 IC Fusion Proteins
Homo sapiens Nogo66 was amplified from a human fetal brain cDNA library (Clontech) and inserted into the pGEX-4T3 plasmid. The GST fusion protein was expressed in Escherichia coli BL21-CodonPlus (DE3) cells (Tiangen, Shanghai, China) and purified by the method of GrandPré et al. (18). The intracellular domain (amino acids 580–620) of human LINGO-1 (LINGO-1 IC) was generated from pEGFP-N1-hLINGO-1 by PCR amplification. To generate the fusion protein TAT-LINGO-1 IC, a sequence containing the minimal translocation domain of the HIV-1 protein TAT (amino acids 47–57, MGSSHHHHHHSSGLVPRGSMASGYGRKKRRQRRRGEF) was inserted in-frame next to the N terminus of the LINGO-1 IC cDNA. The fusion construct was inserted into pET-28a, expressed, and purified using standard recombinant techniques.
WNK3 RNAi
Three siRNA candidates were designed from rat WNK3 DNA sequences: WNK3si-1, CCAACAGGCTCTAAGATTC; WNK3si-2, GCAGGCATGTTCATACCTA; and WNK3si-3, CCTCCAAGTTAGATGGTAA. shRNAs were cloned into the pSUPER vector using the BglII and XhoI sites. To test the effectiveness of the shRNA constructs, WNK levels in PC12 cells were measured by RT-PCR and immunoblotting 48 h after transfection with WNK3 shRNA constructs. For qualitative expression analysis, the High Fidelity PCR system (Roche Applied Science) was used with the following primers: WNK3, 5′-ATCACCACGCAGGCCAAGA-3′/5′-GCTCCCGAAATCCCAACCC-3′; and GAPDH, 5′-ATCACTGCCACCCAGAAGAC-3′/5′-ATGAGGTCCACCACCCTGTT-3′. Empty pSUPER vector was used as a control for immunoblotting and morphology analyses; pSUPER containing an unrelated oligonucleotide (control siRNA) was used as a control for RT-PCR analyses.
Immunoprecipitation and Immunoblotting
Tissue samples and cultured cells were lysed with radioimmune precipitation assay buffer containing protease inhibitors (Roche Applied Science) supplemented with PMSF and centrifuged at 11,200 × g for 20 min at 4 °C. The supernatant (300–500 μl) was incubated with 5 μg of primary antibody for 2 h at 4 °C. Protein G-agarose beads (Roche Applied Science) were then added for another 12 h of rotation at 4 °C, and the immunoprecipitated products were washed three times with lysis buffer, boiled for 3–5 min in loading buffer, resolved by SDS-PAGE, immunoblotted, and visualized by enhanced chemiluminescence (Pierce). The following antibodies were used for immunoblotting: rabbit anti-LINGO-1 (1:500; Upstate); rabbit anti-WNK3 (1:500; Alpha Diagnostics); monoclonal mouse anti-phosphoserine (1:500; Sigma); rabbit anti-caspase-3 (1:1000), monoclonal rabbit anti-cleaved caspase-3 (1:500), monoclonal rabbit anti-GSK3β (1:1000), and rabbit anti-phospho-GSK3α/β (Ser-21/Ser-9; 1:1000) (Cell Signaling Technology); HRP-conjugated anti-GAPDH and HRP-conjugated anti-β-Actin (1:10,000; Kangcheng, Shanghai, China); and HRP-conjugated secondary antibodies (1:10,000; Santa Cruz Biotechnology).
Immunohistochemistry and TUNEL Staining
Cells cultured on coverslips or tissue slices from rat brains were washed with phosphate-buffered saline and fixed for 30 min with 4% paraformaldehyde at room temperature. Fixed samples were permeabilized with 0.1% Triton X-100 for 30 min, subsequently blocked with 1% bovine serum albumin in phosphate-buffered saline, incubated overnight at 4 °C with primary antibody (rabbit anti-LINGO-1 (1:100), goat anti-LINGO-1 (1:100; Santa Cruz Biotechnology), rabbit anti-WNK3 (1:100), mouse anti-Tuj1 (1:100; Chemicon), or monoclonal rabbit anti-cleaved caspase-3 (1:100)), and detected by species-specific FITC- or rhodamine-conjugated secondary antibodies (1:100; Santa Cruz Biotechnology). In rescue assays, Myc staining was detected by Alexa Fluor 647 (1:300; Jackson ImmunoResearch Laboratories). We used an in situ cell death detection kit (Roche Applied Science) to label apoptotic cells and visualized labeled cells with TMR red. Fluorescent images were taken with a Leica SP5 confocal microscope. At least 100 cells were counted under each experimental condition.
Primary Cultures, Transfections, and Survival Assays
Cortical neurons were isolated from embryonic day 18 (E18) Sprague-Dawley rats and transfected by nucleofection (Amaxa) following the manufacturer's instructions. After 3 h, the culture medium was replaced with fresh Neurobasal-A medium with a 2% B-27 supplement (Invitrogen). Three days after transfection, the medium was replaced with DMEM without serum to induce apoptosis. After 24 h, cells were subjected to anti-cleaved caspase-3 antibody or TUNEL staining and observed under an Olympus fluorescence microscope (excitation at 454 nm). Only GFP-positive cells were used for quantitative analysis.
In other apoptosis analyses, E18 cortical neurons were cultured for 3 days in vitro. After serum deprivation (SD) for up to 12 h, cells were subsequently treated with various concentrations of GST or GST-Nogo66 for different time intervals prior to propidium iodide staining. Serum-containing cultures were used in parallel as controls. To test the effect of LINGO-1 on survival, E18 cortical neurons were pretreated with TAT-LINGO-1 IC (2 μm) or an unfused TAT protein (TAT control; 2 μm) for 2 days before subjecting the cells to 24 h of SD. In these assays, survival was measured by cleaved caspase-3 staining.
We determined the rates of apoptosis by counting propidium iodide-positive or caspase-3-positive cells. At least 800 cells were counted in each group. The data were analyzed by one-way analysis of variance and Student's t test. Statistical values are presented as the mean ± S.E. All morphology experiments were repeated at least three times.
In Utero Electroporation
Plasmids were transfected by in utero electroporation as described (19). In brief, multiparous Sprague-Dawley rats at 13 days of gestation were anesthetized with 10% chloral hydrate (3.5 ml/kg, intraperitoneally). Uteruses were exposed, and 15–20 mg of plasmid mixed with Fast Green FCF (2 mg/ml; Sigma) was injected by trans-uterus pressure microinjection into the lateral ventricle of embryos. The plasmid mixture (6 mg/ml) consisted of 1:1 enhanced YFP (EYFP) and shRNA or 1:1:1 EYFP, shRNA, and rescue plasmid. Five electric pulses applied to the cerebral wall (50 ms at 60 V) at 100-ms intervals were generated using an ElectroSquireportator T830 system (BTX). EYFP- and TUNEL-labeled cells were counted in the electroporated cortex, and averages from at least three serial slices per animal were tallied. Statistical significance was determined using one-way analysis of variance and Dunnett's multiple comparisons test or Student's t test.
RESULTS
Effect of LINGO-1 on Neuronal Survival
Nogo66 is a functional domain of Nogo shared by all three family members and is an agonist commonly used for activating the LINGO-1 receptor complex in neuronal cells (1, 20). To investigate a role for LINGO-1 in neuronal apoptosis, we first employed a well established in vitro model of apoptosis in which cultured cortical neurons are challenged with SD (21). To examine whether Nogo66 promotes neuronal apoptosis, cultured cortical neurons were subjected to SD for up to 12 h and then treated with GST alone or GST-Nogo66 at a variety of concentrations and time intervals. Cell viability was evaluated by propidium iodide staining. We found that SD-induced cell death in GST-treated cultures at both 2- and 12-h intervals was significantly increased compared with the serum-containing controls: 100 nm GST for 2 h, 15.11 ± 1.01%, versus serum, 9.41 ± 1.10%; 200 nm GST for 2 h, 17.11 ± 1.94%, versus serum, 8.55 ± 3.46%; 100 nm GST for 12 h, 20.70 ± 1.21%, versus serum, 9.96 ± 1.91%; and 200 nm GST for 12 h, 21.80 ± 1.34%, versus serum, 10.47 ± 1.29%. Importantly, SD-induced cell death was greatly increased by Nogo66 treatment (Fig. 1A). Statistical analyses (Fig. 1C) showed a significantly higher percentage of apoptotic cortical neurons in cultures treated with GST-Nogo66 compared with GST-treated controls: 100 nm GST-Nogo66 for 2 h, 20.87 ± 1.08%, versus 100 nm GST, 15.11 ± 1.01%; 100 nm GST-Nogo66 for 12 h, 32.71 ± 2.30%, versus 100 nm GST, 20.70 ± 1.21%; 200 nm GST-Nogo66 for 2 h, 34.24 ± 3.77%, versus 200 nm GST, 17.11 ± 1.94%; and 200 nm GST-Nogo66 for 12 h, 51.46 ± 2.94%, versus 200 nm GST, 21.80 ± 1.34%. These results are supported by a previously published report indicating that the Nogo66 protein potentiates SD-induced neuronal apoptosis (22).
FIGURE 1.
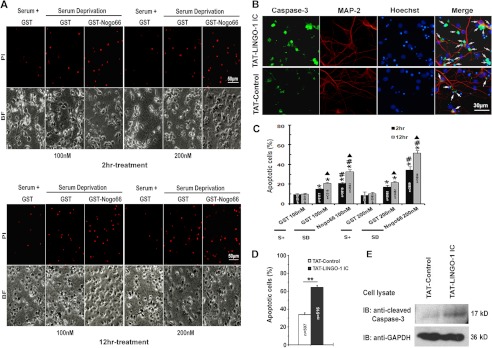
LINGO-1 is involved in regulation of neuronal apoptosis. A, representative images showing the effect of treatment with either GST-Nogo66 or GST alone on apoptosis rates in serum-deprived cultures of E18 primary cortical neurons. Results are compared with serum-containing cultures (Serum+). Cell viability was evaluated by propidium iodide (PI) staining. BF, bright field. Scale bar = 50 μm. B, representative images showing the effects of TAT-LINGO-1 IC on rates of SD-induced apoptosis. The TAT peptide (TAT-control) was used as a negative control. Apoptosis was assessed by immunohistochemical staining for cleaved caspase-3 (green), with the arrows indicating apoptotic cells. Dendrites were visualized using MAP-2 staining (red). Nuclei were stained with Hoechst dye (blue). C, statistical analyses of results in A showing the percentage of propidium iodide-positive cells in each treatment group. S+, serum-containing. The total number of cells counted for each group is indicated in the corresponding column. Values represent the mean ± S.E. *, p < 0.001 versus the serum-containing control; #, p < 0.001 versus the SD/GST control; ▴, p < 0.001 versus 2-h treatment with the same concentration (Student's t test and one-way analysis of variance). D, statistical analyses of results in B showing the percentage of apoptotic cells in each treatment group. The total number of cells counted for each group is indicated in the corresponding column. Values represent the mean ± S.E. **, p < 0.001 versus the control (Student's t test and one-way analysis of variance). E, immunoblot (IB) showing caspase-3 activation in TAT-LINGO-1 IC-treated cortical neurons, with the TAT peptide used as a negative control and GAPDH as a loading control.
To examine whether LINGO-1 is involved in the regulation of neuronal apoptosis, we generated a soluble fusion protein consisting of the intracellular domain of LINGO-1 tagged with TAT (TAT-LINGO-1 IC). As shown in Fig. 2F, TAT-LINGO-1 IC was able to efficiently enter cells as evidenced by immunoblotting with anti-LINGO-1 antibody. Primary cultured cortical neurons pretreated with TAT-LINGO-1 IC (2 μm) or TAT peptide (2 μm; TAT control) were deprived of serum for 24 h, and rates of apoptosis were then measured by cleaved caspase-3 staining. The percentage of cleaved caspase-3-positive neurons was significantly higher in TAT-LINGO-1 IC-treated cultures (64.21 ± 2.08%) than in control cultures (33.99 ± 2.16%) (Fig. 1, B and D). The enhanced activation of caspase-3 in TAT-LINGO-1 IC-treated cultures was also confirmed by immunoblotting (Fig. 1E). These results suggest that intracellular signaling downstream of LINGO-1 has an effect similar to Nogo66 in promoting apoptosis in serum-deprived cortical neurons.
FIGURE 2.

LINGO-1 interacts with endogenous WNK3 in rat brain tissues and regulates its kinase activity. A, immunohistochemical staining showing that WNK3 and LINGO-1 co-localize in E13 rat cortical neurons. Green, LINGO-1 or WNK3; red, neuron-specific Tuj1. Scale bars = 500 μm (upper panels), 100 μm (middle panels), and 50 μm (lower panels). B, immunohistochemical co-staining of LINGO-1 and WNK3 in E18 rat cortical neurons. Scale bar = 20 μm. C, co-immunoprecipitation experiment showing that endogenous WNK3 interacts with LINGO-1 in rat brain tissue. Brain and muscle tissues were used as positive and negative controls, respectively, and GAPDH was used as a loading control. D, immunoprecipitation (IP) showing that endogenous LINGO-1 activation by Nogo66 treatment results in enhanced association with WNK3. Endogenous LINGO-1 expression was used as a loading control. E, co-immunoprecipitation showing phosphorylated WNK3 levels in primary cultured E18 cortical neurons treated with 100 nm GST-Nogo66 or GST alone as a control. F, immunoblotting (IB) of lysates from E18 cortical neurons treated with TAT-LINGO-1 IC or the TAT control using anti-LINGO-1 (10 kDa) and anti-WNK3 (250 kDa) antibodies. GAPDH was used as a loading control and was identified with HRP-conjugated anti-GAPDH antibody (36 kDa) as a loading control. G, co-immunoprecipitation experiment showing decreased WNK3 phosphorylation levels in primary cultured E18 cortical neurons treated with TAT-LINGO-1 IC. H–J, quantification of D, E, and G is presented as -fold of control. Data shown are the mean ± S.D. of three independent experiments. **, p < 0.01 (Student's t test).
LINGO-1 Co-precipitates with WNK3 in Brain Tissues
Given the interaction between LINGO-1 and WNK1 (5) and the anti-apoptotic effect of WNK3 (17), we next asked whether WNK3 activity is responsible for the pro-apoptotic effect of LINGO-1 in cortical neurons. We first examined the expression patterns and cellular localization of WNK3 in rat brain slices and primary cultured neurons. In E13 cortical slices (Fig. 2A), both LINGO-1 and WNK3 were expressed at high levels in neurons, as indicated by Tuj1 co-staining. Furthermore, WNK3, a cytoplasmic protein, and LINGO-1, a transmembrane protein, co-localized in primary cultured E18 cortical neurons (Fig. 2B). We next performed co-immunoprecipitation assays on P1 rat brain tissue lysates and found that LINGO-1 precipitated with WNK3 and vice versa (Fig. 2C). These results suggest that endogenous LINGO-1 and WNK3 can interact in the brain. To further investigate the relevance of Nogo66 to the LINGO-1/WNK3 interaction, primary cultured cortical neurons having undergone 12 h of SD were treated with 100 nm GST or GST-Nogo66 for 3 h. As shown in Fig. 2D, co-immunoprecipitation assays revealed a significantly enhanced interaction between WNK3 and LINGO-1 in Nogo66-treated cultures, suggesting that the LINGO-1/WNK3 interaction is responsive to Nogo66 stimulation.
LINGO-1 Inhibits WNK3 Kinase Activity
Previous studies have revealed that autophosphorylation is an essential step in WNK kinase activity (23–25). Therefore, we examined WNK3 autophosphorylation levels in cortical neurons after Nogo66 treatment. Primary cultured cortical neurons from E18 rats were treated with GST-Nogo66 (100 nm) or GST alone (100 nm), and phospho-WNK3 levels were measured 48 h later by immunoprecipitating WNK3 from the lysate with anti-WNK3 antibody and then immunoblotting the precipitate with anti-phosphoserine antibody. Consistent with a published report (26), serine-phosphorylated WNK3 was abundant in control cultured cortical neurons (Fig. 2, E and I), suggesting that cortical neurons exhibit a high degree of constitutive WNK3 activity. When primary cortical neurons were treated with GST-Nogo66, however, the levels of serine-phosphorylated WNK3 were significantly decreased, indicating that WNK3 kinase activity is negatively regulated by Nogo signaling.
We next examined whether LINGO-1 contributes to the Nogo-dependent inhibition of WNK3 kinase activity. Primary cultured cortical neurons were treated with TAT-LINGO-1 IC (2 μm) or the TAT control (2 μm), and 48 h later, phospho-WNK3 levels were measured. Although total WNK3 concentration was not affected by TAT-LINGO-1 IC treatment, we observed a significant decrease in phospho-WNK3 levels compared with controls (Fig. 2, G and J). These results demonstrate that LINGO-1 can negatively regulate WNK3 kinase activity.
Suppression of Endogenous WNK3 Kinase Sensitizes Cortical Neurons to Apoptosis
To investigate a role for endogenous WNK3 in neuronal survival, we reduced WNK3 levels using RNAi. Three candidate siRNA sequences were designed, and the most effective one was selected and cloned into the pSUPER vector to generate pSUPER-WNK3 shRNA, which we then transfected into PC12 cells. As shown in Fig. 3 (A and B), the levels of WNK3 mRNA and protein were significantly reduced in cells transfected with pSUPER-WNK3 shRNA but were not affected by transfection with a control pSUPER vector containing a scrambled control shRNA sequence or an empty pSUPER vector.
FIGURE 3.

Inhibition of WNK3 increases neuronal apoptosis in vitro. A, RT-PCR analysis of WNK3 mRNA expression in PC12 cells transfected with pSUPER-WNK3 shRNA (WNK3 si), pSUPER-scrambled control shRNA (Control si), or pSUPER vector (Control). GAPDH was used as a loading control. B, immunoblot showing WNK3 expression in pSUPER-WNK3 shRNA- and pSUPER-transfected (control) PC12 cells. C–F, apoptosis of serum-deprived primary E18 cortical neurons transfected with the pSUPER vector (Control) or pSUPER-WNK3 shRNA (WNK3 si) or cotransfected with pSUPER-WNK3 shRNA and pcDNA3-myc-hWNK3 (WNK3 si + hWNK3) as assessed by TUNEL staining (C and G) and caspase-3 activation (E and I). The WNK3 siRNA sequences were designed to interact with rat, but not human, mRNA. Neuronal apoptosis in pEGFP-N1-hWNK3(49–436)K159M (WNK3(49–436)K159M) and pEGFP-N1-hWNK3(49–436) (WNK3(49–436)) was also assessed by TUNEL staining (D and H) and caspase-3 activation (F and J); only transfected neurons were assessed. Arrows indicate apoptosis in transfected/cotransfected neurons. Values in C–F represent the mean ± S.E. **, p < 0.001 versus the control (Dunnett's multiple comparisons test and Student's t test). The total number of cells counted in each group is indicated in the corresponding column. Scale bars = 30 μm.
Next, E18 cortical neurons were transfected with pSUPER-WNK3 shRNA or control vector and cultured for 3 days before being deprived of serum for 24 h. As revealed by TUNEL staining (control, 21.93 ± 2.38%; and WNK3 shRNA group, 44.38 ± 1.99%) (Fig. 3, C and G) and cleaved-caspase-3 staining (control, 21.20 ± 2.53%; and WNK3 shRNA group, 44.88 ± 4.24%) (Fig. 3, E and I), apoptotic GFP-positive cells were significantly increased in WNK3 shRNA-transfected cultures compared with control cultures. Furthermore, when full-length human WNK3, which is resistant to the RNAi approach, was cotransfected with pSUPER-WNK3 shRNA, the effect of WNK3 shRNA on neuronal apoptosis under SD was abolished (TUNEL assay, 20.62 ± 3.34%; and cleaved caspase-3 staining, 23.23 ± 4.87%), as shown in the lower panels of Fig. 3 (G and I), indicating the specificity of the WNK3 RNAi. Finally, we confirmed all of the shRNA results described in Fig. 3, including the reversion of the phenotype experiment, with a second WNK3 shRNA (WNK3si-2; data not shown), thus suggesting that our results cannot be explained by an artifact of off-target siRNA.
To further assess the role of WNK3 kinase activity in neuronal apoptosis, we expressed a transgenic form of WNK3 containing the entire catalytic domain, WNK(49–436) (27), or a kinase-dead mutant form of the same transgene, WNK3(49–436)K159M (17). Both constructs were cloned into pEGFP-N1 and transfected into cultured cortical neurons. TUNEL and cleaved caspase-3 staining revealed that overexpression of the WNK3 catalytic domain had little effect on apoptosis, whereas expression of kinase-dead WNK3 significantly increased apoptosis of cells showing TUNEL labeling (WNK3(49–436)K159M, 66.56 ± 2.94%; and WNK3(49–436), 25.48 ± 2.03%) (Fig. 3, D and H) or cleaved caspase-3 staining (WNK3(49–436)K159M, 61.36 ± 3.30%; and WNK3(49–436), 24.56 ± 2.08%) (Fig. 3, F and J).
WNK3 Kinase Promotes Cortical Neuronal Survival in Vivo
During cortical development, when post-mitotic neurons migrate away from the ventricular zone along radial glial fibers and toward the surface of the cortical plate, many migrating neurons undergo apoptosis. To study the role of WNK3 in neuronal survival during this developmental stage, we transfected E13 ventricular zone neurons with WNK3 shRNA, WNK(49–436)K159M, or the control pSUPER vector together with a plasmid encoding EYFP by in utero electroporation (19). We next prepared coronal sections from the somatosensory cortex (striatum level of the dorsolateral neocortex) on E16 or E19. At E16, no differences in the number of EYFP-positive cells were observed between control cortices and WNK3 shRNA group or WNK3(49–436)K159M cortices (Fig. 4B). In contrast, the number of EYFP-positive cells was markedly reduced on E19 in the WNK3 shRNA group and, to a greater extent, the WNK3(49–436)K159M group compared with the control group (Fig. 4, A and B). We examined this further by TUNEL staining (Fig. 4, A and C); at E16, no difference in the number of TUNEL-labeled EYFP-positive neurons was observed between the two groups. However, in E19 cortices electroporated with WNK3 shRNA or WNK3(49–436)K159M on E13, the proportion of TUNEL-labeled EYFP-positive neurons at E19 was much greater than in controls. The apoptotic effect of WNK3 shRNA was largely abrogated when co-electroporated with full-length wild-type human WNK3, which indicates that the effect of WNK3 shRNA was specific. Taken together, these in vivo results suggest that WNK3 kinase activity plays an essential survival role during cortical development.
FIGURE 4.
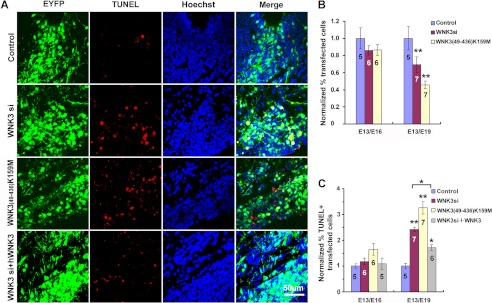
WNK3 promotes cortical neuronal survival in vivo. A, representative images of TUNEL-positive cells in rat cortices electroporated with the indicated constructs plus EYFP on E13 and analyzed on E19. Control, pSUPER vector; WNK3 si, pSUPER-WNK3 shRNA; WNK3(49–436)K159M, pEGFP-WNK3(49–436)K159M; WNK3 si + hWNK3, pSUPER-WNK3 shRNA + pEGFP-hWNK3. Scale bar = 50 μm. B, quantification of EYFP-positive cells in E16 and E19 cortices after transfection as described for A. Values were normalized to those of the control. Data represent the mean ± S.E. from five to seven animals (n values are indicated in the corresponding column). **, p < 0.001 versus the control (Student's t test). C, proportion of TUNEL-labeled EYFP-positive neurons in E16 and E19 cortices after transfection as described for A normalized to those of the control. Data represent the mean ± S.E. *, p < 0.01; **, p < 0.001 versus the Control (Student's t test). The number of cortices analyzed in each group is indicated in the corresponding column.
DISCUSSION
Several recent studies have demonstrated that LINGO-1 is involved in the regulation of neuronal apoptosis. For example, blocking LINGO-1 function improves midbrain dopaminergic neuron survival in an animal model of Parkinson disease (12), increases RGC survival in a rat model of chronic glaucoma (11), and protects cerebellar granule neurons from low potassium-induced apoptosis (13). Moreover, genetic analysis suggests that the LINGO1 gene may be an attractive target when developing therapies for the treatment of neurodegenerative diseases that are thought to be characterized by neuronal apoptosis (28, 29).
The underlying mechanism of LINGO-1 function has been attributed to its association with receptor tyrosine kinases. For example, the binding of LINGO-1 to the EGFR has been shown to directly inhibit EGFR expression. This results in a dampening of PI3K/Akt signaling, which has been shown to protect dopaminergic neurons from apoptosis (12). LINGO-1 has also been shown to associate with TrkB in the regulation of RGC survival (30). Blocking the extracellular domain of LINGO-1 disrupts the interaction between LINGO-1 and these receptor kinases and attenuates inhibition of neuronal survival. In this study, expression of the intracellular domain of LINGO-1 greatly increased the rates of apoptosis in cultured neurons under SD, strongly suggesting that LINGO-1 intracellular signaling contributes to promoting neuronal apoptosis.
Our previous work demonstrated that LINGO-1 interacts with WNK1 to inhibit neurite extension. However, neither knockdown nor overexpression of WNK1 had any effect on neuronal survival, indicating that WNK1 is not involved in the regulation of neuronal apoptosis (5). Among the four known WNK family members, only WNK3 has been reported to regulate cell survival (17). In this study, we found that WNK3 had a potent prosurvival effect on serum-deprived cortical neurons. Suppressing endogenous WNK3 expression by RNAi rendered these neurons far more susceptible to SD-induced apoptosis. We also observed a prosurvival function for WNK3 in vivo; reducing WNK3 expression by siRNA in radially migrating ventricular zone-derived neurons led to higher rates of apoptosis in the E19 cortical plate. This finding is especially interesting in light of the fact that WNK3, which is abundantly expressed in the hippocampus, several cortical layers, and the Purkinje layer of the cerebellum (16), is encoded at Xp11.22, a region of the genome associated with X-linked mental retardation and autism spectrum disorders (31).
It has been reported that WNK members function via both kinase-dependent and kinase-independent mechanisms. For example, kinase-dependent WNK3 activity regulates SLC12A family electroneutral cation/Cl− channels (26) and affects Ca2+ influx by regulating TRPV5 and TRPV6 expression (32). On the other hand, kinase-independent WNK3 activity decreases plasma membrane levels of ROMK1 (33) and inhibits the Cl− channel SLC26A9 (34). The anti-apoptotic role of WNK3 previously observed in HeLa cells was shown to be dependent on its ability to bind procaspase-3 (17). In this study, we found that expression of a transgene consisting of a mutant form of the WNK3 kinase domain had a potent dominant-negative effect on cortical neuron survival both in vitro and in vivo. These results suggest that the kinase activity of WNK3 is essential for promoting cell survival in neuronal cells.
We observed a high degree of constitutive WNK3 kinase activity in cultured neurons, as indicated by high levels of phosphorylated WNK3 under normal culture conditions (23–26). However, when challenged with pro-apoptotic Nogo66, the level of phosphorylated WNK3 was significantly decreased, indicating that WNK3 kinase activity is negatively regulated by Nogo signaling. In support of this, when a TAT peptide containing the intracellular domain of LINGO-1 was introduced into cultured cortical neurons, WNK3 phosphorylation was also significantly attenuated. This strongly suggests that LINGO-1, perhaps acting downstream of Nogo66, negatively regulates WNK3 kinase activity. Given that both LINGO-1 and WNK3 are highly expressed in cortical neurons and can co-precipitate with one another in a manner that is enhanced after Nogo66 stimulation, the influence of LINGO-1 on WNK3 activity may be direct. It is possible that LINGO-1 binds directly to the serine/threonine kinase domain of WNK3, thereby inhibiting its kinase activation.
As a serine/threonine kinase, WNK1 has been reported to be a substrate for Akt-mediated phosphorylation (35) and is dependent on PI3K/Akt-mediated endocytosis of ROMK (36). Recent studies suggest that WNK3 can also be phosphorylated by Akt (37). Growth factor-activated PI3K/Akt signaling pathways have been previously documented as a major pathway enhancing neuronal survival (38). Enhancement of the PI3K/Akt pathway was considered to constitute an important neuroprotective strategy (39).
GSK3β is a Ser/Thr protein kinase. An N-terminal serine residue (Ser-9) is known to inhibit GSK3β activity. Many kinases, including Akt, are known to phosphorylate Ser-9 of GSK3β and therefore retain GSK3β in an inactive state (40, 41). GSK3β has been reported to play a pro-apoptotic role in several models of neuronal cell death, including SD. Inhibition of its phosphorylation, which leads to its activation, has been shown to promote neuronal apoptosis (42). We observed GSK3β to associate with the LINGO-1·WNK3 complex in primary cultured neurons, and the association was also intensified by Nogo66 treatment. Moreover, Nogo66 treatment under SD reduced the phosphorylation of GSK3β (data not shown). Thus, these results strongly indicate that, in the presence of Nogo66, the LINGO-1·WNK3·GSK3β complex might be involved in the negative regulation of neuronal survival, although the precise mechanism still needs further investigation.
In summary, we have provided evidence that LINGO-1 interacts with and inhibits WNK3 kinase activity, thereby increasing the susceptibility of cortical neurons to apoptosis. Our findings shed new light on the mechanism by which Nogo/LINGO-1 signaling promotes cell death during brain development and in response to stress.
Acknowledgments
We are grateful to Prof. Xiao-bin Yuan for providing the pEYFP-N1, pECFP-N1, and pSUPER vectors and Dr. Peter Jordan for providing the pEGFP-C2-hWNK3 and pcDNA3-myc-hWNK3 constructs.
This work was supported by National Key Basic Research Program Grant 2011CB504401 and National Natural Science Foundation Grants 81000524, 31070922, and 31130024.
- EGFR
- EGF receptor
- RGC
- retinal ganglion cell
- LINGO-1 IC
- LINGO-1 intracellular domain
- E18
- embryonic day 18
- SD
- serum deprivation
- EYFP
- enhanced YFP.
REFERENCES
- 1. Mi S., Lee X., Shao Z., Thill G., Ji B., Relton J., Levesque M., Allaire N., Perrin S., Sands B., Crowell T., Cate R. L., McCoy J. M., Pepinsky R. B. (2004) LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat. Neurosci. 7, 221–228 [DOI] [PubMed] [Google Scholar]
- 2. Park J. B., Yiu G., Kaneko S., Wang J., Chang J., He X. L., Garcia K. C., He Z. (2005) A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron 45, 345–351 [DOI] [PubMed] [Google Scholar]
- 3. Mosyak L., Wood A., Dwyer B., Buddha M., Johnson M., Aulabaugh A., Zhong X., Presman E., Benard S., Kelleher K., Wilhelm J., Stahl M. L., Kriz R., Gao Y., Cao Z., Ling H. P., Pangalos M. N., Walsh F. S., Somers W. S. (2006) The structure of the Lingo-1 ectodomain, a module implicated in central nervous system repair inhibition. J. Biol. Chem. 281, 36378–36390 [DOI] [PubMed] [Google Scholar]
- 4. Llorens F., Gil V., Iraola S., Carim-Todd L., Martí E., Estivill X., Soriano E., del Rio J. A., Sumoy L. (2008) Developmental analysis of Lingo-1/Lern1 protein expression in the mouse brain: interaction of its intracellular domain with Myt1l. Dev. Neurobiol. 68, 521–541 [DOI] [PubMed] [Google Scholar]
- 5. Zhang Z., Xu X., Zhang Y., Zhou J., Yu Z., He C. (2009) LINGO-1 interacts with WNK1 to regulate Nogo-induced inhibition of neurite extension. J. Biol. Chem. 284, 15717–15728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mi S., Miller R. H., Lee X., Scott M. L., Shulag-Morskaya S., Shao Z., Chang J., Thill G., Levesque M., Zhang M., Hession C., Sah D., Trapp B., He Z., Jung V., McCoy J. M., Pepinsky R. B. (2005) LINGO-1 negatively regulates myelination by oligodendrocytes. Nat. Neurosci. 8, 745–751 [DOI] [PubMed] [Google Scholar]
- 7. Mi S., Hu B., Hahm K., Luo Y., Kam Hui E. S., Yuan Q., Wong W. M., Wang L., Su H., Chu T. H., Guo J., Zhang W., So K. F., Pepinsky B., Shao Z., Graff C., Garber E., Jung V., Wu E. X., Wu W. (2007) LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat. Med. 13, 1228–1233 [DOI] [PubMed] [Google Scholar]
- 8. Satoh J., Tabunoki H., Yamamura T., Arima K., Konno H. (2007) TROY and LINGO-1 expression in astrocytes and macrophages/microglia in multiple sclerosis lesions. Neuropathol. Appl. Neurobiol. 33, 99–107 [DOI] [PubMed] [Google Scholar]
- 9. Pepinsky R. B., Walus L., Shao Z., Ji B., Gu S., Sun Y., Wen D., Lee X., Wang Q., Garber E., Mi S. (2011) Production of a PEGylated Fab′ of the anti-LINGO-1 Li33 antibody and assessment of its biochemical and functional properties in vitro and in a rat model of remyelination. Bioconjug. Chem. 22, 200–210 [DOI] [PubMed] [Google Scholar]
- 10. Ji B., Li M., Wu W. T., Yick L. W., Lee X., Shao Z., Wang J., So K. F., McCoy J. M., Pepinsky R. B., Mi S., Relton J. K. (2006) LINGO-1 antagonist promotes functional recovery and axonal sprouting after spinal cord injury. Mol. Cell. Neurosci. 33, 311–320 [DOI] [PubMed] [Google Scholar]
- 11. Fu Q. L., Hu B., Wu W., Pepinsky R. B., Mi S., So K. F. (2008) Blocking LINGO-1 function promotes retinal ganglion cell survival following ocular hypertension and optic nerve transection. Invest. Ophthalmol. Vis. Sci. 49, 975–985 [DOI] [PubMed] [Google Scholar]
- 12. Inoue H., Lin L., Lee X., Shao Z., Mendes S., Snodgrass-Belt P., Sweigard H., Engber T., Pepinsky B., Yang L., Beal M. F., Mi S., Isacson O. (2007) Inhibition of the leucine-rich repeat protein LINGO-1 enhances survival, structure, and function of dopaminergic neurons in Parkinson's disease models. Proc. Natl. Acad. Sci. U.S.A. 104, 14430–14435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao X. H., Jin W. L., Wu J., Mi S., Ju G. (2008) Inactivation of glycogen synthase kinase-3β and up-regulation of LINGO-1 are involved in LINGO-1 antagonist regulated survival of cerebellar granular neurons. Cell. Mol. Neurobiol. 28, 727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lööv C., Fernqvist M., Walmsley A., Marklund N., Erlandsson A. (2012) Neutralization of LINGO-1 during in vitro differentiation of neural stem cells results in proliferation of immature neurons. PLoS ONE 7, e29771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Veríssimo F., Jordan P. (2001) WNK kinases, a novel protein kinase subfamily in multi-cellular organisms. Oncogene 20, 5562–5569 [DOI] [PubMed] [Google Scholar]
- 16. Kahle K. T., Rinehart J., de Los Heros P., Louvi A., Meade P., Vazquez N., Hebert S. C., Gamba G., Gimenez I., Lifton R. P. (2005) WNK3 modulates transport of Cl− in and out of cells: implications for control of cell volume and neuronal excitability. Proc. Natl. Acad. Sci. U.S.A. 102, 16783–16788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Veríssimo F., Silva E., Morris J. D., Pepperkok R., Jordan P. (2006) Protein kinase WNK3 increases cell survival in a caspase-3-dependent pathway. Oncogene 25, 4172–4182 [DOI] [PubMed] [Google Scholar]
- 18. GrandPré T., Li S., Strittmatter S. M. (2002) Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature 417, 547–551 [DOI] [PubMed] [Google Scholar]
- 19. Saito T., Nakatsuji N. (2001) Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev. Biol. 240, 237–246 [DOI] [PubMed] [Google Scholar]
- 20. Liu X., Wang Y., Zhang Y., Zhu W., Xu X., Niinobe M., Yoshikawa K., Lu C., He C. (2009) Nogo-A inhibits necdin-accelerated neurite outgrowth by retaining necdin in the cytoplasm. Mol. Cell. Neurosci. 41, 51–61 [DOI] [PubMed] [Google Scholar]
- 21. Bonni A., Brunet A., West A. E., Datta S. R., Takasu M. A., Greenberg M. E. (1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286, 1358–1362 [DOI] [PubMed] [Google Scholar]
- 22. Qi B., Qi Y., Watari A., Yoshioka N., Inoue H., Minemoto Y., Yamashita K., Sasagawa T., Yutsudo M. (2003) Pro-apoptotic ASY/Nogo-B protein associates with ASYIP. J. Cell. Physiol. 196, 312–318 [DOI] [PubMed] [Google Scholar]
- 23. Rinehart J., Kahle K. T., de Los Heros P., Vazquez N., Meade P., Wilson F. H., Hebert S. C., Gimenez I., Gamba G., Lifton R. P. (2005) WNK3 kinase is a positive regulator of NKCC2 and NCC, renal cation-Cl− cotransporters required for normal blood pressure homeostasis. Proc. Natl. Acad. Sci. U.S.A. 102, 16777–16782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu B., English J. M., Wilsbacher J. L., Stippec S., Goldsmith E. J., Cobb M. H. (2000) WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J. Biol. Chem. 275, 16795–16801 [DOI] [PubMed] [Google Scholar]
- 25. Xu B. E., Min X., Stippec S., Lee B. H., Goldsmith E. J., Cobb M. H. (2002) Regulation of WNK1 by an autoinhibitory domain and autophosphorylation. J. Biol. Chem. 277, 48456–48462 [DOI] [PubMed] [Google Scholar]
- 26. de Los Heros P., Kahle K. T., Rinehart J., Bobadilla N. A., Vázquez N., San Cristobal P., Mount D. B., Lifton R. P., Hebert S. C., Gamba G. (2006) WNK3 bypasses the tonicity requirement for K-Cl cotransporter activation via a phosphatase-dependent pathway. Proc. Natl. Acad. Sci. U.S.A. 103, 1976–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holden S., Cox J., Raymond F. L. (2004) Cloning, genomic organization, alternative splicing and expression analysis of the human gene WNK3 (PRKWNK3). Gene 335, 109–119 [DOI] [PubMed] [Google Scholar]
- 28. Lorenzo-Betancor O., Samaranch L., García-Martín E., Cervantes S., Agúndez J. A., Jiménez-Jiménez F. J., Alonso-Navarro H., Luengo A., Coria F., Lorenzo E., Irigoyen J., Pastor P., and Iberian Parkinson's Disease Genetics Study Group Researchers (2011) LINGO1 gene analysis in Parkinson's disease phenotypes. Mov. Disord. 26, 722–727 [DOI] [PubMed] [Google Scholar]
- 29. Gerin C. G., Madueke I. C., Perkins T., Hill S., Smith K., Haley B., Allen S. A., Garcia R. P., Paunesku T., Woloschak G. (2011) Combination strategies for repair, plasticity, and regeneration using regulation of gene expression during the chronic phase after spinal cord injury. Synapse 65, 1255–1281 [DOI] [PubMed] [Google Scholar]
- 30. Fu Q. L., Hu B., Li X., Shao Z., Shi J. B., Wu W., So K. F., Mi S. (2010) LINGO-1 negatively regulates TrkB phosphorylation after ocular hypertension. Eur. J. Neurosci. 31, 1091–1097 [DOI] [PubMed] [Google Scholar]
- 31. Qiao Y., Liu X., Harvard C., Hildebrand M. J., Rajcan-Separovic E., Holden J. J., Lewis M. E. (2008) Autism-associated familial microdeletion of Xp11.22. Clin. Genet. 74, 134–144 [DOI] [PubMed] [Google Scholar]
- 32. Zhang W., Na T., Peng J. B. (2008) WNK3 positively regulates epithelial calcium channels TRPV5 and TRPV6 via a kinase-dependent pathway. Am. J. Physiol. Renal Physiol. 295, F1472–F1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leng Q., Kahle K. T., Rinehart J., MacGregor G. G., Wilson F. H., Canessa C. M., Lifton R. P., Hebert S. C. (2006) WNK3, a kinase related to genes mutated in hereditary hypertension with hyperkalaemia, regulates the K+ channel ROMK1 (Kir1.1). J. Physiol. 571, 275–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dorwart M. R., Shcheynikov N., Wang Y., Stippec S., Muallem S. (2007) SLC26A9 is a Cl− channel regulated by the WNK kinases. J. Physiol. 584, 333–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jiang Z. Y., Zhou Q. L., Holik J., Patel S., Leszyk J., Coleman K., Chouinard M., Czech M. P. (2005) Identification of WNK1 as a substrate of Akt/protein kinase B and a negative regulator of insulin-stimulated mitogenesis in 3T3-L1 cells. J. Biol. Chem. 280, 21622–21628 [DOI] [PubMed] [Google Scholar]
- 36. Cheng C. J., Huang C. L. (2011) Activation of PI3-kinase stimulates endocytosis of ROMK via Akt1/SGK1-dependent phosphorylation of WNK1. J. Am. Soc. Nephrol. 22, 460–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garzon-Muvdi T., Schiapparelli P., ap Rhys C., Guerrero-Cazares H., Smith C., Kim D. H., Kone L., Farber H., Lee D. Y., An S. S., Levchenko A., Quinones-Hinojosa A. (2012) Regulation of brain tumor dispersal by NKCC1 through a novel role in focal adhesion regulation. PLoS Biol. 10, e1001320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brunet A., Datta S. R., Greenberg M. E. (2001) Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr. Opin. Neurobiol. 11, 297–305 [DOI] [PubMed] [Google Scholar]
- 39. Arboleda G., Morales L. C., Benítez B., Arboleda H. (2009) Regulation of ceramide-induced neuronal death: cell metabolism meets neurodegeneration. Brain Res. Rev. 59, 333–346 [DOI] [PubMed] [Google Scholar]
- 40. Cross D. A., Alessi D. R., Cohen P., Andjelkovich M., Hemmings B. A. (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789 [DOI] [PubMed] [Google Scholar]
- 41. Stambolic V., Woodgett J. R. (1994) Mitogen inactivation of glycogen synthase kinase-3β in intact cells via serine 9 phosphorylation. Biochem. J. 303, 701–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ambacher K. K., Pitzul K. B., Karajgikar M., Hamilton A., Ferguson S. S., Cregan S. P. (2012) The JNK- and AKT/GSK3β-signaling pathways converge to regulate Puma induction and neuronal apoptosis induced by trophic factor deprivation. PLoS ONE 7, e46885. [DOI] [PMC free article] [PubMed] [Google Scholar]