

Background: Antifreeze proteins bind to ice crystals and inhibit their growth.
Results: The crystal structure of a potent beetle antifreeze protein was determined by direct methods.
Conclusion: Ordered crystallographic waters on the protein surface match several planes of hexagonal ice.
Significance: The structure is the largest determined ab initio without heavy atoms, and its ordered waters suggest a molecular basis for ice binding.
Keywords: Biophysics, Computer Modeling, Crystallography, Protein Structure, X-ray Crystallography
Abstract
Antifreeze proteins (AFPs) help some organisms resist freezing by binding to ice crystals and inhibiting their growth. The molecular basis for how these proteins recognize and bind ice is not well understood. The longhorn beetle Rhagium inquisitor can supercool to below −25 °C, in part by synthesizing the most potent antifreeze protein studied thus far (RiAFP). We report the crystal structure of the 13-kDa RiAFP, determined at 1.21 Å resolution using direct methods. The structure, which contains 1,914 nonhydrogen protein atoms in the asymmetric unit, is the largest determined ab initio without heavy atoms. It reveals a compressed β-solenoid fold in which the top and bottom sheets are held together by a silk-like interdigitation of short side chains. RiAFP is perhaps the most regular structure yet observed. It is a second independently evolved AFP type in beetles. The two beetle AFPs have in common an extremely flat ice-binding surface comprising regular outward-projecting parallel arrays of threonine residues. The more active, wider RiAFP has four (rather than two) of these arrays between which the crystal structure shows the presence of ice-like waters. Molecular dynamics simulations independently reproduce the locations of these ordered crystallographic waters and predict additional waters that together provide an extensive view of the AFP interaction with ice. By matching several planes of hexagonal ice, these waters may help freeze the AFP to the ice surface, thus providing the molecular basis of ice binding.
Introduction
Antifreeze proteins (AFPs)3 enable the survival of many organisms that inhabit subzero environments, from vertebrates to bacteria (1). It is generally accepted that AFPs adsorb to the surface of nascent ice crystals. Upon adsorption to ice, AFPs block water molecules from accessing the ice surface at the bound location. The ice front thus becomes convex toward the solution between the surface-bound AFPs, which is energetically unfavorable for ice growth by the Gibbs-Thompson-Herring effect (2–4). This results in thermal hysteresis, or the noncolligative depression of the freezing point of the solution without altering its melting temperature. Although the adsorption-inhibition mechanism for ice growth inhibition has been generally accepted for >30 years, the mechanism by which AFPs recognize and bind ice has gone through several radical revisions without a consensus emerging. Recent simulations reveal that in contrast to other inorganic surfaces to which proteins may bind, an ice surface is disordered at the molecular level, with a 1–2-nm transition region separating ice from the surrounding bulk water solution (5).
The accepted structure of ordinary hexagonal ice consists of sheets of tessellating hexagonal rings composed of water molecules (6) with the angle between hydrogen bonds in the crystal lattice approximating the tetrahedral angle of 109.5°. By slicing this crystalline lattice at different angles, various two-dimensional ice surfaces are exposed that represent different planes of ice. It has been proposed recently that the ice-binding residues of antifreeze proteins might organize waters into an ice-like configuration that matches and merges with the quasiliquid layer at the ice/water interface (7, 8). The ordered waters match specific ice planes, facilitating interaction between the AFP and ice and thereby influencing ice crystal morphology. Molecular dynamics and a solution NMR study reveal that these organized waters are highly mobile, exchanging with bulk water on a subnanosecond time scale (9).
Crystal structures have been determined for six different types of AFPs from freeze-avoiding organisms: three from fish (10–13) and three from insects (14–16). Despite their common function, AFPs display remarkable diversity in their tertiary architectures. The richness in structural folds found in AFPs represents an exceptional example of radically different proteins serving the same function through convergent evolution. Although the most active (hyperactive) AFPs thus far are found in insects, they have proven difficult to produce in sufficient quantities for crystallographic determination (17). The difficulty of recombinant expression has also limited the use of the hyperactive AFPs as cryoprotective agents for applications outside of basic science (18).
The longhorn beetle, Rhagium inquisitor, survives harsh winter temperatures in Siberia and can supercool to below −25 °C (19). The ability of the beetle to withstand these temperatures is due in part to the presence of an AFP in the hemolymph. The only AFP identified in R. inquisitor, RiAFP, is a 13-kDa protein with one of the highest antifreeze activities measured for any AFP (20). Its amino acid sequence compared with those of other insect hyperactive AFPs from Choristoneura fumiferana (spruce budworm, CfAFP), Dendroides canadensis (fire-colored beetle, DcAFP), Hypogastrura harveyi (snow flea, sfAFP), and Tenebrio molitor (yellow mealworm, TmAFP) (14–16, 21, 22) suggests that RiAFP is a distinct type of AFP with a novel fold. Because the mechanism of ice recognition by antifreeze proteins is not well understood, a structural study of RiAFP could provide new insights into the molecular basis for antifreeze activity as well as the adaptations which proteins have acquired to fold and function at temperatures as low as −25 °C. Structural comparisons of RiAFP with other insect AFPs may also provide important molecular insights into their convergent evolution and the basis for antifreeze hyperactivity.
Here, we report the crystal structure of RiAFP determined at 1.21 Å resolution by ab initio direct methods. Direct methods provide an alternative for macromolecular crystal structure determination at atomic resolution (beyond 1.2 Å) in the absence of experimental phases or an adequate molecular replacement search model. However, proteins with >1,000 nonhydrogen atoms have been recalcitrant to structure determination by direct methods, especially without anomalous scatterers in the crystal (23). The RiAFP structure presented here is the largest determined ab initio without heavy atoms.
Our structure has a square prism shape and reveals a novel β-solenoid architecture with an ice-binding surface (IBS) containing a remarkably regular and extensive array of threonine residues. The IBS is exceptionally flat and holds crystallographic waters in a pattern that matches the primary prism plane of hexagonal ice. Based on simulations these waters occupy natural positions and help recruit other waters, some of which match the basal plane. A fluorescence-based ice etching experiment reveals that GFP-tagged RiAFP can bind the entire surface of a single crystal hemisphere, suggesting affinity to basal, primary prism and additional planes of ice.
EXPERIMENTAL PROCEDURES
Structure Determination of RiAFP
Cloning, expression, purification, and crystallization of RiAFP were reported previously (24). Diffraction data for RiAFP were collected using a 1.1 Å synchrotron radiation source at Brookhaven National Laboratory (beamline X25) (24) and processed to 1.147 Å resolution with XDS (25). Data collected on a 1.54 Å copper anode x-ray source were not of sufficient quality for structure determination ab initio or by more conventional methods. Molecular replacement using other insect antifreeze protein crystal structures as search models did not yield any obvious solutions. The high resolution diffraction (1.15 Å resolution, albeit with only 8.3% completeness in the highest resolution shell, see Table 1) prompted us to consider ab initio methods. Initial phases were obtained by applying the charge flipping program SUPERFLIP, which is mostly used for small molecule crystal structure determination (26, 27). The resulting electron density map was input into PEAKMAX in the CCP4 (28) suite for peak picking. The all-water “dummy atom” model produced by PEAKMAX was then used as the starting model for ACORN (29). The observed data were artificially extended to 1.0 Å with anisotropy corrections in ACORN. The phases derived from ACORN were input into Arp/wArp (30) for automatic model building. The β-strands built from Arp/wArp runs (Fig. 1A) were used as a molecular replacement search model in PHENIX (31). Iterative cycles of model building in COOT (32) and refinement in REFMAC (33) were performed. Notably, even with most of the structure built (20 of the total 26 β-strands, Fig. 1B), electron density for the surrounding loops was weak or absent, prompting us to reexamine the data and reprocess it in space group P31 and P1. Molecular replacement in P31 followed by automated model building with Arp/wArp yielded a nearly complete structure. The structure was ultimately refined in space group P3121 with Rwork = 12.76% and Rfree = 15.10%. The six C-terminal histidine residues from the affinity purification tag in chain A were clearly visible in the electron density map. However, residues 90–91 and the polyhistidine tag in chain B were disordered but with some residual electron density in the area. There are two RiAFP molecules in the asymmetric unit with a solvent content of ∼42.8%. Complete crystallographic and refinement statistics are listed in Table 1.
TABLE 1.
Crystallographic data collection and refinement statistics for RiAFP
| Crystal parameters | |
| Space group | P3121 |
| Unit cell dimensions (Å) | a = b = 46.46, c = 193.21 |
| Data collection | |
| Resolution (Å)a | 52.96-1.147 (1.18-1.147) |
| Number of reflections | |
| Total | 541,809b |
| Unique | 136,527b |
| Redundancy | 3.9 |
| Completeness (%) | 81.8% (8.3%) |
| Rmerge (%)c | 6.8% (20.1%) |
| I/σ | 10.93 (1.58) |
| Refinement statistics | |
| Reflections | 76,298d |
| Rwork (%)e | 12.76 |
| Rfree(%)f | 15.10 |
| Final model | |
| Nonhydrogen atoms | 2,322 |
| Riding hydrogens | 1,828 |
| SO42+ions | 4 |
| Glycerols | 3 |
| r.m.s.d.g | |
| Bond length (Å) | 0.006 |
| Bond angles (°) | 1.116 |
| PDB ID code | 4DT5 |
a At the reported resolution of 1.21 Å, completeness is 52.8%, Rmerge is 0.195, I/σ is 2.54 in the highest resolution shell (1.24–1.21 Å). The highest resolution shell statistics are in parentheses when applicable.
b Data were processed using XDS through the xia2 interface.
c Rmerge = <ΣhklΣj|Ij(hkl)-> − <I(hkl)>|>/<I(hkl)>, merging statistics for all symmetry-mates.
d Approximately 2-fold differences are due to unmerged and merged Friedel-mates.
e Rwork = Σhkl|Fobs(hkl) − Fcalc(hkl)|/Σhkl|Fobs(hkl), standard crystallographic R-factor.
f Rfree is the cross-validation R-factor for ∼5% of the total unique reflections that were randomly selected.
g r.m.s.d., root mean square deviation to ideal values.
FIGURE 1.

Structure determination of RiAFP. A, preliminary electron density map generated by Arp/wArp from initial phases. The 2Fo − 2Fc map is in blue and contoured at 1.3σ, whereas the Fo − Fc map is in green and red (indicating positive and negative peaks, respectively) and contoured at +3.0σ. B, model used in P31 molecular replacement. Despite building most of the final model, no connectivity was observed between individual β-strands in the higher (P3121) space group.
Thermal Hysteresis Experiments and Fluorescence-based Ice Plane Affinity Analysis
Fluorescence-based ice plane affinity analysis and thermal hysteresis measurements were performed as described in (7).
Multiangle Light Scattering
Purified RiAFP (∼10 mg/ml) and GFP-RiAFP (5 mg/ml) were injected onto a Superdex 75 (10/300) column (GE Healthcare) equilibrated in 20 mm Tris-HCl, pH 7.5, 0.1 m NaCl, coupled to a DAWN EOS spectrometer and OPTILAB DSP interferometric refractometer (Wyatt Technologies) at 25 °C. Peaks were detected as they eluted off the column with a UV detector at 280 nm, a light scattering detector at 690 nm, and a refractive index detector. The molar mass of the protein sample was determined from the Debye plot of light scattering intensity versus scattering angle. Data processing was performed with ASTRA software (Wyatt Technology Corp.).
Molecular Dynamics Simulations to Locate Surface Waters
A simulation to locate water molecule positions on the IBS of RiAFP was performed as described previously (34) with slight differences. The simulation consisted of an energy minimization step and two position-restrained molecular dynamics steps. The solvent in the box contained 6,220 water molecules and 5 Cl− ions to offset the charge of the protein. The first position-restrained step was a 100-ps simulation in which the system volume remained constant. The second position-restrained step involved a 20-ns simulation in which the system pressure remained constant, saving data at 0.02-ns intervals. The trajectory of the constant-pressure position-restrained simulation was used to calculate the water density around the protein after doing a least squares fit of the protein backbone using the first frame as a reference. Water density was calculated using the VolMap plugin (version 1.1) of VMD (version 1.9.1) with a resolution of 0.5 Å, atom size 1 Å, weights as mass, computed as the average for all saved frames. Waters were manually built into peaks in the VolMap density using PyMOL.
RESULTS AND DISCUSSION
Structure Determination of RiAFP
The RiAFP structure, which contains 1,914 nonhydrogen protein atoms and no atom heavier than sulfur in the asymmetric unit (26 kDa), is the largest structure determined to date by direct methods without anomalous scattering data. It also represents the first structure determined by direct methods at a resolution limit lower than 1.0 Å that has >1,000 nonhydrogen atoms. The procedure for ab initio structure determination presented here significantly extends the capability of direct methods and provides an alternative to traditional heavy atom-based methods for proteins of up to 26-kDa when crystallographic data are available to 1.2 Å or beyond. Data and refinement statistics are listed in Table 1.
Overall Structure of RiAFP
The structure of RiAFP reveals a new β-solenoid architecture in which the polypeptide chain follows a mixed handed superhelical trajectory to form a β-sandwich of two parallel 6- and 7-stranded β-sheets (Fig. 2, A and B). The central section forms a left handed superhelix of remarkable regularity where the β-sheets lie on top of each other with the upper and lower strands parallel but in the opposite orientation. The only deviations from this handedness and helix regularity occur at either end to form β-helix capping structures (Fig. 2B). At the N terminus there is a reversal of the handedness of the first two strands (β1 and β2), which results in β1 and β2 being antiparallel with their neighboring strands. At the C terminus, β-strands 12 and 13 loop back to insert between strands 10 and 11. These capping structures help to prevent end-to-end associations that would spoil the solubility of RiAFP and lead to oligomerization and aggregation. The three residues in β-strand 11 at the C terminus (Gln110, Gln112, and Ile114) that are too bulky to be accommodated into the core may also contribute to the capping structure to prevent amyloid-like polymerization.
FIGURE 2.

Crystal structure of RiAFP. A, overall fold of RiAFP. β-Strands are shown in blue and yellow. The threonine side chains and coordinated water molecules (red) on the IBS are shown in ball-and-stick representation. The disulfide bond is indicated in green. B, secondary structure diagram with sequentially numbered ice-binding β-strands in blue, hydrophilic β-strands in yellow. N and C represent the protein termini. C and D, flat ice-binding surface of RiAFP (C) and TmAFP (D), represented by a hydrophobic surface diagram produced in Chimera, which relies on the Kyte-Doolittle scale to rank amino acid hydrophobicity. Water molecules coordinated by the threonine hydroxyls on the IBS are shown in red. E, RiAFP dimers in the crystallographic asymmetric unit, with the ice-binding surfaces packed face-to-face.
Several AFPs have independently evolved to have a β-solenoid fold, including CfAFP from spruce budworm (21), TmAFP from yellow mealworm beetles (14), and MpAFP from an Antarctic bacterium (7). Also, the AFP from an inchworm (a different moth species from the spruce budworm) has been modeled as a β-solenoid (17). Whereas β-solenoids often form multimers (35), the AFPs are distinctly monomeric and are essentially devoid of twist. The lack of twist exemplified in RiAFP (Fig. 2, B and C) is essential for forming a flat IBS. A novel feature of the RiAFP solenoid is its compressed nature. In the core of RiAFP, the side chains within apposed β-strands from the two β-sheets are staggered, allowing the side chains to interdigitate and pack tightly against one another (Fig. 3). Similar interdigitation was observed in silk fibers (36) and was predicted in an inchworm AFP model (17). Most of the side chains in the core are from alanine and serine, with no residue larger than threonine. With this configuration, the average distance between the sheets is only 6 Å, compared with 10 Å across for TmAFP (14) and 14 Å across for CfAFP (21, 22), creating a more compact fold that may contribute to the high stability and antifreeze activity of RiAFP. In addition to the hydrophobic interactions within the core, hydrogen bonds between Thr65-Ser55, Thr85-Ser75, and Thr132-Ser124, along with a single disulfide bond between Cys4 and Cys21, link the two sheets together to further stabilize the β-sandwich fold. The β-turns in the structure contain mostly glycine or proline residues.
FIGURE 3.

The thin core of RiAFP spans less than 6 Å and is tightly packed with interdigitating Ala, Ser, and Thr residues. A, end-on and 180° view of the core residues in RiAFP contributing to the capping motifs. B, close up of the interdigitating alanine, serine, and threonine residues within the tightly packed, thin, stable core.
Two types of insect AFPs (TmAFP and CfAFP) contain repeated TXT motifs organized into arrays that form the ice-binding sites of the proteins (Figs. 2D and 4, C and F). Mutations of the threonines within these motifs decrease the thermal hysteresis activity of the AFPs (15, 37). RiAFP contains five expanded TXTXTXT motifs within the top (blue) β-sheet (β-strands 2, 3, 5, 7, 9, 11) (Fig. 2, A and C). These motifs are remarkably regular, allowing any row(s) (or column(s)) of TXTXTXT motifs to be exactly superposed onto any other row(s) (or column(s)). The threonine hydroxyls in these motifs define a flat IBS of 420 Å2 (Figs. 2C and 4D), a much larger surface area than the ice-binding sites of TmAFP (150 Å2) (14), CfAFP (190 Å2) (21, 22), sfAFP (80 Å2) (16), LpAFP (180 Å2) (38), and MpAFP (300 Å2) (7) (Fig. 4). The larger IBS of RiAFP (Figs. 2C and 4) may contribute to its greater thermal hysteresis activity compared with these other AFPs. Indeed, several examples are known through the observation of natural isoforms (21, 39) or protein engineering (40, 41) where an increase in the IBS correlates with higher antifreeze activity. The other β-sheet (β-strands 1, 4, 6, 8, 10, and 12) forms a more irregular, hydrophilic surface consistent with its role of interacting with the solvent as opposed to ice.
FIGURE 4.

Comparisons of ice-binding surfaces, colored blue, among sfAFP (A), LpAFP (B), TmAFP (C), RiAFP (D), MpAFP (E) and CfAFP (F).
Surface Complementarity to Ice
The adsorption-inhibition mechanism of AFPs was proposed by Raymond and DeVries 35 years ago (2). The driving force for AFP binding in the adsorption process was at one time thought to be the entropic gain from the liberation of the water molecules coordinated on the IBS, together with van der Waals interactions and hydrogen bonding (42). Adsorption of the AFP ice-binding surface to ice could be facilitated by the flatness of the IBS of the AFP. Indeed, like other insect AFPs, RiAFP has exceptional flatness along its ice-binding site. The flatness can be quantified by a “flatness function” (43) or by calculating the difference between the Cα-Cα and Cβ-Cβ distances of the first and last threonine residues within each row of TXT(XTXT) motifs. Smaller distances reflect reduced curvature of the surface. In the RiAFP structure, this difference is limited to 0.01 Å, whereas in T. molitor AFP the difference reaches 0.28 Å. By aligning ice-binding surface atoms of RiAFP to hexagonal ice, we identified four possible interactions of RiAFP with the primary prism plane of ice (Fig. 5). The shape complementarity (44) between protein and ice interfaces is comparable for all four binding modes, with Sc values of 0.75, 0.68, 0.70, and 0.78, respectively. A Sc value of 1.0 indicates perfect shape complementarity between two partners. For comparison, antigen-antibody complexes usually have their Sc values in the range of 0.64–0.68 (44). Although it is possible that RiAFP directly binds to ice along its entire IBS, a closer examination of the structure suggests a more indirect binding mechanism.
FIGURE 5.

Docking of RiAFP to the primary prism plane of a hexagonal ice lattice (light cyan) based on Sc calculations. Dashed lines (black) represent potential hydrogen bonds (within 2.4–3.2 Å). A, end-on view of one of four possible binding modes (Sc = 0.78) shown for illustrative purposes. The c-axis of the ice lattice is perpendicular to the plane of the page. B, 90° side view corresponding to all four binding modes of RiAFP to the primary prism plane. The c-axis of the ice lattice runs from left to right.
Ordered RiAFP-bound Waters and the Molecular Basis for Ice Binding
In the RiAFP structure, the threonine hydroxyls on the IBS bind three ranks of six water molecules with equivalent spacing between the four ranks of threonine side chains (Fig. 2A). These hydrogen-bonded water molecules have lost both translational and rotational freedom and resemble those in an ice lattice. They have lower average temperature factors (B factors) than the protein side chains (6.1 Å2 versus 9.7 Å2, respectively), indicating that the water molecules are bound tightly. The two molecules in the asymmetric unit are juxtaposed with their ice-binding surfaces in contact through the bound water (Fig. 2E). However, it is not clear from the crystal structure alone whether the positions of bound waters on the IBS of RiAFP are influenced by crystal contacts between the proteins. To evaluate this concern, we performed molecular dynamics simulations using the Gromacs MD software package (45) with a single molecule of RiAFP in a box of waters represented by the TIP5P model. After 20 ns the average positions of waters on the IBS of RiAFP matched very closely the 18 regularly spaced waters seen in the crystal structure (Fig. 6A). After restraining the protein during the simulation a second layer of waters became apparent. Indeed, many of the waters on the IBS of the second RiAFP molecule can be found in this layer (Fig. 6B). Overall, the waters observed in the simulation appear to be organized in an ice-like formation, with close matches to the primary prism and basal planes of ice. This supports an emerging idea that an AFP ice-binding surface is responsible for ordering an ice-like array of anchored “clathrate” water molecules to promote adsorption to ice, so that the IBS merges with and freezes to the ice surface (7, 8). An advantage of this indirect binding mechanism is that the organized waters are still fluid enough to make flexible matches to the ice-like quasi-liquid layer around the ice before becoming rigidified as the junction layer freezes. At this point, the protein is in direct contact with ice or an ice-like lattice.
FIGURE 6.

Ordered surface waters on the IBS of RiAFP. Crystallographic waters are in red, simulation waters are in pink. A, correspondence of water simulation and crystallographic waters in the threonine layer (layer 1) representing bound water molecules considered intrinsic to RiAFP. B, correspondence of water simulation and crystallographic waters in layer 2.
The average distance between the threonine hydroxyls within the TXTXTXT motifs (or between the bound waters between the threonine rows) is 6.66 Å (6.19–6.93 Å in chain A). The average distance between hydroxyls of these threonine residues in adjacent TXTXTXT motifs is 4.73 Å (4.44–5.38 Å). The two-dimensional array of residues formed by these threonine hydroxyls is a close match to the primary prism plane of the hexagonal ice lattice (7.35 Å along the c-axis and 4.52 Å along the a-axis), but a less good match to the basal plane (7.83 Å and 4.52 Å). However, the discrepancy in the longer distance is compounded by the wider IBS in RiAFP than in TmAFP or CfAFP, and it is unlikely that more than two ranks of threonine residues can fit well to either of these planes. What then is the advantage of the wider IBS? We suggest that having several ranks of matching hydroxyls/waters will give the RiAFP more than one opportunity to recruit a quorum of ice-like waters that can match the ice crystal lattice (Fig. 7). A corollary to this argument is that if the waters on the ice-binding site were to make a perfect match to one particular ice plane, then the AFP would only bind this plane.
FIGURE 7.
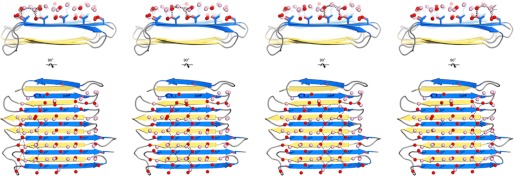
Dashed lines indicate a quorum of anchored clathrate waters organized by each of the four ranks of threonine residues. All four water patterns, any of which could potentially independently bind the AFP to ice, are illustrated. Crystallographic waters are in red; simulation waters are in pink. Dashed lines (black) represent potential hydrogen bonds (within 2.4–3.5 Å). It is unlikely that more than two ranks of anchored clathrate waters can simultaneously fit well to either the primary prism or basal plane of ice.
All the very active (hyperactive) AFPs bind to more than one ice plane, necessarily including the basal plane (42, 46). Aside from the superior thermal hysteresis activity of RiAFP, an early indication of protein hyperactivity and basal plane ice binding is provided by the dendritic pattern of ice growth seen after the nonequilibrium freezing point is exceeded (Fig. 8, A and B). Ice growth occurs from six prism surfaces and not out of the basal plane. However, when hyperactive AFPs are tested, most of them, including TmAFP, sfAFP (47), and MpAFP (7), bind to multiple planes as shown by adsorption to all surfaces of a single crystal hemisphere. GFP-tagged RiAFP also uniformly binds the entire surface of a single crystal hemisphere during fluorescence-based ice plane affinity analysis (Fig. 8, C and D). In contrast, an antifreeze protein from grass LpAFP clearly has a more limited ability to bind ice planes and only targeted the basal plane and primary prism plane (Fig. 8, E and F). It is possible that the ordered waters observed in the crystal structure of RiAFP extend for several additional layers in nature, providing options to bind to multiple other planes of ice, thereby resulting in total ice crystal hemisphere coverage.
FIGURE 8.

Ice-binding characteristics of RiAFP. A, an ice crystal formed in a solution containing RiAFP has a rounded, oval shape with the c-axis perpendicular to (coming out of) the plane of the page. B, at temperatures below the thermal hysteresis gap, the ice crystal “bursts” laterally along the a-axes in a 6-point dendritic pattern. C and D, top-down and side views of a single ice crystal hemisphere grown in the presence of GFP-RiAFP are shown. The direction of the c-axis is indicated by a white arrow. Green color represents the binding to ice and overgrowth of the GFP-tagged RiAFP, which in this experiment occurs over the entire hemisphere with no indication of preferential ice plane binding. E, an ice hemisphere grown in GFP-LpIBP with its c-axis perpendicular to the page shows binding to the basal plane (center) and six equivalent primary prism planes. F, an ice hemisphere grown in GFP-LpIBP reveals binding to three of the six equivalent primary prism planes, with the other three planes being hidden by the hemisphere. The images in E and F are reprinted with permission from Middleton et al. (38).
Like other hyperactive insect AFPs (14, 16, 21, 22), RiAFP crystallized with the ice-binding surfaces of two molecules facing each other and with extensive hydrogen bonding between the ordered water molecules on the two IBSs (Fig. 2E). However, multiangle light scattering and size-exclusion chromatography profiles show that RiAFP is a monomer in solution and that the ice-binding site is therefore fully accessible under physiological conditions (Fig. 9, B–D). Because the IBS mimics the ice lattice that it binds, it is not surprising that AFPs tend to associate through their ice-binding surfaces. However, the shape complementarity (44) between the two subunits in the RiAFP crystallographic dimer (Sc = 0.62) is not as high as between RiAFP and ice (Sc = 0.68 to 0.78), which explains why a RiAFP dimer is not stable in solution.
FIGURE 9.

Biophysical analysis of RiAFP in solution. A, the thermal hysteresis activities of recombinant RiAFP (black) and GFP-RiAFP (green) compared as a function of concentration. B, monodisperse peak of RiAFP elution from a Superdex 200 (10/300) column at 4 °C. C and D, multiangle light scattering profile of recombinant RiAFP (C) and GFP-RiAFP (D) at 25 °C.
Biological Implications
The longhorn beetle has a supercooling point of −28 °C (19). Recombinant RiAFP purified from Escherichia coli has a thermal hysteresis activity of up to 6 °C at 0.5 mg/ml (Fig. 9A), which closely matches the activity of RiAFP purified from insect hemolymph (20) and is the highest reported activity of any antifreeze polypeptide. The activity measured in vitro is likely to be a significant underestimation of the natural activity because the cooling rate and the initial ice crystal size are greater than those occurring in nature. It has been demonstrated that these two parameters are inversely proportional to the thermal hysteresis activity measured by experiment (48). Importantly, whereas other hyperactive antifreeze proteins have proven difficult to produce in large quantities, RiAFP is expressed in E. coli at concentrations of up to 50 mg/liter of cell culture (24). Hyperactive AFPs have not yet been used for applications outside basic science due to the difficulty and high cost of producing them. The hyperactivity and efficient recombinant production of RiAFP make it a suitable candidate for further study for applications requiring freeze resistance or the control of ice growth and morphology. The crystal structure reported here also provides a springboard from which to design increasingly active antifreeze proteins.
Notably, Rhagium mordax, a longhorn beetle from northern Europe and a close relative of R. inquisitor, produces several isoforms of a hyperactive AFP (RmAFP1–8), each with significant sequence identity (75–82%) to RiAFP (49). A molecular dynamics model of RmAFP1 reveals a β-solenoid prediction that is similar to the architecture RiAFP. However, the absence of capping motifs in the RmAFP model compared with our crystal structure reveals that our understanding of the complexity of protein folding, although advanced, remains incomplete.
Compared with AFPs from the Tenebrinoid beetles T. molitor and D. canadensis, which share a 12-residue consensus sequence TCTXSXXCXXAX and similar tertiary structures, our crystal structure of RiAFP revealed a tight, two-layered β-sandwich with a more extensive IBS, suggesting that not all AFPs from beetles share a common architecture. Indeed, the primary sequences of RiAFP and RmAFP are unlike those from any other protein, suggesting a different genetic origin and prompting RiAFP to be considered the second distinct type of AFP found in beetles. The surprising structural similarity of RiAFP to the unrelated inchworm antifreeze protein (iwAFP) provides another striking example of convergent evolution and of the paradigm of a regular and repetitive ice-binding site as Nature's preferred solution to reduce ice damage.
Acknowledgments
We thank N. Grindley and C. Joyce for the use of laboratory space.
This work was supported, in whole or in part, by National Institutes of Health Grant P01 GM022778 (to Y. M.) and Training Grant T32 GM08383. This work was also supported by a Burroughs Wellcome Investigator in the Pathogenesis of Infectious Disease award (to Y. M.), Yale College Dean's fellowships (to A. H. and D. F. Z.), the Arnold and Mabel Beckman Foundation and Barry M. Goldwater scholarships (to D. T.), funds from the Canadian Institutes for Health Research and the Canada Research Chair Program (to P. L. D.) and by the Raymond and Beverly Sackler Institute for Biological, Physical, and Engineering Sciences, the Yale Science and Engineering Association, Yale University West Campus, and the Yale Departments of Molecular Biophysics and Biochemistry, Molecular, Cellular, and Developmental Biology, Biomedical Engineering, and Physics.
The atomic coordinates and structure factors (code 4DT5) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- AFP
- antifreeze protein
- CfAFP
- Choristoneura fumiferana AFP
- DcAFP
- Dendroides canadensis AFP
- IBS
- ice-binding surface
- iwAFP
- inchworm AFP
- RiAFP
- Rhagium inquisitor AFP
- RmAFP
- Rhagium mordax AFP
- Sc
- shape complementarity
- sfAFP
- Hypogastura harveyi AFP
- TmAFP
- Tenebrio molitor AFP
- Mp
- Marinomonas primoryensis
- Lp
- Lolium perenne.
REFERENCES
- 1. Jia Z., Davies P. L. (2002) Antifreeze proteins: an unusual receptor-ligand interaction. Trends Biochem. Sci. 27, 101–106 [DOI] [PubMed] [Google Scholar]
- 2. Raymond J. A., DeVries A. L. (1977) Adsorption inhibition as a mechanism of freezing resistance in polar fishes. Proc. Natl. Acad. Sci. U.S.A. 74, 2589–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Knight C. A. (2000) Structural biology: adding to the antifreeze agenda. Nature 406, 249, 251 [DOI] [PubMed] [Google Scholar]
- 4. Nada H., Furukawa Y. (2011) Growth inhibition at the ice prismatic plane induced by a spruce budworm antifreeze protein: a molecular dynamics simulation study. Phys. Chem. Chem. Phys. 13, 19936–19942 [DOI] [PubMed] [Google Scholar]
- 5. Hayward J. A., Haymet A. D. J. (2001) Ice/water interface: molecular dynamics simulations of the basal, prism, {20–21}, and {2–1-10} interfaces of ice Ih. J. Chem. Phys. 114, 3713–3726 [Google Scholar]
- 6. Hobbes P. V. (1974) Ice Physics, pp. 525–725, Oxford University Press, Oxford [Google Scholar]
- 7. Garnham C. P., Campbell R. L., Davies P. L. (2011) Anchored clathrate waters bind antifreeze proteins to ice. Proc. Natl. Acad. Sci. U.S.A. 108, 7363–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nutt D. R., Smith J. C. (2008) Dual function of the hydration layer around an antifreeze protein revealed by atomistic molecular dynamics simulations. J. Am. Chem. Soc. 130, 13066–13073 [DOI] [PubMed] [Google Scholar]
- 9. Modig K., Qvist J., Marshall C. B., Davies P. L., Halle B. (2010) High water mobility on the ice-binding surface of a hyperactive antifreeze protein. Phys. Chem. Chem. Phys. 12, 10189–10197 [DOI] [PubMed] [Google Scholar]
- 10. Sicheri F., Yang D. S. (1995) Ice-binding structure and mechanism of an antifreeze protein from winter flounder. Nature 375, 427–431 [DOI] [PubMed] [Google Scholar]
- 11. Nishimiya Y., Kondo H., Takamichi M., Sugimoto H., Suzuki M., Miura A., Tsuda S. (2008) Crystal structure and mutational analysis of Ca2+-independent type II antifreeze protein from longsnout poacher, Brachyopsis rostratus. J. Mol. Biol. 382, 734–746 [DOI] [PubMed] [Google Scholar]
- 12. Antson A. A., Smith D. J., Roper D. I., Lewis S., Caves L. S., Verma C. S., Buckley S. L., Lillford P. J., Hubbard R. E. (2001) Understanding the mechanism of ice binding by type III antifreeze proteins. J. Mol. Biol. 305, 875–889 [DOI] [PubMed] [Google Scholar]
- 13. Liu Y., Li Z., Lin Q., Kosinski J., Seetharaman J., Bujnicki J. M., Sivaraman J., Hew C. L. (2007) Structure and evolutionary origin of Ca2+-dependent herring type II antifreeze protein. PloS One 2, e548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liou Y. C., Tocilj A., Davies P. L., Jia Z. (2000) Mimicry of ice structure by surface hydroxyls and water of a beta-helix antifreeze protein. Nature 406, 322–324 [DOI] [PubMed] [Google Scholar]
- 15. Graether S. P., Kuiper M. J., Gagné S. M., Walker V. K., Jia Z., Sykes B. D., Davies P. L. (2000) β-Helix structure and ice-binding properties of a hyperactive antifreeze protein from an insect. Nature 406, 325–328 [DOI] [PubMed] [Google Scholar]
- 16. Pentelute B. L., Gates Z. P., Tereshko V., Dashnau J. L., Vanderkooi J. M., Kossiakoff A. A., Kent S. B. (2008) X-ray structure of snow flea antifreeze protein determined by racemic crystallization of synthetic protein enantiomers. J. Am. Chem. Soc. 130, 9695–9701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin F. H., Davies P. L., Graham L. A. (2011) The Thr- and Ala-rich hyperactive antifreeze protein from inchworm folds as a flat silk-like β-helix. Biochemistry 50, 4467–4478 [DOI] [PubMed] [Google Scholar]
- 18. Venketesh S., Dayananda C. (2008) Properties, potentials, and prospects of antifreeze proteins. Crit. Rev. Biotechnol. 28, 57–82 [DOI] [PubMed] [Google Scholar]
- 19. Zachariassen K. E., Li N. G., Laugsand A. E., Kristiansen E., Pedersen S. A. (2008) Is the strategy for cold hardiness in insects determined by their water balance? A study on two closely related families of beetles: Cerambycidae and Chrysomelidae. J. Comp. Physiol. B 178, 977–984 [DOI] [PubMed] [Google Scholar]
- 20. Kristiansen E., Ramløv H., Højrup P., Pedersen S. A., Hagen L., Zachariassen K. E. (2011) Structural characteristics of a novel antifreeze protein from the longhorn beetle Rhagium inquisitor. Insect Biochem. Mol. Biol. 41, 109–117 [DOI] [PubMed] [Google Scholar]
- 21. Leinala E. K., Davies P. L., Doucet D., Tyshenko M. G., Walker V. K., Jia Z. (2002) A β-helical antifreeze protein isoform with increased activity: structural and functional insights. J. Biol. Chem. 277, 33349–33352 [DOI] [PubMed] [Google Scholar]
- 22. Leinala E. K., Davies P. L., Jia Z. (2002) Crystal structure of β-helical antifreeze protein points to a general ice binding model. Structure 10, 619–627 [DOI] [PubMed] [Google Scholar]
- 23. Mooers B. H., Matthews B. W. (2006) Extension to 2268 atoms of direct methods in the ab initio determination of the unknown structure of bacteriophage P22 lysozyme. Acta Crystallogr. D Biol. Crystallogr. 62, 165–176 [DOI] [PubMed] [Google Scholar]
- 24. Hakim A., Thakral D., Zhu D. F., Nguyen J. B. (2012) Expression, purification, crystallization and preliminary crystallographic studies of Rhagium inquisitor antifreeze protein. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 547–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oszlányi G., Süto A. (2004) Ab initio structure solution by charge flipping. Acta Crystallogr. A 60, 134–141 [DOI] [PubMed] [Google Scholar]
- 27. Palatinus L. (2004) Ab initio determination of incommensurately modulated structures by charge flipping in superspace. Acta Crystallogr. A 60, 604–610 [DOI] [PubMed] [Google Scholar]
- 28. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 29. Jia-xing Y., Woolfson M. M., Wilson K. S., Dodson E. J. (2005) A modified ACORN to solve protein structures at resolutions of 1.7 Å or better. Acta Crystallogr. D 61, 1465–1475 [DOI] [PubMed] [Google Scholar]
- 30. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for x-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 32. Emsley P., Cowtan K. (2004) COOT: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 33. Winn M. D., Murshudov G. N., Papiz M. Z. (2003) Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374, 300–321 [DOI] [PubMed] [Google Scholar]
- 34. Garnham C. P., Campbell R. L., Walker V. K., Davies P. L. (2011) Novel dimeric β-helical model of an ice nucleation protein with bridged active sites. BMC Struct. Biol. 11, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kajava A. V., Steven A. C. (2006) β-Rolls, β-helices, and other β-solenoid proteins. Adv. Protein Chem. 73, 55–96 [DOI] [PubMed] [Google Scholar]
- 36. Asakura T., Yao J. (2002) 13C CP/MAS NMR study on structural heterogeneity in Bombyx mori silk fiber and their generation by stretching. Protein Sci. 11, 2706–2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marshall C. B., Daley M. E., Graham L. A., Sykes B. D., Davies P. L. (2002) Identification of the ice-binding face of antifreeze protein from Tenebrio molitor. FEBS Lett. 529, 261–267 [DOI] [PubMed] [Google Scholar]
- 38. Middleton A. J., Marshall C. B., Faucher F., Bar-Dolev M., Braslavsky I., Campbell R. L., Walker V. K., Davies P. L. (2012) Antifreeze protein from freeze-tolerant grass has a β-roll fold with an irregularly structured ice-binding site. J. Mol. Biol. 416, 713–724 [DOI] [PubMed] [Google Scholar]
- 39. Chao H., Hodges R. S., Kay C. M., Gauthier S. Y., Davies P. L. (1996) A natural variant of type I antifreeze protein with four ice-binding repeats is a particularly potent antifreeze. Protein Sci. 5, 1150–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Baardsnes J., Kuiper M. J., Davies P. L. (2003) Antifreeze protein dimer: when two ice-binding faces are better than one. J. Biol. Chem. 278, 38942–38947 [DOI] [PubMed] [Google Scholar]
- 41. Marshall C. B., Daley M. E., Sykes B. D., Davies P. L. (2004) Enhancing the activity of a β-helical antifreeze protein by the engineered addition of coils. Biochemistry 43, 11637–11646 [DOI] [PubMed] [Google Scholar]
- 42. Scotter A. J., Marshall C. B., Graham L. A., Gilbert J. A., Garnham C. P., Davies P. L. (2006) The basis for hyperactivity of antifreeze proteins. Cryobiology 53, 229–239 [DOI] [PubMed] [Google Scholar]
- 43. Yang D. S., Hon W. C., Bubanko S., Xue Y., Seetharaman J., Hew C. L., Sicheri F. (1998) Identification of the ice-binding surface on a type III antifreeze protein with a “flatness function” algorithm. Biophys. J. 74, 2142–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lawrence M. C., Colman P. M. (1993) Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234, 946–950 [DOI] [PubMed] [Google Scholar]
- 45. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 46. Bar-Dolev M., Celik Y., Wettlaufer J. S., Davies P. L., Braslavsky I. (2012) New insights into ice growth and melting modifications by antifreeze proteins. J. R. Soc. Interface 9, 3249–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mok Y. F., Lin F. H., Graham L. A., Celik Y., Braslavsky I., Davies P. L. (2010) Structural basis for the superior activity of the large isoform of snow flea antifreeze protein. Biochemistry 49, 2593–2603 [DOI] [PubMed] [Google Scholar]
- 48. Takamichi M., Nishimiya Y., Miura A., Tsuda S. (2007) Effect of annealing time of an ice crystal on the activity of type III antifreeze protein. FEBS J. 274, 6469–6476 [DOI] [PubMed] [Google Scholar]
- 49. Kristiansen E., Wilkens C., Vincents B., Friis D., Lorentzen A. B., Jenssen H., Løbner-Olesen A., Ramløv H. (2012) Hyperactive antifreeze proteins from longhorn beetles: Some structural insights. J. Insect Physiol. 58, 1502–1510 [DOI] [PubMed] [Google Scholar]