

Background: The mitochondrial fission enzyme dynamin-related protein 1 (Drp1) is regulated via reversible phosphorylation of Ser-656.
Results: The Drp1 LXVP motif mediates dephosphorylation and activation by calcineurin (CaN), which influences mitochondrial morphology and survival post-injury in neurons.
Conclusion: The CaN-Drp1 signaling axis can be detrimental to injured neurons.
Significance: The CaN-Drp1 complex may be a target for neuroprotective therapeutic intervention.
Keywords: Calcineurin, GTPase, Ischemia, Membrane Fusion, Mitochondria, Protein-Protein Interactions, Serine-Threonine Protein Phosphatase, Dynamin-related Protein 1, Mitochondrial Fission
Abstract
Fission and fusion events dynamically control the shape and function of mitochondria. The activity of the mitochondrial fission enzyme dynamin-related protein 1 (Drp1) is finely tuned by several post-translational modifications. Phosphorylation of Ser-656 by cAMP-dependent protein kinase (PKA) inhibits Drp1, whereas dephosphorylation by a mitochondrial protein phosphatase 2A isoform and the calcium-calmodulin-dependent phosphatase calcineurin (CaN) activates Drp1. Here, we identify a conserved CaN docking site on Drp1, an LXVP motif, which mediates the interaction between the phosphatase and mechanoenzyme. We mutated the LXVP motif in Drp1 to either increase or decrease similarity to the prototypical LXVP motif in the transcription factor NFAT, and assessed stability of the mutant Drp1-CaN complexes by affinity precipitation and isothermal titration calorimetry. Furthermore, we quantified effects of LXVP mutations on Drp1 dephosphorylation kinetics in vitro and in intact cells. With tools for bidirectional control of the CaN-Drp1 signaling axis in hand, we demonstrate that the Drp1 LXVP motif shapes mitochondria in neuronal and non-neuronal cells, and that CaN-mediated Drp1 dephosphorylation promotes neuronal death following oxygen-glucose deprivation. These results point to the CaN-Drp1 complex as a potential target for neuroprotective therapy of ischemic stroke.
Introduction
Mitochondria exist in a spectrum of morphologies with individual spheres and highly interconnected networks at the extremes. Mitochondrial shape influences rates of respiration (1–4), calcium homeostasis (5–7), and transport of the organelle especially in highly polarized cells such as neurons (8). In addition, mitochondrial fission contributes to efficient cellular execution by apoptosis (9). Mitochondrial morphology is established by the opposing processes of mitochondrial fission and fusion, both of which are catalyzed by large GTPases of the dynamin family. The mechanoenzyme dynamin-related protein 1 (Drp1)2 catalyzes mitochondrial fission and this activity is dynamically regulated by several post-translational modifications (10).
As the best characterized post-translational modification, reversible phosphorylation of Drp1 at a conserved serine residue in the GTPase effector domain, Ser-656 (also notated as residue 617 or 637 depending on the Drp1 splice variant and species of origin), modulates mitochondrial fission activity with phosphorylation inhibiting fission. Phosphorylation of Ser-656 is catalyzed by the cAMP-dependent protein kinase (PKA) (11, 12) and has been shown to influence many interconnected facets of Drp1 activity including intermolecular assembly of larger Drp1 oligomers (13, 14), GTPase activity (12), and translocation to the outer mitochondrial membrane (15). Two phosphatases have been identified to be active at Drp1 Ser-656, protein phosphatase 2A in complex with its regulatory subunit Bβ2 (PP2A/Bβ2) (16), and calcineurin (CaN) (11, 15). In neurons, PP2A/Bβ2-mediated dephosphorylation of Drp1 regulates neuronal maturation (4) and survival following injury (16, 17). In cardiac tissue, expression of CaN negatively correlates with phosphorylation of Drp1 Ser-656 and survival of cardiomyocytes following ischemic injury (18).
CaN is a Ca2+/calmodulin (CaM)-dependent protein phosphatase ubiquitously expressed, but found at high levels in the mammalian central nervous system (19). CaN is composed of two subunits, a catalytic calcineurin A subunit (CNA) and a calcium-sensitive regulatory calcineurin B subunit (CNB) (20). Upon binding of Ca2+ to the EF hands of CNB and Ca2+/CaM to CNA, CNA undergoes a conformational change that dislodges its autoinhibitory domain from the catalytic cleft (21–23). This conformational change also exposes a composite hydrophobic docking surface made of the interface between CNA and CNB, which mediates binding to substrates containing an LXVP motif (24, 25). Signaling by CaN has long been recognized as contributing to the response of neurons to injury (26, 27). A number of pro-apoptotic and pro-survival targets of CaN-mediated phosphatase activity following neuronal injury have been described and include: the Bcl-2 family member Bad (28), the delayed rectifying K+ channel Kv2.1 (29), and the NMDA receptor (30). The activity of another pro-apoptotic Bcl-2 family member, Bax, was recently found to intersect with Drp1-mediated mitochondrial fission (31, 32). The possibility therefore exists that activation of Drp1 by CaN may be an integral part of a larger cellular response to injury.
Here, we tested the hypothesis that Ca2+/CaN signaling to Drp1 is partially responsible for neuronal death following ischemic injury. To this end, we developed methodologies to specifically disrupt or strengthen the CaN-Drp1 signaling axis while preserving other functions of the phosphatase and mechanoenzyme. We identified a conserved region on Drp1, a LXVP motif, similar to known CaN docking sites present in the transcription factor family NFAT (24, 33), the RIIα subunit of PKA (34), and the yeast protein regulator of calcineurin 1 (35). Through modification of the Drp1 LXVP motif we achieved bidirectional control of the enzyme-substrate interaction and CaN-mediated Drp1 dephosphorylation at Ser-656. Our results show that gain- and loss-of-function mutations of the LXVP motif influence mitochondrial morphology in HeLa cells and primary hippocampal neurons. Finally, we demonstrate that disruption of the CaN-Drp1 complex promotes survival of hippocampal neurons in a model of ischemic injury. Collectively these data show that Ser-656 dephosphorylation and activation of Drp1 by CaN participates in ischemic neuronal injury.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
The following commercially available antibodies were used: rabbit anti-calcineurin A (Cell Signaling), mouse anti-PP2A catalytic subunit (BD Transduction Laboratories), rabbit anti-ERK1/2 (Santa Cruz Biotechnology), mouse anti-DLP1/Drp1 (BD Transduction Laboratories), rabbit anti-GFP (ab290, Abcam), mouse anti-MTCO2 (cytochrome oxidase II, NeoMarkers), mouse anti-MAP2B (BD Transduction Laboratories), Alexa fluorophore-coupled secondary antibodies (Invitrogen), and infrared fluorophore-coupled secondary antibodies (Licor). Rabbit anti-GST was a kind gift of Dr. J. Hell (University of California at Davis, CA) (36), whereas the chicken anti-PP1 antibody was a generous gift of David Brautigan (University of Virginia). The mouse antibody specific for Drp1 phosphorylated at Ser-656 (pDrp1) was previously described (11). Hoechst 33342 was from Invitrogen. H89 and tautomycetin were from Tocris, calyculin A was from Calbiochem, and okadaic acid was from Axxora. Unless otherwise indicated all other reagents were purchased from Sigma.
Vectors
Drp1 replacement plasmids that allows for simultaneous knockdown of endogenous Drp1 by shRNA and expression of shRNA-resistant GFP-tagged, wild-type (WT) and Ser-656-mutant Drp1 were previously described (11). The Rattus norvegicus-derived Drp1 cDNA is the neuron-enriched splice variant containing all three alternatively spliced exons (GenBankTM accession number AF019043). Residues 585–662 were from the variable domain and GED was amplified from the above plasmid by PCR and fused to the C terminus of GST via BamHI and EcoRI restriction sites in the pGEX-2T plasmid (Amersham Biosciences). Mutations of the Drp1 LXVP motif were introduced by site-directed mutagenesis following the QuikChange protocol (Stratagene). The pGEX-4T2-based vector for recombinant expression of the GST-tagged tyrosine hydroxylase regulatory domain (residues 31–164) was a kind gift of Li-Huei Tsai (Harvard) (37). pDsRed2/mito was from Clontech. A vector for simultaneous expression of recombinant calcineurin Aα (CNA) and calcineurin B1 (CNB) from Homo sapiens was obtained from Addgene, Inc. (38). A vector for recombinant expression of CaM from R. norvegicus was a kind gift from Madeline Shea (University of Iowa) (39).
Expression and Purification of Recombinant Proteins
To prepare GST, GST-TH-(31-164), GST-Drp1-(585-662) WT, or LXVP motif mutants, and recombinant CNA/CNB, Escherichia coli BL21(DE3) (Invitrogen) were clonally grown in 1 liter of SOB (ampicillin) at 37 °C while shaking; A600 was monitored periodically. At A600 ≥ 0.6, isopropyl β-d-1-thiogalactoside and lactose were added to final concentrations of 0.5 mm and 5 g/liter, respectively, and growth was continued for an additional 8 h followed by isolation by centrifugation. Cells were resuspended in 50 ml of buffer containing 50 mm Tris, pH 7.5, 150 mm NaCl, 0.1 mm PMSF, 2 mm benzamidine, 2 mm CaCl2, sonicated, centrifuged, and subjected to affinity purification with glutathione-agarose or Calmodulin Affinity Resin (Agilent Technologies) in Interaction Buffer (20 mm Tris, pH 8.0, 100 mm NaCl, 6 mm MgCl2, 1.5 mm CaCl2). When necessary, GST-Drp1-(585–662) was eluted from the glutathione-agarose with Interaction Buffer + 10 mm glutathione. CNA/CNB was eluted with 20 mm Tris, pH 8.0, 100 mm NaCl, 10 mm EGTA. Protein purity was assessed by SDS-PAGE and protein concentration was determined by BCA analysis (Pierce, Thermo Scientific). Recombinant CaM was prepared exactly as previously described (39).
Recombinant Drp1 Dephosphorylation Assays
The contribution of all major Ser/Thr protein phosphatases to turnover of phosphorylated Drp1 Ser-656 was assessed using GST-Drp1-(585–662) as a model substrate. Recombinant GST-Drp1-(585–662) constructs immobilized on glutathione-agarose were prepared as described above and phosphorylated on Ser-656 with [γ-32P]ATP and recombinant PKA (gift of Susan Taylor, University of California, San Diego) and used for phosphatase reactions as previously described (16).
Soluble proteins from adult Sprague-Dawley rat (Harlan) brain and heart were used as a source of the major Ser/Thr protein phosphatase families. Tissue samples were surgically isolated, homogenized in Tissue Lysis Buffer (0.5 mg/ml of digitonin, 100 mm KCl, 20 mm HEPES, pH 7.5, 1 mm EDTA, 1 mm EGTA, 1 mm benzamidine, 5 μg/μl of leupeptin, 1 mm DTT, 1 mm PMSF) at 4 °C with 10 ml of Tissue Lysis Buffer per 1 g of tissue followed by centrifugation and the supernatant was buffer-exchanged using a Zeba Spin Column (Pierce, Thermo Scientific) into Reaction Buffer (50 mm Tris, pH 7.5, 100 mm NaCl, 0.2 mm EGTA, 2 mg/ml of BSA, 1 mm benzamidine, 0.1% (v/v) β-mercaptoethanol). The activities of the major Ser/Thr protein phosphatase families in the tissue samples were isolated by the addition of specific activators or inhibitors as follows (40): all Ser/Thr phosphatases (Reaction Buffer + 2 mm CaCl2, 20 mm MgCl2), no Ser/Thr phosphatases (Reaction Buffer + 2 mm EDTA + 10 nm tautomycetin + 20 nm calyculin A), PP2A (Reaction Buffer + 10 nm tautomycetin), CaN (Reaction Buffer + 10 nm tautomycetin + 2 mm CaCl2 + 20 nm calyculin A), PP2C (Reaction Buffer + 10 nm tautomycetin + 20 mm MgCl2 + 20 nm calyculin A), and PP1 (Reaction Buffer + 2.5 nm okadaic acid).
Phosphopeptide Dephosphorylation Assays
To quantitatively describe the effect of the Drp1 LXVP motif on the CaN catalytic cycle we performed kinetic studies using small phosphopeptides from Selleck Chemicals as model substrates. Phosphopeptides were dissolved in buffer containing 100 mm Tris, pH 7.5, 100 mm NaCl, 0.5 mm DTT, 1 mm MnCl2, 0.4 mm CaCl2, 100 μg/ml of BSA and concentrations were determined from mass. Phospho-Drp1 turnover by 25 nm CNA/CNB (prepared as described above) and 250 nm CaM (Sigma) after 1–30 min following addition of enzyme was measured using the PiColorLock Gold kit (Innova Biosciences) and a Synergy 4 plate reader (BioTek) equipped with a monochromator unit. Initial velocity data from three independent experiments were pooled and fit to the Michaelis-Menten equation using the NLStools package for the statistical software R (41).
Calcineurin Affinity Precipitation
To qualitatively assess the interaction between Drp1 and CaN, the GST-Drp1-(585–662) proteins described above were used to pull down CaN from crude brain lysate prepared in 0.5% Triton X-100, 25 mm Tris, pH 7.5, 50 mm NaCl, 2 mm DTT, 0.5 mm EDTA, 1 mm PMSF, ±2 mm EGTA, ±2 mm CaCl2. CaN bound to the immobilized protein was detected by Western blot analysis.
Isothermal Titration Calorimetry
To quantitatively describe the interaction between Drp1 and CaN, the dissociation constants (Kd) for the CaN-Drp1 complexes were determined by isothermal titration calorimetry (ITC). Free GST-Drp1-(585–662), CNA/CNB, and CaM were prepared as described above, concentrated with Amicon Ultra-15 Centrifugal Units (EMD Millipore), buffer exchanged into Interaction Buffer with Zeba Spin Columns, and co-dialyzed with 2 × 4 liters of Interaction Buffer for 24 h at 4 °C. Final protein concentrations were adjusted with filtered dialysate. Trimeric complexes of CNA-CNB-CaM were prepared by co-incubating CNA-CNB with CaM at a 20% molar excess. Protein samples were degassed prior to experimentation. ITC measurements were conducted in a VP-ITC (Micro-Cal) in Interaction Buffer at 25 °C with GST-Drp1-(585–662) constructs as the injector sample and CNA-CNB-CaM as the cell sample. A small, but not insignificant, heat of dilution was observed upon injection of the GST-Drp1-(582–662) into Interaction Buffer alone and was removed by subtraction. The observed integrated heats of interaction were fit to a single-site model of interaction and the Kd was determined using the ORIGIN software package (MicroCal). The average Kd and standard deviation were determined from three independent experiments.
Drp1 Dephosphorylation in Intact Cells
To assess the effect of the LXVP motif mutations on the phosphorylation state of Drp1 Ser-656 in intact cells we performed Western blot analysis to directly measure phospho-Drp1 in a cell line model system. COS1 cells were transfected with the indicated GFP-Drp1 construct using Lipofectamine 2000 (Invitrogen) (0.8%, 2 μg/ml of total DNA). 24 h later cells were treated with the adenylate cyclase agonist forskolin (10 μm) and the phosphodiesterase IV inhibitor rolipram (2 μm) to stimulate PKA-dependent phosphorylation of Drp1 at Ser-656. Drp1 Ser-656 phosphorylation was then assessed as previously described (16).
To assess the decay of phospho-Ser-656, COS1 cells transfected as above were pretreated with forskolin/rolipram (10/2 μm) for 30 min followed by treatment with the PKA inhibitor H89 (20 μm) for 5–120 min. Cells were lysed in SDS sample buffer 2.5 h after the initial forskolin/rolipram treatment and Drp1 Ser-656 phosphorylation was assessed as described above.
Phosphorylation was expressed as the ratio of pDrp1 to total Drp1 by densitometry using the gel analysis plug-in for ImageJ (National Institutes of Health). Time-dependent decay of the pDrp1 signal data was fit to a generalized four parameter logistic function (Equation 1) first described by David Rodbard of the NIH (42) and the area under the normalized and scaled curves was determined by integration.
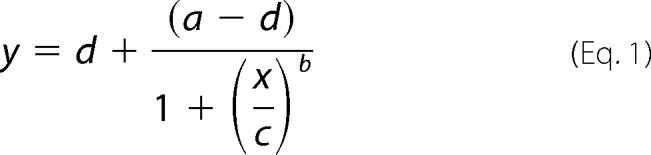 |
For analysis of the initial Drp1 Ser-656 phosphorylation state after forskolin/rolipram treatment the results were pooled from six independent experiments. For analysis of the time-dependent decay of the Drp1 Ser-656 phosphorylation signal the area under the normalized and scaled curves from four independent experiments was pooled.
Mitochondrial Morphology
To investigate the consequences of modification of the CaN-Drp1 signaling axis by mutation of the Drp1 LXVP motif we assessed mitochondrial morphology in a heterologous cell line model system and cultured primary hippocampal neurons. HeLa cells were transfected with the indicated GFP-Drp1 construct using Lipofectamine 2000 (0.6%, 2 μg/ml of total DNA). 24–48 h after transfection cells were fixed with 4% paraformaldehyde, labeled for immunofluorescence microscopy as previously described (14), and imaged with a Leica DMI4000B epifluorescence microscope with a ×100 oil-immersion objective.
Rat primary hippocampal neurons were cultured as previously described (4) and on day in vitro (DIV) 10 neurons were transfected with Lipofectamine 2000 (0.1%, 1 μg/ml of total DNA) containing 75% of the indicated GFP-Drp1 plasmid and 25% pDsRed2/mito, by mass. Three days later neurons were fixed with 4% paraformaldehyde, nuclei were stained with Hoechst 33342 (1 μg/ml), and imaged with a Leica DMI4000B epifluorescence microscope with a ×40 oil-immersion objective.
Images were pre-processed and analyzed for mitochondrial morphology as described (14). The morphology of mitochondria was assessed using a previously described macro (43) for ImageJ (NIH) and expressed in terms the average form factor (ff = perimeter2/(4 × π × area)), aspect ratio (ar = major axis/minor axis), or length of the mitochondria in each imaged cell. For analysis of the mitochondrial morphology in neurons, only mitochondria within neuronal processes were analyzed after masking out-of-focus soma fluorescence. In all experiments, cells were imaged, processed, and analyzed by an observer blinded to the experimental conditions.
Neuronal Survival
To investigate the effect of disrupting the CaN-Drp1 signaling axis on the survival of cultured neurons following injury we used oxygen-glucose deprivation (OGD) as a model of ischemic injury. Hippocampal neurons were prepared, maintained, and transfected with GFP-Drp1 constructs as described above. On DIV 13 neurons were challenged with OGD by replacement of the growth medium with Neurobasal medium without glucose (formula 05–0128DJ, Invitrogen) supplemented with B27, glutamine (0.5 mm), and HEPES (10 mm) and maintained in a Billups-Rothenberg chamber with 95% N2 and 5% CO2 for 20 min followed by media exchange with conditioned growth medium. Sham treatment consisted of replacement of glucose-containing culture medium and incubation in an environment of 95% air, 5% CO2 for 20 min followed by media exchange with conditioned growth medium. After a 24-h recovery period, neurons were fixed with 4% paraformaldehyde and labeled for GFP and MAP2B by immunofluorescence, and nuclei were stained with Hoechst 33342 (1 μg/ml). As the OGD injury paradigm was found to cause rapid detachment of dead neurons a methodology similar to that previously described was used to assess survival (44). Briefly, live transfected neurons in each well were identified in all conditions as those that met the following criteria: positive for GFP, positive for MAP2B, non-fragmented, non-condensed nuclear morphology. The total number of live transfected neurons in the sham and OGD-treated conditions was counted by an observer blinded to the experimental conditions. Survival of neurons following OGD treatment was expressed as a percentage of total neurons exposed to sham treatment.
Statistical Analyses
Intra-group variations were assessed by analysis of variance. Individual comparisons between a single experimental group and Drp1 WT were made by one-tailed two-sample Student's t test with the Welch correction applied as appropriate, *, p < 0.05; **, p < 0.01; ***, p < 0.005. Simultaneous comparison of multiple experimental groups to Drp1 WT were made by one-tailed Dunnett's test, */#, p < 0.05; **/##, p < 0.01; ***/###, p < 0.005, using the statistical software R (41).
RESULTS
CaN Is the Dominant Protein Phosphatase toward Drp1 Ser-656 in Brain and Heart
We and others have previously implicated CaN as a phosphatase responsible for dephosphorylating Drp1 Ser-656 and subsequent activation of Drp1-mediated mitochondrial fission (11, 15, 18, 45, 46). However, we have since found that another protein phosphatase, PP2A/Bβ2, is capable of dephosphorylating Drp1 Ser-656 as well (16). We therefore assessed the relative contribution of the four major families of protein Ser/Thr phosphatases toward turnover of the phosphate at Drp1 Ser-656. Soluble proteins were isolated from rat brain (Fig. 1A, top) and heart tissue (Fig. 1A, bottom), the latter selected for purposes of comparison and because Drp1 phosphoregulation was previously implicated in cardiomyocyte injury (18). A GST fusion protein of the Drp1 variable domain, GST-Drp1-(585–662), was phosphorylated at Ser-656 with [γ-32P]ATP by PKA (11). Relative phosphatase activities toward 32P-labeled Drp1-(585–662) were determined by co-incubating the substrate and tissue samples in the presence or absence of phosphatase-specific activators and inhibitors (40), as indicated in Fig. 1A. Western blotting of extracts used in phosphatase assays showed higher levels of CaN in brain than in heart, and roughly equivalent levels of PP1 and PP2A in both organs (Fig. 1B). As the PP2C family consists of 22 divergent catalytic subunits (47), PP2C levels were not assessed by Western blot. Notwithstanding that important substrate specificity determinants such as subcellular localization are not preserved in in vitro assays, these experiments point to CaN as the dominant Drp1 Ser-656 phosphatase in both brain (Fig. 1A, top) and heart (Fig. 1A, bottom).
FIGURE 1.
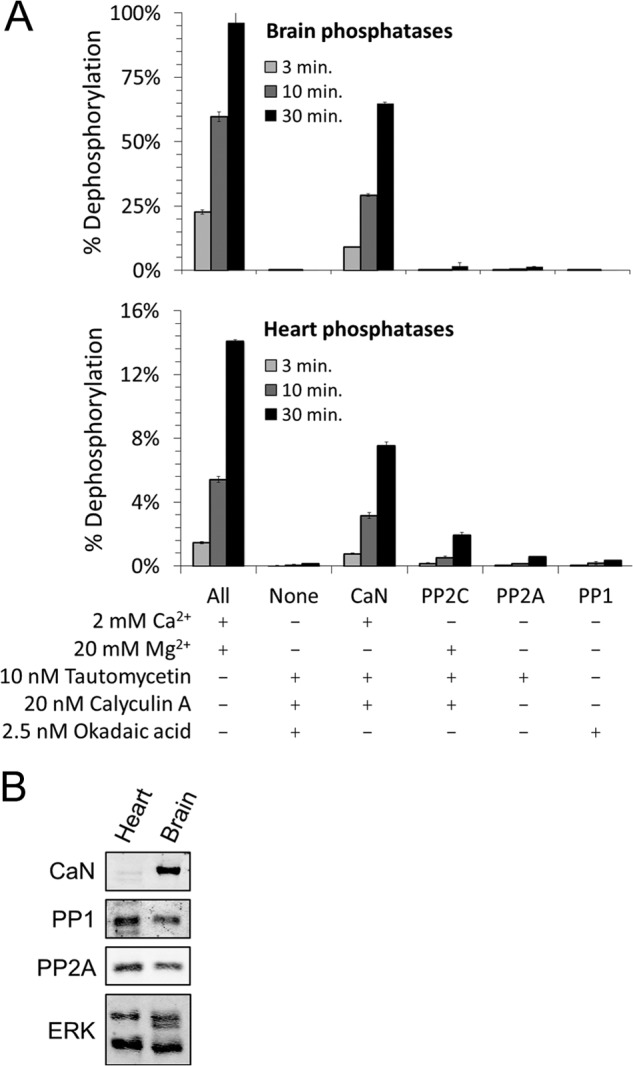
CaN is the major Drp1 Ser-656 phosphatase in brain and heart tissue. A, GST-Drp1-(585–662) was phosphorylated in vitro with [γ-32P]ATP and PKA and used as a substrate for phosphatases in digitonin extracts from adult rat brain (top) and heart (bottom). The activities of PP1, PP2A, PP2B (CaN), and PP2C were isolated by the addition of specific activators or inhibitors as indicated. Reactions were terminated after 3, 10, and 30 min by the addition of 10% TCA. Soluble 32P was quantified by liquid scintillation counting and expressed as percent of input (mean ± S.E. from four independent experiments). B, Western blots of the above tissue extracts for the catalytic subunits of CaN (CNAα/β), PP1, and PP2A; ERK is a loading control.
The LXVP Motif of Drp1 Influences CaN-Drp1 Complex Formation
Sequence alignments revealed a phylogenetically conserved region in the variable domain of Drp1 just upstream of the Ser-656 site (Fig. 2A). Residues 645–648 of Drp1 are identical to residues upstream of an autophosphorylation site in the RIIα subunit of PKA, which were previously shown to mediate avid dephosphorylation by CaN (34). They also align with a calcium-dependent CaN docking site found in the transcription factor family NFAT. Cooperating with a second, calcium-independent CaN docking motif (PXIXIT (48)), the NFAT LXVP motif is required for efficient for multisite dephosphorylation and nuclear translocation of NFAT (24, 33). We therefore speculated that the LXVP motif of Drp1 may play a similar role for recruitment of CaN and subsequent dephosphorylation of Ser-656.
FIGURE 2.
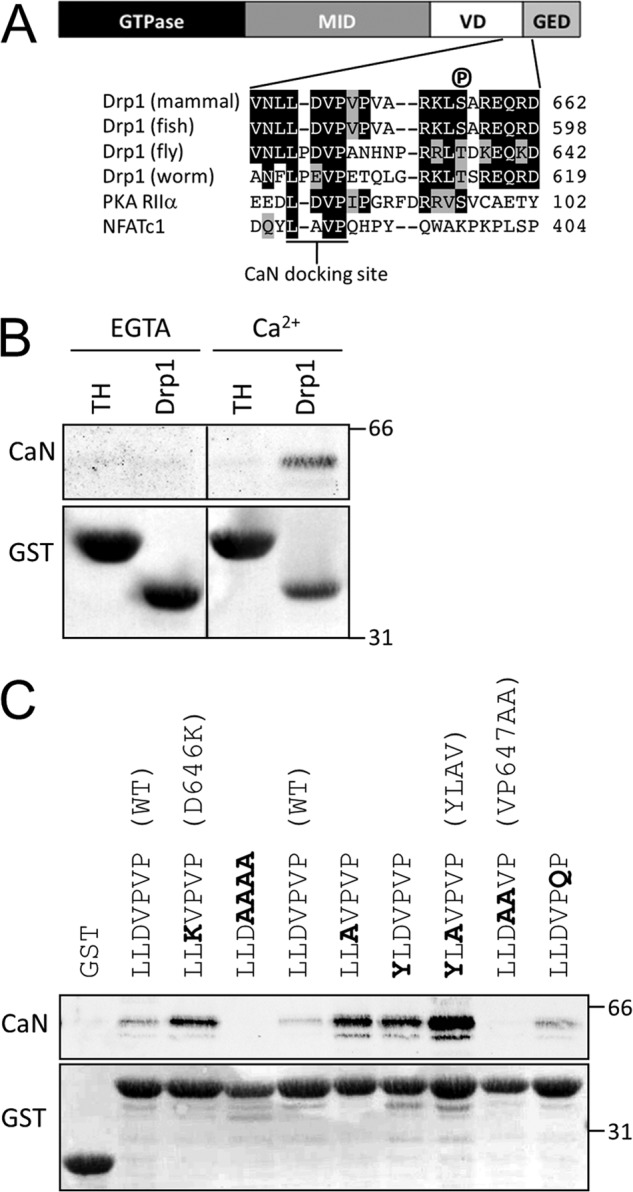
A LXVP motif in Drp1 mediates Ca2+-dependent binding of CaN. A, a Drp1 schematic indicates the GTPase (GTP), middle (MID), variable (VD), and GTPase effector domains (GED). Drp1 sequences spanning the VD-GED from mammals (e.g. R. norvegicus, NP_446107.2), fish (Danio rerio, NP_957216.1), fly (Drosophila melanogaster, NP_608694.2), and worm (Caenorhabditis elegans, NP_741403.2) are shown aligned with the cAMP-dependent protein kinase A regulatory subunit IIα (PKA RIIα) from Bos taurus (NP_001178296.1) and nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1) from H. sapiens (NP_765978.1). B, immobilized GST-Drp1-(585–662) (Drp1), but not an irrelevant GST fusion protein (TH), pulls down CaN from rat brain lysates in a Ca2+-dependent manner. C, Drp1 LXVP motif mutations in the context of GST-Drp1-(585–662) increase or decrease the interaction between Drp1 and CaN by GST pulldown from rat brain lysate in the presence of Ca2+ (1 mm). The position of molecular mass markers (kDa) is indicated on the right. Shown in parentheses are mutant designations used in later studies. Blots are representative of at least three independent experiments.
To begin investigating the role of the Drp1 LXVP motif in the interaction between CaN and Drp1, we affinity isolated endogenous CaN from rat brain lysate with GST-Drp1-(585–662). GST-Drp1, but not an unrelated GST fusion protein containing the regulatory domain of tyrosine hydroxylase (GST-TH-(31–164)), isolated CaN from brain lysate in a calcium-dependent manner (Fig. 2B). To show that the interaction between CaN and Drp1 depends on the LXVP motif, we introduced a series of point mutations into Drp1-(585–662) and assessed CaN affinity by GST pulldown from brain lysates in the presence of calcium (Fig. 2C). Replacement of the Val-Pro dipeptide in the LXVP motif with Ala (V647A/P648A = VP647AA) eliminated CaN binding. On the other hand, Asp-646 (residue “X” in “LXVP”) appears to be a negative binding determinant, because substitution with either Lys or Ala (D646K, D646A) enhanced CaN binding compared with wild-type GST-Drp1-(585–662). Replacing both Asp-646 and Leu-644 to increase similarity to the LXVP motif of NFATc1 resulted in a further increase in Drp1-CaN complex formation (Fig. 2C, L644Y/D646A = YLAV mutant).
To quantitatively describe the interaction between CaN and Drp1, we turned to ITC. Wild-type or LXVP-mutant GST-Drp1-(585–662) was injected into a calorimetric cell containing recombinant CNA/CNB dimer complexed to Ca2+/CaM, and the dissociation constant (Kd) was determined from the evolved heat (see “Experimental Procedures”). The complex of CaN with wild-type GST-Drp1-(585–662) had a Kd of 27 μm (Fig. 3 top, n = 3 experiments). Consistent with the GST pulldown results, the NFATc1-mimicking YLAV mutation enhanced the stability of the Drp1-CaN complex by ∼3-fold (Kd = 8.8 μm, Fig. 3, middle), whereas complex formation between CaN and Drp1 VP647AA was not detectable (Fig. 3, bottom). These studies show that the NFAT-like LXVP motif in Drp1 mediates association with CaN, and that the YLAV and VP647AA mutations allow for bidirectional control of the association strength.
FIGURE 3.
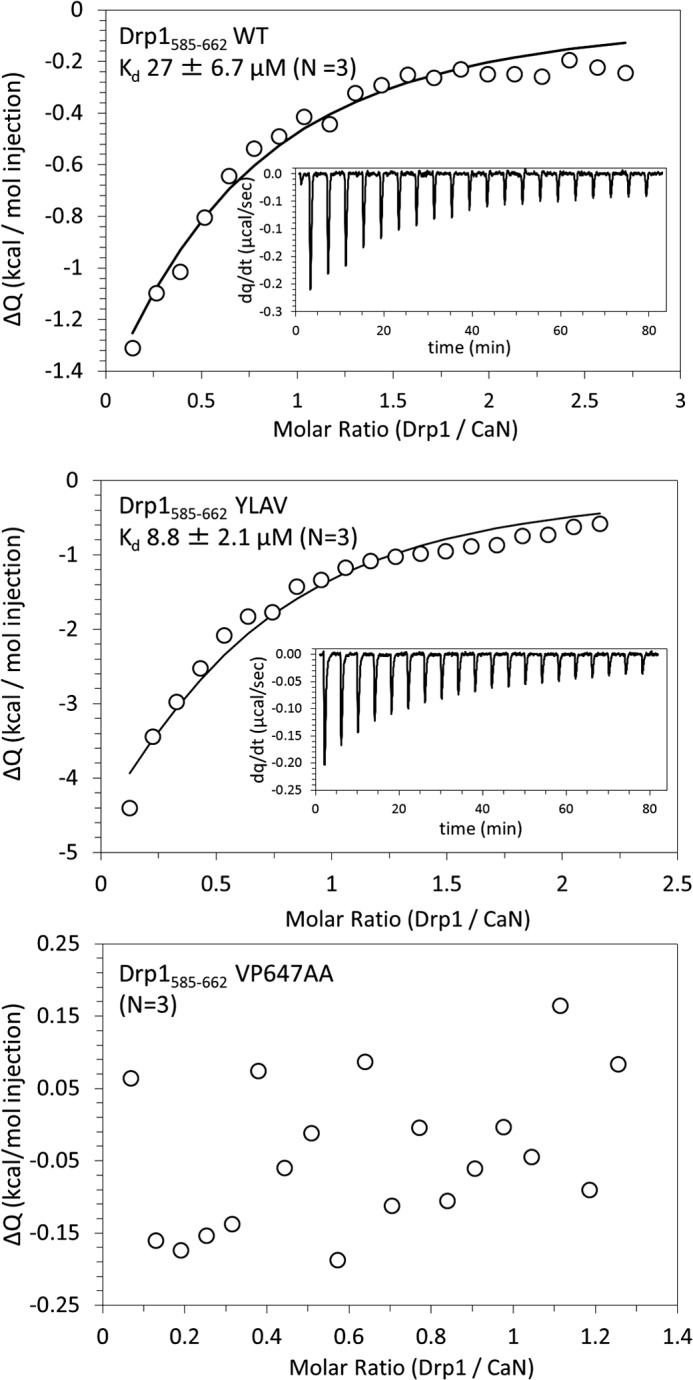
Stability of wild-type and LXVP mutant CaN-Drp1 complexes. Isothermal titration calorimetry was used to measure dissociation constants (Kd) of CaN complexed to wild-type Drp1 (top), Drp1 YLAV (middle), and Drp1 VP647AA (bottom). GST-Drp1(585–662) (100–450 μm) was used as the injected sample and CNA/CNB/CaM (10–20 μm) was used as the cell sample at 25 °C. Integrated heats of interaction (circles), best fit line (solid line), and the evolved heat of interaction as a function of time (inset) from one of three independent experiments for each condition are shown. The Kd (mean ± S.D.) is shown for Drp1 WT and Drp1 YLAV with CaN; no interaction between Drp1 VP647AA and CaN was detectable.
The LXVP Motif Influences CaN-catalyzed Drp1 Dephosphorylation in Vitro
Our GST pulldown and ITC experiments measured the stability of a complex between CaN and its substrate Drp1 in its dephosphorylated form. It is conceivable that increased stability of the CaN-Drp1 complex could reduce catalytic turnover of CaN by slowing dissociation of the enzyme-product complex (i.e. product inhibition). To examine the role of the Drp1 LXVP motif in Ser-656 dephosphorylation by CaN, we phosphorylated wild-type and mutant GST-Drp1-(585–662) with [γ-32P]ATP and PKA in vitro, and measured 32P release upon incubation with brain lysate under conditions that isolate CaN activity (1 mm CaCl2, 2.5 μm okadaic acid). The stability of the CaN-Drp1 complex correlated with rates of Drp1 Ser-656 dephosphorylation, with YLAV and VP647AA mutants displaying, respectively, accelerated and decelerated 32P release compared with wild-type Drp1 (Fig. 4A).
FIGURE 4.

The Drp1 LXVP motif determines Ser-656 dephosphorylation by CaN in vitro. A, GST-Drp1-(585–662) WT, YLAV, and VP647AA were phosphorylated with [γ-32P]ATP and PKA in vitro and used to assay CaN activity isolated from brain lysates by the inclusion of 2 mm Ca2+ and 2.5 μm okadaic acid. At the indicated times, phosphatase reactions were stopped by addition to 10% TCA and released 32P was expressed as a percentage of initial substrate (mean ± S.E. from three independent experiments). Linear portions of the plots were fit by linear least squares regression (solid line), the nonlinear portion of the plot continues as a dashed line. B shows sequences of Drp1-derived phosphopeptides that were used as substrates for enzyme kinetics studies. C–F, dephosphorylation kinetics were determined with CNA/CNB (25 nm) and Ca2+/CaM (1 mm/250 nm) incubated (1–30 min, 25 °C) with the indicated concentrations of Drp1 phosphopeptides using a malachite green-based colorimetric assay. Initial velocities from three independent experiments were pooled (mean ± S.D. shown as circles) and fit to the Michaelis-Menten equation (solid line). Kinetic parameters are listed in Table 1.
We next examined kinetic parameters of Drp1 phosphopeptide dephosphorylation by CaN as a function of the LXVP motif. We first tested phosphatase activity of our recombinant CNA/CNB dimer toward the model substrate para-nitrophenyl phosphate in the presence of Ca2+/CaM. Fitting the initial velocity to the Michaelis-Menten equation yielded a Michaelis-Menten constant (Km) of 124 mm and a maximal velocity (kcat) of 1.98 s−1 (supplemental Fig. S1), in good agreement with the literature (38, 49). Listed in Fig. 4B, phosphopeptides derived from wild-type and LXVP-mutant Drp1 were assayed next. The “Drp1 short” peptide consists of Drp1 residues 653–663, thus excluding the LXVP motif. “WT”, “YLAV” and “VP647AA” peptides correspond to phospho-Ser-656 Drp1-(643–660) with respective substitutions in the LXVP motif (Fig. 4B). Using recombinant CaN (CNA/CNB dimer), Ca2+/CaM, and variable phosphopeptide concentrations in a fixed time point assay based on the colorimetric reaction of free phosphate with malachite green, we observed that inclusion of the LXVP motif enhanced catalytic efficiency (kcat/Km) by more than 2 orders of magnitude (Fig. 4, C and D, and Table 1). The YLAV mutation increased kcat/Km by an additional 30%, as a consequence of a 4-fold decrease in Km and a 3-fold decrease in kcat compared with wild-type Drp1-(643–660). In contrast, the VP647AA substitution lowered catalytic efficiency by ∼30%, as a result of a 2-fold increase in Km and a slight increase in kcat with respect to the wild-type peptide (Fig. 4, D and F, and Table 1). Thus, effects of the LXVP mutation on Kd are positively correlated with Km, but also kcat, the latter presumably because LXVP mutations affect product release from as well as substrate binding to the enzyme. Altogether, these studies indicate that the LXVP docking sequence dictates catalytic efficiency of Drp1 Ser-656 dephosphorylation by CaN.
TABLE 1.
Kinetics of Drp1 phosphopeptide dephosphorylation by CaN
CaN (CNAα/CNBα, 25 nm) and Ca2+/CaM (1 mm/250 nm) were incubated (30 min, 25 °C ) with Drp1 phosphopeptides (see Fig. 4B for sequences) at varying concentrations (Drp1 short: 94–3000 μm, others: 6–750 μm) and phosphate release was quantified using a colorimetric assay. Initial velocity data was fit to the Michaelis-Menten equation to determine the parameters, Km, kcat, and kcat/Km; values in parentheses represent the S.E. of the fit.
| Substrate peptide | kcat | Km | kcat/Km | n |
|---|---|---|---|---|
| s−1 | μm | μm−1 s−1 (×10−2) | ||
| Drp1 short | 0.67 (0.047) | 2160 (280) | 0.031 (0.005) | 3 |
| Drp1 WT | 7.6 (0.34) | 160 (22) | 4.8 (0.69) | 3 |
| Drp1 YLAV | 2.5 (0.056) | 41 (3.5) | 6.1 (0.54) | 3 |
| Drp1 VP647AA | 9.5 (0.37) | 290 (26) | 3.3 (0.32) | 3 |
The Drp1 LXVP Motif Influences Drp1 Dephosphorylation Kinetics in Intact Cells
To examine the role of the LXVP motif in intact COS1 cells, we replaced endogenous Drp1 with mutant GFP-Drp1 by transient transfection of a plasmid that expresses shRNA targeting endogenous Drp1 together with RNAi-resistant Drp1 cDNA (11). Ser-656 phosphorylation of GFP-Drp1 was measured by dual-channel immunoblotting with a phosphospecific and a total Drp1 antibody (11). To allow for accurate quantification, the phospho-Ser-656 Drp1 signal was boosted by raising [cAMP] via concomitant stimulation of adenylate cyclase (forskolin, 10 μm) and inhibition of phosphodiesterases (rolipram, 2 μm) for 1 h prior to cell lysis (13, 16). Two Drp1 mutations that enhanced CaN association in vitro (D646K, YLAV) were found to lower steady-state phosphorylation of GFP-Drp1. Conversely, blocking CaN binding with the VP647AA substitution resulted in Drp1 hyperphosphorylation at Ser-656 (Fig. 5, A and B).
FIGURE 5.

The Drp1 LXVP motif determines Ser-656 dephosphorylation by CaN in intact cells. A and B, COS1 cells were transfected with the indicated GFP-Drp1 plasmids, which simultaneously silence endogenous Drp1 via shRNA. After 24 h cells were treated for 1 h with forskolin/rolipram (10 μm/2 μm) followed by cell lysis and immunoblotting for phospho-Ser-656 (p) Drp1 and total Drp1 (GFP antibody, representative blot in A). B shows densitometric quantification of pDrp1/Drp1 (mean ± S.E. of six independent experiments). C, Drp1 dephosphorylation was monitored in intact cells, treating COS1 cells prepared as in A with the PKA inhibitor H89 (20 μm) for 5–120 min prior to cell lysis and assessing Drp1 Ser-656 phosphorylation as above (C, representative blots). D shows pDrp1/Drp1 densitometry, scaled to maximum (0 min) and minimum (120 min) phosphorylation, with the inset plotting area under the curve determined by integration of the curve fit (mean ± S.E. of four independent experiments). For statistics, gain-of-function (YLAV and D646K) and loss-of-function (VP647AA) mutations were grouped separately and compared with Drp1 WT first by analysis of variance to identify intragroup variation followed by one-tailed Dunnett's test for Drp1 YLAV and Drp1 D646K (#, p < 0.05; ##, p < 0.01; ###, p < 0.005) or one-tailed Student's t test with the Welch correction for Drp1 VP647AA (*, p < 0.05; **, p < 0.01).
We next employed an intact cell phosphatase assay, following Drp1 dephosphorylation for up to 2 h after adding the PKA inhibitor H89 (20 μm) to medium containing forskolin/rolipram. Immunoblotting total cell lysates with phospho- and total Drp1 antibodies revealed faster decay of Drp1 phosphorylation when affinity of the LXVP motif was increased by the D646K or YLAV mutations. Conversely, decreasing the stability of the CaN-Drp1 complex with the VP647AA mutation slowed Drp1 dephosphorylation (Fig. 5, C and D). Measuring the area under the decay curve revealed significant effects (Fig. 5D, inset). These results further validate Drp1 LXVP mutants as tools for bidirectional manipulation of the CaN-Drp1 signaling axis.
The Drp1 LXVP Motif Controls Mitochondrial Morphology via Ser-656
The Ser-656 phosphorylation state of Drp1 is a robust determinant of mitochondrial morphology (11–13, 15, 16), with phosphorylation arresting the Drp1 translocation cycle either in the cytosol or at the OMM to shift the balance toward mitochondrial fusion (13, 14). We exploited the LXVP-mutant Drp1 to interrogate the role of CaN in shaping mitochondria, initially by Drp1 replacement in HeLa cells, a cell line amenable to facile imaging of the mitochondrial network by epifluorescence microscopy. Images of GFP-Drp1 positive cells processed for immunofluorescence with an antibody against cytochrome oxidase subunit II were subjected to mitochondrial morphometry (13, 14). Compared with wild-type GFP-Drp1 and as reported previously in several cell types (11, 13, 15), phospho-mimetic (Asp) and phosphorylation-blocking (Ala) substitutions of Drp1 Ser-656 increased and decreased, respectively, mitochondrial form factor, a measure of mitochondrial elongation with a minimum value of 1 for perfectly round mitochondria (Fig. 6B). Promoting CaN association via the YLAV substitution in Drp1 phenocopied mitochondrial fragmentation by the Drp1 S656A mutant. On the other hand, destabilizing the CaN-Drp1 complex (Drp1 VP647AA) reproduced mitochondrial elongation by pseudophosphorylated Drp1 (S656D), falling slightly short of GTPase-deficient, K38A-mutant Drp1 (Fig. 6, A and B). We next set out to confirm that LXVP motif modifications impact mitochondrial shape via Ser-656, rather than via other phosphorylation sites (50) or nonspecific effects on protein folding. To this end, we combined mutations of the LXVP motif and Ser-656 in the same polypeptide. Indeed, blocking phosphorylation (S656A) overrode mitochondrial elongation by Drp1 that cannot bind CaN (VP647AA), whereas constitutive phosphorylation (S656D) overcame mitochondrial fragmentation due to enhanced stability of the Drp1-CaN complex (YLAV, Fig. 6, A and B). Because Ser-656 mutations rendered LXVP mutations completely ineffectual, CaN associated with Drp1 appears to promote mitochondrial fission exclusively by virtue of Ser-656 dephosphorylation.
FIGURE 6.

The CaN docking motif in Drp1 influences mitochondrial shape via Ser-656. A and B, HeLa cells were substituted with WT and LXVP-mutant GFP-Drp1 (green), fixed, and labeled for mitochondria (mito, red) by immunofluorescence (cytochrome oxidase II) and for nuclei with Hoechst 33342 (blue). A shows representative images, whereas B plots morphology (form factor) of mitochondria (mean ± S.E. of 258–412 cells from three to four independent transfections). For statistics, gain- and loss-of-function mutants were grouped separately and compared with Drp1 WT first by analysis of variance to identify intragroup variation followed by one-tailed Dunnett's test (*/#, p < 0.05; **/##, p < 0.01; ***/###, p < 0.005).
CaN Regulation of Drp1 Influences Mitochondrial Morphology in Neuronal Processes
We previously showed that reversible phosphorylation of Drp1 at Ser-656 dictates mitochondrial morphology in neurons, influencing survival, dendrite outgrowth, and synaptogenesis likely via bioenergetic mechanisms (4, 13). To assess the contribution of CaN to Drp1 activity in neurons, Drp1 endogenous to hippocampal neurons cultured from embryonic rats was replaced with wild-type or mutant GFP-Drp1. As determined by quantitative immunofluorescence microscopy, GFP-Drp1 replacement did not change cellular Drp1 levels (supplemental Fig. S2). Mitochondria were labeled with a matrix-targeted fluorophore, DsRed2/mito, by co-transfection, imaged by fluorescence microscopy, and length, form factor, and aspect ratio of mitochondria in neuronal processes was determined by automated image analysis (Fig. 7). Compared with wild-type Drp1, enhanced CaN recruitment by the Drp1 YLAV mutant resulted in a trend toward mitochondrial fission; however, the latter effect was not statistically significant (Fig. 7, B and C), possibly because of low basal Ser-656 phosphorylation stoichiometry. Given high levels of CaN in the central nervous system (Fig. 2C) (19), it is also possible that CaN saturates even the suboptimal LXVP docking site on Drp1. In contrast, destabilization of the CaN-Drp1 complex with the VP647AA mutant resulted in highly significant mitochondrial elongation (Fig. 7C).
FIGURE 7.

CaN docking to Drp1 influences mitochondrial shape in primary hippocampal neurons. A–C, hippocampal neuron cultures were co-transfected at DIV 10 with GFP-Drp1 (green) replacement plasmids and DsRed2/mito to label mitochondria (mito, red). After 72 h, neurons were fixed and imaged by epifluorescence microscopy. A shows representative images; B and C plot mitochondrial morphology (form factor, aspect ratio, and length) in neuronal processes (mean ± S.E. of 271–297 neurons from 3 independent transfections). Comparison to Drp1 WT by one-tailed Student's t test with the Welch correction was used (***, p < 0.005).
Disruption of the CaN-Drp1 Complex Is Neuroprotective
Mitochondrial recruitment of PKA via protein kinase A anchoring protein 1 (AKAP1) promotes neuronal survival by way of Drp1 phosphorylation at Ser-656 and mitochondrial elongation (13). Conversely, CaN signaling has been shown to increase susceptibility to injury and subsequent neuronal death by apoptosis (27, 28). The relevant survival-opposing substrates of CaN have not been established, although the proapoptotic Bcl-2 family member Bad was implicated (28). To examine whether the CaN-Drp1 signaling axis promotes cell death following ischemic neuronal injury, we exposed cultured hippocampal neurons expressing wild-type or LXVP-mutant GFP-Drp1 to OGD (0 glucose, 95% N2, 5% CO2 for 20 min). Because this injury paradigm causes rapid detachment of dead cells from the substrate, we quantified percent neuronal survival by counting the total number of GFP/MAP2B double-positive cells with intact nuclei and neurites (Fig. 8A) following sham and OGD treatments for each transfection condition essential as described (44). Percent survival from 4 independent experiments and culture dates is shown in Fig. 8B. Although trends toward neuroprotection by Drp1 VP647AA and harm by Drp1 YLAV are apparent, effects are not significant because of culture to culture variations. However, normalizing survival to Drp1 wild-type before averaging experiments revealed significant neuroprotection by inhibiting CaN-mediated activation of Drp1 (VP647AA, Fig. 8C). We conclude from these results that CaN contributes to ischemic neuronal injury by docking to Drp1 and dephosphorylating Ser-656.
FIGURE 8.

The Drp1-CaN complex promotes neuronal demise following oxygen-glucose deprivation. A–C, endogenous Drp1 in DIV 10 hippocampal neurons was replaced with WT or LXVP mutant GFP-Drp1 followed by 20 min OGD or sham treatment at 13 DIV. After a 24-h recovery period, cultures were fixed, immunofluorescently labeled for GFP (green) and MAP2B (red), and assessed for survival by counting all healthy, transfected (GFP and MAP2B positive) neurons (arrow in A). B and C plot the results from four independent experiments (50–200 transfected neurons in the sham group) either as a combined scatter/box plot of percent survival (B, #OGD/#sham × 100%) or as mean ± S.E. (n = 4) after normalizing percent survival to Drp1 WT (C). Comparison to Drp1 WT by one-tailed Student's t test with the Welch correction (*, p < 0.05).
DISCUSSION
Our studies have identified a LXVP motif in the mitochondrial fission enzyme Drp1 that mediates an interaction with the Ca2+/CaM-dependent protein phosphatase CaN. As a consequence of this interaction, Drp1 is dephosporylated at Ser-656, which results in increased Drp1-mediated mitochondrial fission. Disrupting the CaN-Drp1 signaling axis leads to mitochondrial elongation and promotes survival of cultured hippocampal neurons following OGD.
Phosphorylated by PKA in response to cAMP elevation, Drp1 Ser-656 is a pivotal regulatory site targeted by at least two phosphatases, CaN and PP2A/Bβ2 (11, 15, 16). Dephosphorylation of Drp1 Ser-656 by PP2A/Bβ2 requires translocation of the PP2A holoenzyme to the OMM, a process that is regulated by phosphorylation of the mitochondrial targeting sequence of Bβ2 (16). Our in vitro analysis using total brain extracts indicates that, compared with CaN, PP2A contributes relatively little to the total phosphatase activity toward Drp1 Ser-656 (Fig. 1, A and B). In apparent contrast, our previous studies showed that knockdown of Bβ2 preserves mitochondrial networks and protects hippocampal neurons against several types of injury (17). The discrepancy between in vitro and intact cell studies emphasizes the importance of subcellular localization in phosphatase substrate specificity, in particular of PP2A (51). Mechanisms targeting CaN to different subcellular locales exist and have been shown to be an important part of CaN signaling. For example, AKAP79/150 targets CaN to the post-synaptic density where it regulates ionotropic glutamate receptors and l-type Ca2+ channels (52–55). It is therefore possible that similar scaffolding interactions target CaN to the OMM to amplify Drp1 Ser-656 dephosphorylation.
Formation of the CaN-Drp1 complex requires the LXVP motif of Drp1 (Fig. 2C). The LXVP motif is a common feature of CaN substrates (25) and has been extensively characterized in the context of the transcription factor NFAT (24, 33). Paralleling previous studies using peptide derived from the PKA regulatory subunit RIIα (34), our enzyme kinetic studies showed that the LXVP motif of Drp1 is critical for efficient Ser-656 dephosphorylation by CaN (Fig. 4, C and D, Table 1). As no calcium-independent CaN docking site conforming to the PXIXIT motif is apparent in the Drp1 sequence, we propose that Drp1 does not function as a CaN anchoring protein. Instead, the LXVP motif defines Drp1 as a high-affinity CaN substrate, in analogy to RIIα (34) and the mitogen-activated protein kinase scaffold KSR2 (56).
Replacing two amino acids to increase similarity to the LXVP motif of NFATc1 (LLDVP → YLAVP) resulted in a 3-fold increase in CaN-Drp1 binding affinity as measured by ITC, whereas changing two signature residues (LLDVP → LLDAA) reduced affinity below the detection limit (Fig. 3). These Drp1 mutations had corresponding bidirectional effects on phospho-Ser-656 turnover both in vitro and in cells (Table 1, Figs. 4 and 5) and altered mitochondrial morphology in non-neuronal and neuronal cells as predicted from their effects on Ser-656 phosphorylation (Figs. 6 and 7). Beyond these strong correlations, we demonstrated causation, using double mutants of the LXVP motif and Ser-656 to show that CaN regulates mitochondrial fission exclusively via this phosphorylation site (Fig. 6).
Previous studies by us and other laboratories showed that Drp1 Ser-656 phosphorylation and resulting mitochondrial elongation is predictive of enhanced cell survival (11, 13, 16, 18). Results presented in Fig. 8 align with these studies, extending neuroprotection by PKA-mediated phosphorylation of Drp1 at Ser-656 to OGD, an in vitro model of ischemic stroke. Ischemic and excitotoxic neuronal death is caused by massive Ca2+ influx, predominantly via NMDA-type glutamate receptors. Toxic levels of intracellular Ca2+ damage mitochondria by overloading mitochondrial Ca2+ stores (57), and by activating Ca2+-dependent enzymes, among them proteases of the calpain family and the Ser/Thr phosphatase CaN (58). Supporting a pro-injury role of the phosphatase, the immunosuppressants and CaN inhibitors cyclosporine A and FK506 were reported to reduce infarct volume in rodent and non-human primate models of focal cerebral ischemia (59–64). Specifically manipulating the dephosphorylation of one of many cellular CaN substrates, our experiments indicate that Drp1 dephosphorylation at Ser-656 at least in part mediates ischemic injury downstream to CaN activation.
How does Drp1 activation by CaN promote neuronal injury? Ischemic and excitotoxic insults result in rapid mitochondrial restructuring, described in the literature as swelling, fragmentation, or a combination of both (65–67). It is therefore tempting to speculate that neuroprotection following disruption of the CaN-Drp1 signaling axis is related to the canonical role of Drp1-mediated mitochondrial fragmentation in cytochrome c release and apoptosis (9). However, excitotoxic mitochondrial fragmentation was reported to occur independent of Drp1 activity (65, 68), driven instead primarily by ion and water fluxes across the inner mitochondrial membrane (67, 69–71). As well, our preliminary results support a Drp1-independent mechanism of glutamate-induced mitochondrial fragmentation in primary hippocampal neurons (data not shown).
Several alternative hypotheses regarding the role of CaN-mediated Drp1 activation in neuronal injury can be advanced. For instance, CaN could limit rebound hyperphosphorylation of Drp1 following ischemia (72), thereby slowing recovery of pathologically fragmented mitochondria by fusion events. Also, Drp1 stimulation by tonic CaN activity may influence mitochondrial ATP production, Ca2+ buffering, and generation/sequestration of reactive oxygen species, all of which can impact the outcome of subsequent insults (73). In support of such a bioenergetic mechanism, we recently reported that mitochondrial fusion hyperpolarizes, whereas fission depolarizes mitochondria in hippocampal neurons (4). However, further studies are needed to unravel the precise mechanisms by which regulation of the mitochondrial fission/fusion equilibrium influences neuronal survival in injury and disease.
This work was supported, in whole or in part, by National Institutes of Health Grants NS056244 and NS057714 (to S. S.) and CA136729 (to J. C. D. H.) and Predoctoral Fellowship F31 NS077563 (to A. M. S.).

This article contains supplemental Figs. S1 and S2.
- Drp1
- dynamin-related protein 1
- CaN
- calcineurin
- CaM
- calmodulin
- CNA
- calcineurin A subunit
- CNB
- calcineurin B subunit
- OGD
- oxygen-glucose deprivation
- ITC
- isothermal titration calorimetry
- DIV
- day in vitro.
REFERENCES
- 1. Li Z., Okamoto K., Hayashi Y., Sheng M. (2004) The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887 [DOI] [PubMed] [Google Scholar]
- 2. Parone P. A., Da Cruz S., Tondera D., Mattenberger Y., James D. I., Maechler P., Barja F., Martinou J. C. (2008) Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS One 3, e3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gomes L. C., Di Benedetto G., Scorrano L. (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dickey A. S., Strack S. (2011) PKA/AKAP1 and PP2A/Bβ2 regulate neuronal morphogenesis via Drp1 phosphorylation and mitochondrial bioenergetics. J. Neurosci. 31, 15716–15726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Szabadkai G., Simoni A. M., Chami M., Wieckowski M. R., Youle R. J., Rizzuto R. (2004) Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol. Cell 16, 59–68 [DOI] [PubMed] [Google Scholar]
- 6. Frieden M., James D., Castelbou C., Danckaert A., Martinou J. C., Demaurex N. (2004) Ca2+ homeostasis during mitochondrial fragmentation and perinuclear clustering induced by hFis1. J. Biol. Chem. 279, 22704–22714 [DOI] [PubMed] [Google Scholar]
- 7. Saotome M., Safiulina D., Szabadkai G., Das S., Fransson A., Aspenstrom P., Rizzuto R., Hajnóczky G. (2008) Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc. Natl. Acad. Sci. U.S.A. 105, 20728–20733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen H., Chan D. C. (2009) Mitochondrial dynamics, fusion, fission, movement, and mitophagy, in neurodegenerative diseases. Hum. Mol. Genet. 18, R169–R176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frank S., Gaume B., Bergmann-Leitner E. S., Leitner W. W., Robert E. G., Catez F., Smith C. L., Youle R. J. (2001) The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 1, 515–525 [DOI] [PubMed] [Google Scholar]
- 10. Wilson T. J., Slupe A. M., Strack S. (2013) Cell signaling and mitochondrial dynamics. Implications for neuronal function and neurodegenerative disease. Neurobiol. Dis. 51, 13–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cribbs J. T., Strack S. (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 8, 939–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chang C. R., Blackstone C. (2007) Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 282, 21583–21587 [DOI] [PubMed] [Google Scholar]
- 13. Merrill R. A., Dagda R. K., Dickey A. S., Cribbs J. T., Green S. H., Usachev Y. M., Strack S. (2011) Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1. PLoS Biol. 9, e1000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Strack S., Cribbs J. T. (2012) Allosteric modulation of Drp1 mechanoenzyme assembly and mitochondrial fission by the variable domain. J. Biol. Chem. 287, 10990–11001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cereghetti G. M., Stangherlin A., Martins de Brito O., Chang C. R., Blackstone C., Bernardi P., Scorrano L. (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. U.S.A. 105, 15803–15808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Merrill R. A., Slupe A. M., Strack S. (2013) N-terminal phosphorylation of protein phosphatase 2A/Bβ2 regulates translocation to mitochondria, dynamin-related protein 1 dephosphorylation, and neuronal survival. FEBS J. 280, 662–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dagda R. K., Merrill R. A., Cribbs J. T., Chen Y., Hell J. W., Usachev Y. M., Strack S. (2008) The spinocerebellar ataxia 12 gene product and protein phosphatase 2A regulatory subunit Bβ2 antagonizes neuronal survival by promoting mitochondrial fission. J. Biol. Chem. 283, 36241–36248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang J. X., Jiao J. Q., Li Q., Long B., Wang K., Liu J. P., Li Y. R., Li P. F. (2011) miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med. 17, 71–78 [DOI] [PubMed] [Google Scholar]
- 19. Klee C. B., Draetta G. F., Hubbard M. J. (1988) in Advances in Enzymology and Related Areas of Moledular Biology, pp. 149–200, John Wiley & Sonc, Inc., New York: [DOI] [PubMed] [Google Scholar]
- 20. Rusnak F., Mertz P. (2000) Calcineurin. Form and function. Physiol. Rev. 80, 1483–1521 [DOI] [PubMed] [Google Scholar]
- 21. Klee C. B., Ren H., Wang X. (1998) Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J. Biol. Chem. 273, 13367–13370 [DOI] [PubMed] [Google Scholar]
- 22. Ye Q., Wang H., Zheng J., Wei Q., Jia Z. (2008) The complex structure of calmodulin bound to a calcineurin peptide. Proteins Struct. Funct. Bioinformat. 73, 19–27 [DOI] [PubMed] [Google Scholar]
- 23. O'Donnell S. E., Yu L., Fowler C. A., Shea M. A. (2011) Recognition of β-calcineurin by the domains of calmodulin. Thermodynamic and structure evidence for distinct roles. Proteins Struct. Funct. Bioinformat. 79, 765–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rodríguez A., Roy J., Martínez-Martínez S., López-Maderuelo M. D., Niño-Moreno P., Ortí L., Pantoja-Uceda D., Pineda-Lucena A., Cyert M. S., Redondo J. M. (2009) A conserved docking surface on calcineurin mediates interaction with substrates and immunosuppressants. Mol. Cell 33, 616–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roy J., Cyert M. S. (2009) Cracking the phosphatase code. Docking interactions determine substrate specificity. Sci. Signal 2, re9. [DOI] [PubMed] [Google Scholar]
- 26. Ankarcrona M., Dypbukt J. M., Orrenius S., Nicotera P. (1996) Calcineurin and mitochondrial function in glutamate-induced neuronal cell death. FEBS Lett. 394, 321–324 [DOI] [PubMed] [Google Scholar]
- 27. Asai A., Qiu Jh., Narita Y., Chi S., Saito N., Shinoura N., Hamada H., Kuchino Y., Kirino T. (1999) High level calcineurin activity predisposes neuronal cells to apoptosis. J. Biol. Chem. 274, 34450–34458 [DOI] [PubMed] [Google Scholar]
- 28. Wang H. G., Pathan N., Ethell I. M., Krajewski S., Yamaguchi Y., Shibasaki F., McKeon F., Bobo T., Franke T. F., Reed J. C. (1999) Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 284, 339–343 [DOI] [PubMed] [Google Scholar]
- 29. Misonou H., Mohapatra D. P., Menegola M., Trimmer J. S. (2005) Calcium- and metabolic state-dependent modulation of the voltage-dependent Kv2.1 channel regulates neuronal excitability in response to ischemia. J. Neurosci. 25, 11184–11193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wood A. M., Bristow D. R. (1998) N-Methyl-d-aspartate receptor desensitisation is neuroprotective by inhibiting glutamate-induced apoptotic-like death. J. Neurochem. 70, 677–687 [DOI] [PubMed] [Google Scholar]
- 31. Arnoult D., Rismanchi N., Grodet A., Roberts R. G., Seeburg D. P., Estaquier J., Sheng M., Blackstone C. (2005) Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr. Biol. 15, 2112–2118 [DOI] [PubMed] [Google Scholar]
- 32. Montessuit S., Somasekharan S. P., Terrones O., Lucken-Ardjomande S., Herzig S., Schwarzenbacher R., Manstein D. J., Bossy-Wetzel E., Basañez G., Meda P., Martinou J. C. (2010) Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 142, 889–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martinez-Martinez S., Rodriguez A., Lopez-Maderuelo M. D., Ortega-Perez I., Vazquez J., Redondo J. M. (2006) Blockade of NFAT activation by the second calcineurin binding site. J. Chem. Biol. 281, 6227–6235 [DOI] [PubMed] [Google Scholar]
- 34. Blumenthal D. K., Takio K., Hansen R. S., Krebs E. G. (1986) Dephosphorylation of cAMP-dependent protein kinase regulatory subunit (type II) by calmodulin-dependent protein phosphatase. Determinants of substrate specificity. J. Biol. Chem. 261, 8140–8145 [PubMed] [Google Scholar]
- 35. Mehta S., Li H., Hogan P. G., Cunningham K. W. (2009) Domain architecture of the regulators of calcineurin (RCANs) and identification of a divergent RCAN in yeast. Mol. Cell Biol. 29, 2777–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leonard A. S., Davare M. A., Horne M. C., Garner C. C., Hell J. W. (1998) SAP97 is associated with the α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J. Biol. Chem. 273, 19518–19524 [DOI] [PubMed] [Google Scholar]
- 37. Moy L. Y., Tsai L. H. (2004) Cyclin-dependent kinase 5 phosphorylates serine 31 of tyrosine hydroxylase and regulates its stability. J. Biol. Chem. 279, 54487–54493 [DOI] [PubMed] [Google Scholar]
- 38. Mondragon A., Griffith E. C., Sun L., Xiong F., Armstrong C., Liu J. O. (1997) Overexpression and purification of human calcineurin α from Escherichia coli and assessment of catalytic functions of residues surrounding the binuclear metal center. Biochemistry 36, 4934–4942 [DOI] [PubMed] [Google Scholar]
- 39. Pedigo S., Shea M. A. (1995) Quantitative endoproteinase GluC footprinting of cooperative Ca2+ binding to calmodulin. Proteolytic susceptibility of E31 and E87 indicates interdomain interactions. Biochemistry 34, 1179–1196 [DOI] [PubMed] [Google Scholar]
- 40. McAvoy T., Nairn A. C. (2010) Serine/threonine protein phosphatase assays. Curr. Protoc. Mol. Bol. 92, 18.18.1–18.18.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. R Development Core Team (2011) R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 42. Rodbard D. (1974) Statistical quality control and routine data processing for radioimmunoassays and immunoradiometric assays. Clin. Chem. 20, 1255–1270 [PubMed] [Google Scholar]
- 43. Cribbs J. T., Strack S. (2009) Functional characterization of phosphorylation sites in dynamin-related protein 1. Methods Enzymol. 457, 231–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jahani-Asl A., Pilon-Larose K., Xu W., MacLaurin J. G., Park D. S., McBride H. M., Slack R. S. (2011) The mitochondrial inner membrane GTPase, optic atrophy 1 (Opa1), restores mitochondrial morphology and promotes neuronal survival following excitotoxicity. J. Biol. Chem. 286, 4772–4782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sandebring A., Thomas K. J., Beilina A., van der Brug M., Cleland M. M., Ahmad R., Miller D. W., Zambrano I., Cowburn R. F., Behbahani H., Cedazo-Mínguez A., Cookson M. R. (2009) Mitochondrial alterations in PINK1-deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS One 4, e5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cho S.-G., Du Q., Huang S., Dong Z. (2010) Drp1 dephosphorylation in ATP depletion-induced mitochondrial injury and tubular cell apoptosis. Am. J. Physiol. Renal Physiol. 299, F199-F206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ogata T., Machida S., Oishi Y., Higuchi M., Muraoka I. (2009) Differential cell death regulation between adult-unloaded and aged rat soleus muscle. Mech. Ageing Dev. 130, 328–336 [DOI] [PubMed] [Google Scholar]
- 48. Li H., Rao A., Hogan P. G. (2011) Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol. 21, 91–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Martin B. L., Jurado L. A., Hengge A. C. (1999) Comparison of the reaction progress of calcineurin with Mn2+ and Mg2+. Biochemistry 38, 3386–3392 [DOI] [PubMed] [Google Scholar]
- 50. Taguchi N., Ishihara N., Jofuku A., Oka T., Mihara K. (2007) Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282, 11521–11529 [DOI] [PubMed] [Google Scholar]
- 51. Slupe A. M., Merrill R. A., Strack S. (2011) Determinants for substrate specificity of protein phosphatase 2A. Enzyme Res. 2011, 398751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Coghlan V. M., Perrino B. A., Howard M., Langeberg L. K., Hicks J. B., Gallatin W. M., Scott J. D. (1995) Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science 267, 108–111 [DOI] [PubMed] [Google Scholar]
- 53. Oliveria S. F., Gomez L. L., Dell'Acqua M. L. (2003) Imaging kinase–AKAP79–phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J. Cell Biol. 160, 101–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sanderson J. L., Gorski J. A., Gibson E. S., Lam P., Freund R. K., Chick W. S., Dell'Acqua M. L. (2012) AKAP150-anchored calcineurin regulates synaptic plasticity by limiting synaptic incorporation of Ca2+-permeable AMPA receptors. J. Neurosci. 32, 15036–15052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Oliveria S. F., Dittmer P. J., Youn D. H., Dell'Acqua M. L., Sather W. A. (2012) Localized calcineurin confers Ca2+-dependent inactivation on neuronal l-type Ca2+ channels. J. Neurosci. 32, 15328–15337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dougherty M. K., Ritt D. A., Zhou M., Specht S. I., Monson D. M., Veenstra T. D., Morrison D. K. (2009) KSR2 is a calcineurin substrate that promotes ERK cascade activation in response to calcium signals. Mol. Cell 34, 652–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stout A. K., Raphael H. M., Kanterewicz B. I., Klann E., Reynolds I. J. (1998) Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat. Neurosci. 1, 366–373 [DOI] [PubMed] [Google Scholar]
- 58. Bano D., Young K. W., Guerin C. J., Lefeuvre R., Rothwell N. J., Naldini L., Rizzuto R., Carafoli E., Nicotera P. (2005) Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell 120, 275–285 [DOI] [PubMed] [Google Scholar]
- 59. Sharkey J., Butcher S. P. (1994) Immunophilins mediate the neuroprotective effects of FK506 in focal cerebral ischaemia. Nature 371, 336–339 [DOI] [PubMed] [Google Scholar]
- 60. Bochelen D., Rudin M., Sauter A. (1999) Calcineurin inhibitors FK506 and SDZ ASM 981 alleviate the outcome of focal cerebral ischemic/reperfusion injury. J. Pharmacol. Exp. Ther. 288, 653–659 [PubMed] [Google Scholar]
- 61. Furuichi Y., Katsuta K., Maeda M., Ueyama N., Moriguchi A., Matsuoka N., Goto T., Yanagihara T. (2003) Neuroprotective action of tacrolimus (FK506) in focal and global cerebral ischemia in rodents. Dose dependency, therapeutic time window and long-term efficacy. Brain Res. 965, 137–145 [DOI] [PubMed] [Google Scholar]
- 62. Furuichi Y., Maeda M., Moriguchi A., Sawamoto T., Kawamura A., Matsuoka N., Mutoh S., Yanagihara T. (2003) Tacrolimus, a potential neuroprotective agent, ameliorates ischemic brain damage and neurologic deficits after focal cerebral ischemia in nonhuman primates. J. Cereb. Blood Flow Metab. 23, 1183–1194 [DOI] [PubMed] [Google Scholar]
- 63. Furuichi Y., Maeda M., Matsuoka N., Mutoh S., Yanagihara T. (2007) Therapeutic time window of tacrolimus (FK506) in a nonhuman primate stroke model. Comparison with tissue plasminogen activator. Exp. Neurol. 204, 138–146 [DOI] [PubMed] [Google Scholar]
- 64. Sharifi Z. N., Abolhassani F., Zarrindast M. R., Movassaghi S., Rahimian N., Hassanzadeh G. (2012) Effects of FK506 on hippocampal CA1 cells following transient global ischemia/reperfusion in Wistar rat. Stroke Res. Treat. 809417, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Barsoum M. J., Yuan H., Gerencser A. A., Liot G., Kushnareva Y., Gräber S., Kovacs I., Lee W. D., Waggoner J., Cui J., White A. D., Bossy B., Martinou J. C., Youle R. J., Lipton S. A., Ellisman M. H., Perkins G. A., Bossy-Wetzel E. (2006) Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 25, 3900–3911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rintoul G. L., Filiano A. J., Brocard J. B., Kress G. J., Reynolds I. J. (2003) Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J. Neurosci. 23, 7881–7888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shalbuyeva N., Brustovetsky T., Bolshakov A., Brustovetsky N. (2006) Calcium-dependent spontaneously reversible remodeling of brain mitochondria. J. Biol. Chem. 281, 37547–37558 [DOI] [PubMed] [Google Scholar]
- 68. Young K. W., Piñon L. G., Bampton E. T., Nicotera P. (2010) Different pathways lead to mitochondrial fragmentation during apoptotic and excitotoxic cell death in primary neurons. J. Biochem. Mol. Toxicol. 24, 335–341 [DOI] [PubMed] [Google Scholar]
- 69. Pivovarova N. B., Nguyen H. V., Winters C. A., Brantner C. A., Smith C. L., Andrews S. B. (2004) Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. J. Neurosci. 24, 5611–5622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Greenwood S. M., Mizielinska S. M., Frenguelli B. G., Harvey J., Connolly C. N. (2007) Mitochondrial dysfunction and dendritic beading during neuronal toxicity. J. Biol. Chem. 282, 26235–26244 [DOI] [PubMed] [Google Scholar]
- 71. Brustovetsky T., Li V., Brustovetsky N. (2009) Stimulation of glutamate receptors in cultured hippocampal neurons causes Ca2+-dependent mitochondrial contraction. Cell Calcium 46, 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu W., Tian F., Kurata T., Morimoto N., Abe K. (2012) Dynamic changes of mitochondrial fusion and fission proteins after transient cerebral ischemia in mice. J. Neurosci. Res. 90, 1183–1189 [DOI] [PubMed] [Google Scholar]
- 73. Nicholls D. G. (2008) Oxidative stress and energy crises in neuronal dysfunction. Ann. N.Y. Acad. Sci. 1147, 53–60 [DOI] [PubMed] [Google Scholar]