

Abstract
Background
Apixaban is an oral, selective, direct factor Xa inhibitor approved for thromboprophylaxis after orthopedic surgery and stroke prevention in patients with atrial fibrillation, and under development for treatment of venous thromboembolism. This study investigated the effect of a gastric acid suppressant, famotidine (a histamine H2-receptor antagonist), on the pharmacokinetics of apixaban in healthy subjects.
Methods
This two-period, two-treatment crossover study randomized 18 healthy subjects to receive a single oral dose of apixaban 10 mg with and without a single oral dose of famotidine 40 mg administered 3 hours before dosing with apixaban. Plasma apixaban concentrations were measured up to 60 hours post-dose and pharmacokinetic parameters were calculated.
Results
Famotidine did not affect maximum apixaban plasma concentration (Cmax) or area under the plasma concentration-time curve from zero to infinite time (AUC∞). Point estimates for ratios of geometric means with and without famotidine were close to unity for Cmax (0.978) and AUC∞ (1.007), and 90% confidence intervals were entirely contained within the 80%–125% no-effect interval. Administration of apixaban alone and with famotidine was well tolerated.
Conclusion
Famotidine does not affect the pharmacokinetics of apixaban, consistent with the physicochemical properties of apixaban (lack of an ionizable group and pH-independent solubility). Apixaban pharmacokinetics would not be affected by an increase in gastrointestinal pH due to underlying conditions (eg, achlorhydria), or by gastrointestinal pH-mediated effects of other histamine H2-receptor antagonists, antacids, or proton pump inhibitors. Given that famotidine is also an inhibitor of the human organic cation transporter (hOCT), these results indicate that apixaban pharmacokinetics are not influenced by hOCT uptake transporter inhibitors. Overall, these results support that apixaban can be administered without regard to coadministration of gastric acid modifiers.
Keywords: apixaban, factor Xa inhibitor, famotidine, H2-receptor antagonists, hOCT inhibitor, drug–drug interaction
Introduction
Apixaban is an oral, potent, reversible, direct, and highly selective inhibitor of the coagulation factor Xa,1,2 which plays a pivotal role in the clotting cascade by decreasing the conversion of prothrombin to thrombin.3 Apixaban is approved as a fixed dose in a number of countries for thromboprophylaxis in patients who have undergone elective hip or knee replacement surgery4–6 and for stroke prevention in patients with nonvalvular atrial fibrillation.7,8 Apixaban is also being developed for the treatment of deep vein thrombosis and/or pulmonary embolism.9,10
Clinical studies show that apixaban has a predictable pharmacokinetic profile across a wide range of doses. The oral bioavailability of apixaban is approximately 50%, and its elimination half-life is approximately 12 hours. The presence of food has no relevant effect on apixaban exposure.11,12 Apixaban is eliminated by both renal and nonrenal pathways, and is a substrate for the P-glycoprotein and breast cancer resistance protein transporters. Nonrenal elimination pathways include metabolism by cytochrome P450 (CYP) enzymes, primarily CYP3A4.13 Renal excretion of apixaban accounts for approximately 27% of total clearance.14–16
Given the high prevalence of gastric acid secretion disorders and related conditions such as reflux esophagitis and gastroesophageal reflux disease in the general population,17,18 as well as the broad use of different classes of drugs in the treatment of these disorders, it is likely that apixaban will be coadministered with gastric acid modifiers. Over-the-counter availability of gastric acid suppressants further increases the likelihood that coadministration of these agents with apixaban will occur. While a significant pharmacokinetic interaction was not expected between apixaban and drugs that modify gastric pH, because apixaban has no ionizable groups, it was important to confirm in a clinical trial whether alterations in gastric pH would affect the pharmacokinetics of apixaban.
Famotidine is a commonly prescribed histamine H2-receptor antagonist that suppresses secretion of gastric acid by parietal cells.19,20 Famotidine was selected for this study because it is a widely used gastric acid suppressant with a well established safety and pharmacokinetic profile, in addition to a rapid onset of action following single-dose administration. Maximal plasma famotidine concentrations occur within 2–3 hours after oral administration, and coincide with maximal increases in gastric pH (effects are seen approximately 1–3 hours post-dose).19–21 There is no cumulative effect with repeated dosing, and gastric pH returns to baseline 10–12 hours after cessation of administration.19–21 Famotidine is minimally metabolized and primarily eliminated unchanged in the urine.21 Famotidine has minimal potential for CYP-mediated drug–drug interactions.22–24
Famotidine is a potent inhibitor of the uptake transporter protein, human organic cation transporter (hOCT)-3, and a moderate inhibitor of hOCT-1 and hOCT-2,25 and thus has the potential for hOCT-mediated drug–drug interactions. This study investigated the effect of famotidine on the pharmacokinetics of apixaban in healthy subjects as a primary objective. The safety and tolerability of apixaban when given alone and when coadministered with famotidine in healthy subjects were also assessed as a secondary objective.
Materials and methods
Subjects and study design
This was an open-label, randomized, two-period, two-treatment crossover study (Figure 1) conducted in healthy subjects at MDS Pharma Services, St Laurent, Quebec, Canada. Men and women 18–45 years of age with a body mass index of 18–30 kg/m2 and no clinically significant deviation from normal in medical history, physical examination, electrocardiography, and clinical laboratory determinations were enrolled. Women of childbearing potential could not be pregnant or nursing, and were required to have a negative pregnancy test within 24 hours prior to the first dose of study medication and use an acceptable method of contraception throughout the duration of the study. Key exclusion criteria included any history, signs, or symptoms that indicated a high risk of bleeding, including but not limited to: a history of upper gastrointestinal tract ulcer disease within the last 6 months; a history or evidence of any abnormal bleeding or anticoagulation disorder, or a first-degree relative ≤50 years of age with a history of such a disorder; a history of significant head injury within the previous 2 years; and blood transfusion or any major surgery within the 4 weeks prior to dosing. In addition, subjects were excluded if they had a history of disease or surgery that might impact absorption of either drug; any contraindications to the administration of famotidine; inability to tolerate oral medication or to tolerate venipuncture; and drug or alcohol abuse, as defined by the DSM-IV-TR (Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision) within 6 months or smoking more than 10 cigarettes a day.
Figure 1.
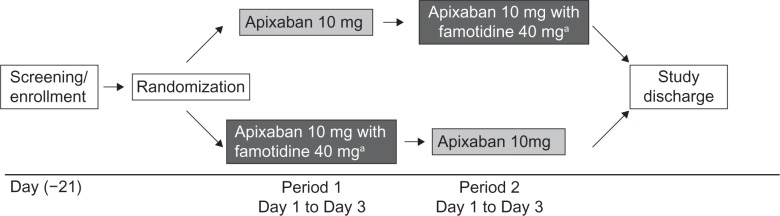
Study design.
Notes: aFamotidine was given as a single 40 mg tablet 3 hours prior to administration of a single 10 mg dose of apixaban. Each study period was separated by a 3-day washout period.
The study was conducted in accordance with the principles stated in the Declaration of Helsinki and consistent with Good Clinical Practice, as defined by the International Conference on Harmonisation and in accordance with the ethical principles underlying European Union Directive 2001/20/EC and the United States Code of Federal Regulations, Title 21, Part 50 (21CFR50). The protocol and subject consent forms were approved by the institutional review board before the study began.
Subjects were screened for eligibility within 21 days prior to administration of study medication. Subjects who met the eligibility criteria and provided written informed consent were admitted to the clinical facility the day before the first dose of study medication was administered and stayed in the clinic until study discharge. On day 1 of period 1, subjects were randomly assigned in a 1:1 ratio to receive either a single 10 mg oral dose of apixaban (two 5 mg tablets) only or a single 10 mg dose of apixaban (two 5 mg tablets) administered 3 hours after a single 40 mg dose of famotidine (one tablet). Both treatments were given after an 8-hour fast. Subjects then received the alternative treatment on day 1 of the second period. Each period was separated by a 3-day washout. Randomization was carried out according to a computer-generated randomization scheme provided by the Drug Supply Management Department, Bristol-Myers Squibb Research and Development, Princeton, NJ, USA.
Given that there is no cumulative effect with multiple dosing,19–21 the maximum effect of famotidine on gastric pH was expected at 3 hours following a single 40 mg dose, which is the highest recommended dose for the management of duodenal ulcers.20 A single 10 mg oral dose of apixaban was chosen for this study because it represents the high end of the dose range tested in Phase III clinical trials.
Sample collection
Blood samples for pharmacokinetic analysis of apixaban were collected on day 1 of both treatment periods at the following time points: predose, and at 30 minutes and 1, 2, 3, 4, 6, 12, 24, 36, 48, and 60 hours following administration of apixaban. Blood samples were collected in 2.7 mL tubes containing 3.2% sodium citrate and were centrifuged for 15 minutes at approximately 1500 × g to separate plasma. Plasma samples were stored at −20°C until analyzed.
Sample analysis
Frozen plasma samples were shipped to the bioanalytical laboratory (Intertek Pharmaceutical Services [previously known as Alta Analytical Laboratory], El Dorado Hills, CA, USA). Apixaban was quantitatively determined in plasma samples using a validated high-performance liquid chromatography tandem mass spectrometry method. Sample extraction for plasma utilized protein precipitation. A stable-label apixaban (C2413CH22D3N5O4) was used as the internal standard for the assay. The lower limit of quantification was 1.0 ng/mL for apixaban. The coefficient of variation for between-run and within-run variability was ≤5.95% and ≤ 11.9%, respectively, with deviations from the nominal concentration of no more than ±9.55%. Stability of apixaban in human plasma was established for at least 589 days at −20°C, and all samples were analyzed within this period of analyte stability.
Pharmacokinetic analysis
Apixaban single-dose pharmacokinetic parameters, the maximum observed plasma concentration of apixaban (Cmax), time to reach Cmax (Tmax), area under the plasma concentration-time curve from time zero to the last quantifiable concentration (AUC0–T), elimination half-life (t1/2), and AUC from time zero to infinity (AUC∞) were determined using established noncompartmental methods implemented in the program Kinetica™ (version 4.4.1, Thermo Electron Corporation, Philadelphia, PA, USA).
Safety assessments
Safety was evaluated by assessing subject-reported or directly observed adverse events, and by investigator review of vital signs, laboratory tests, and electrocardiographic data. All adverse events were coded according to system organ class and preferred term using MeDRA (Medical Dictionary for Regulatory Activities, version 11.1), and reviewed for severity, relationship to study drugs, and clinical importance. Twelve-lead electrocardiography and physical examinations were performed at screening, predose on day 1 of each study period and before study discharge. Blood and urine samples for clinical laboratory tests were collected on day −1, prior to study drug administration in the second treatment period and prior to study discharge on day 4 of the second treatment period, and analyzed at Life Labs, Quebec, Canada, and MDS Pharma Services, Mississauga, Canada.
Statistical analysis
The pharmacokinetic parameters of apixaban were analyzed in a descriptive manner by summary statistics for each treatment (apixaban with and without famotidine). Geometric means and coefficients of variation were presented for apixaban Cmax, AUC0–T, and AUC∞; medians, minima, and maxima were reported for Tmax; and means and standard deviations were reported for t1/2. Plasma samples with concentrations below the lower limit of quantification were treated as missing for the calculation of mean and standard deviation values.
To assess the effect of coadministration of famotidine on the pharmacokinetics of apixaban, log-transformed apixaban pharmacokinetic parameters (Cmax, AUC0–T, and AUC∞) were analyzed using a general linear mixed model. The factors in the analyses were treatment sequence, period, and treatment as fixed effects, and subject within sequence as a random effect. No adjustments were made for multiplicity. Point estimates and 90% confidence intervals (CIs) for ratios of geometric means of apixaban Cmax, AUC0–T, and AUC∞ with and without famotidine were estimated. An absence of effect of famotidine was concluded if the 90% CIs for the ratio of geometric means for apixaban Cmax, AUC0–T, and AUC∞ with and without famotidine were contained within the prespecified equivalence interval of 80%–125%. Data from 18 subjects would provide 85% power to conclude an absence of effect of famotidine with respect to apixaban Cmax and 91% power to conclude an absence of effect of famotidine with respect to apixaban AUC∞.26 These sample size calculations assumed Cmax and AUC∞ were log-normally distributed with intrasubject standard deviation of 0.20 for natural logarithm (ln) Cmax and 0.19 for ln AUC∞, and assumed an absence of effect.
Results
Study population
Eighteen subjects were randomized to treatment. All 18 subjects completed the study and none withdrew because of adverse events. The demographic characteristics of the study population are shown in Table 1.
Table 1.
Subject demographics
| Characteristic | Subjects (n = 18) |
|---|---|
| Gender [n (%)] | |
| Male | 14 (78) |
| Female | 4 (22) |
| Age (years) | |
| Mean (SD) | 34 (8.0) |
| Range | 20–44 |
| Race [n (%)] | |
| White | 18 (100) |
| Body weight (kg) | |
| Mean (SD) | 73.7 (10.7) |
| Range | 55.7–91.7 |
| Height (cm) | |
| Mean (SD) | 172.7 (8.7) |
| Range | 152.0–192.0 |
| BMI (kg/m2) | |
| Mean (SD) | 24.7 (2.5) |
| Range | 19.7–28.4 |
Abbreviations: SD, standard deviation; BMI, body mass index.
Pharmacokinetics
Plasma concentration-time profiles for apixaban were similar following administration of apixaban alone or with famotidine (Figure 2). Coadministration of famotidine had no effect on apixaban Cmax, AUC0–T, or AUC∞ (Table 2 and Figure 3). Individual subject pharmacokinetic parameters for apixaban with and without famotidine are shown in Figure 3. Point estimates for the ratio of geometric mean with and without famotidine were close to unity for Cmax, AUC∞, and AUC0–T of apixaban, and the 90% CIs for the ratio of the geometric mean with and without famotidine were entirely contained within the prespecified 80%–125% no-effect interval (Table 2).
Figure 2.

Mean (+standard deviation) plasma concentration-time profiles for apixaban in 18 healthy subjects following a single 10 mg dose of apixaban alone or administered 3 hours after a single 40 mg dose of famotidine.
Table 2.
Summary statistics and statistical analysis for apixaban pharmacokinetic parameters
| Pharmacokinetic variablea | Treatment | Geometric mean | Point estimateb of ratio of geometric mean (90% CI) |
|---|---|---|---|
| Cmax (ng/mL) | Apixaban | 230 (28) | 0.978 (0.875–1.093) |
| Apixaban + famotidine | 225 (20) | ||
| Tmax (h), median (range) | Apixaban | 3.00 (1.00–5.98) | |
| Apixaban + famotidine | 2.01 (1.00–4.02) | ||
| AUC0–T (ng ⋅ h/mL) | Apixaban | 2193 (26) | 1.003 (0.934–1.077) |
| Apixaban + famotidine | 2198 (20) | ||
| AUC∞ (ng ⋅ h/mL) | Apixaban | 2222 (25) | 1.007 (0.937–1.082) |
| Apixaban + famotidine | 2237 (20) | ||
| t1/2 (h), mean (SD) | Apixaban | 9.2 (4.42) | |
| Apixaban + famotidine | 11.0 (4.61) |
Notes:
Data are shown as the geometric mean and % coefficient of variation unless otherwise stated;
estimated from a general linear mixed-effect model.
Abbreviations: AUC∞, area under the plasma concentration-time curve from zero to infinite time; AUC0–T, area under the plasma concentration-time curve from zero to last quantifiable measurement; CI, confidence interval; Cmax, peak observed plasma apixaban concentration; SD, standard deviation; t1/2, half-life; Tmax, time to peak observed plasma apixaban concentration; h, hours.
Figure 3.

Individual pharmacokinetic parameters for apixaban following a single 10 mg dose of apixaban alone or administered 3 hours after a single 40 mg dose of famotidine.
Six subjects (two administered apixaban alone, four administered apixaban with famotidine) had measurable apixaban concentrations (≤2.98 ng/mL) prior to study drug administration in period 2. These values were near the lower limit of quantification (1 ng/mL) of the assay and represented <1.5% of the Cmax in these subjects. Therefore, these concentrations did not contribute any meaningful carryover effect and did not impact the results or conclusions of the study.
Safety and tolerability
No deaths, serious adverse events, discontinuations due to adverse events, or clinically relevant bleeding-related adverse events were reported during the study. Seven adverse events occurred in five subjects, with hematoma at the vessel puncture site being the most frequently reported (two subjects [11.1%], one subject during each treatment period). All adverse events were mild in nature and resolved by either the time of study discharge or subject follow-up. All adverse events were considered unrelated to the study medication except for one, ie, an elevated international normalized ratio of 3.5 observed in one subject at study discharge, 3 days following a single dose of apixaban with famotidine. Prothrombin time and activated partial thromboplastin time were within normal limits in this subject throughout the study. The plasma apixaban concentration in this subject was below the lower limit of quantification at least 24 hours prior to the event. No changes in vital signs or physical examination results were observed. No other adverse events were reported for this subject. There were no clinically relevant changes in electrocardiography, vital signs, or laboratory tests observed during the study.
Discussion
This study was designed to investigate the effect of coadministration of famotidine, a typical gastric acid suppressant, on the pharmacokinetics of apixaban. Upon administration of a single 10 mg dose of apixaban 3 hours after a single 40 mg dose of famotidine, the geometric means for apixaban Cmax, AUC∞, and AUC0–T were unchanged, and the 90% CIs for their ratio of geometric means, with and without famotidine, were entirely contained within the prespecified no-effect interval of 80%–125%, indicating no effect of famotidine on the pharmacokinetics of apixaban.
The elimination half-life of apixaban has been well characterized in numerous clinical pharmacology studies and is approximately 12 hours. Accordingly, the 60-hour, intensively sampled pharmacokinetic monitoring period for apixaban and a 3-day washout period (approximately five elimination half-lives for apixaban) were considered appropriate in this study.14–16,27 The famotidine dose (40 mg) and time interval (administered 3 hours prior to apixaban) in combination with the dose of apixaban (10 mg) were considered to represent the largest potential for an interaction due to gastric acid suppression.
The lack of a pharmacokinetic drug–drug interaction observed between apixaban and famotidine is in agreement with the physicochemical properties of apixaban. Apixaban is a compound that lacks ionizable groups in its molecular structure, and thus its aqueous solubility is not affected by changes in pH. The results of this study suggest that increases in gastric pH due to other gastric acid modifiers (such as other H2-receptor antagonists, proton pump inhibitors, and antacids) or the presence of abnormally elevated gastric pH (eg, achlorhydria) are unlikely to affect the pharmacokinetics of apixaban.
It is important to consider the potential for metabolic or transporter drug–drug interactions between famotidine and apixaban. While apixaban is metabolized primarily by CYP3A4 and is a substrate for P-glycoprotein and breast cancer resistance protein, famotidine is minimally metabolized, is primarily eliminated unchanged in urine, and is a substrate for P-glycoprotein.20 Famotidine has minimal potential for CYP450-mediated drug–drug interactions.22–24 However, it is an inhibitor of hOCT uptake transporters.25 The data from the current study indicate that the pharmacokinetics of apixaban are not influenced by hOCT-3, hOCT-2, or hOCT-1 inhibitors such as famotidine, and confirm the in vitro findings, indicating that apixaban is not a substrate for hOCT-2 transporters (data on file, Bristol-Myers Squibb, Princeton, NJ, USA).
In summary, the gastric acid inhibitor, famotidine, did not affect the pharmacokinetics of apixaban, consistent with the pH-independent solubility properties of apixaban. In addition, no significant drug–drug interaction between apixaban and famotidine occurred via the human organic cation transporter system (hOCT-1, hOCT-2, and hOCT-3). Administration of apixaban alone and with famotidine was well tolerated in healthy subjects. Overall, these results suggest that increases in gastric pH due to other gastric acid modifiers such as antacids, proton pump inhibitors, or other H2-receptor antagonists are unlikely to affect the pharmacokinetics of apixaban by means of their influence on pH or inhibition of hOCT. Other inhibitors of hOCT are also not expected to affect the pharmacokinetics of apixaban.
Acknowledgments
The authors gratefully acknowledge the contributions of Matthew Farmer, Tong Li, and Zhigang Yu to this study. We thank the investigator, Gaetano Morelli, and the staff at MDS Pharma Services, Quebec, Canada, for the conduct of this study.
Footnotes
Disclosure
VVU, YS, JW, JMP, FL, and CEF are employees of Bristol-Myers Squibb and received salaries and benefits commensurate with employment at the time of the study. WB and RAB are employees of Pfizer and received salaries and benefits commensurate with employment at the time of the study. This study was funded by Bristol-Myers Squibb and Pfizer. Professional medical writing and editorial assistance was provided by Andy Shepherd and Dana Fox at Caudex Medical, and was funded by Bristol-Myers Squibb and Pfizer.
References
- 1.Pinto DJ, Orwat MJ, Koch S, et al. Discovery of 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (apixaban, BMS-562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa. J Med Chem. 2007;50(22):5339–5356. doi: 10.1021/jm070245n. [DOI] [PubMed] [Google Scholar]
- 2.Wong PC, Crain EJ, Xin B, et al. Apixaban, an oral, direct and highly selective factor Xa inhibitor: in vitro, antithrombotic and antihemostatic studies. J Thromb Haemost. 2008;6(5):820–829. doi: 10.1111/j.1538-7836.2008.02939.x. [DOI] [PubMed] [Google Scholar]
- 3.Kaiser B. Factor Xa – a promising target for drug development. Cell Mol Life Sci. 2002;59(2):189–192. doi: 10.1007/s00018-002-8415-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lassen MR, Raskob GE, Gallus A, Pineo G, Chen D, Portman RJ. Apixaban or enoxaparin for thromboprophylaxis after knee replacement. N Engl J Med. 2009;361(6):594–604. doi: 10.1056/NEJMoa0810773. [DOI] [PubMed] [Google Scholar]
- 5.Lassen MR, Raskob GE, Gallus A, Pineo G, Chen D, Hornick P. Apixaban versus enoxaparin for thromboprophylaxis after knee replacement (ADVANCE-2): a randomised double-blind trial. Lancet. 2010;375(9717):807–815. doi: 10.1016/S0140-6736(09)62125-5. [DOI] [PubMed] [Google Scholar]
- 6.Lassen MR, Gallus A, Raskob GE, Pineo G, Chen D, Ramirez LM. Apixaban versus enoxaparin for thromboprophylaxis after hip replacement. N Engl J Med. 2010;363(26):2487–2498. doi: 10.1056/NEJMoa1006885. [DOI] [PubMed] [Google Scholar]
- 7.Connolly SJ, Eikelboom J, Joyner C, et al. Apixaban in patients with atrial fibrillation. N Engl J Med. 2011;364(9):806–817. doi: 10.1056/NEJMoa1007432. [DOI] [PubMed] [Google Scholar]
- 8.Granger CB, Alexander JH, McMurray JJ, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981–992. doi: 10.1056/NEJMoa1107039. [DOI] [PubMed] [Google Scholar]
- 9.Buller H, Deitchman D, Prins M, Segers A. Efficacy and safety of the oral direct factor Xa inhibitor apixaban for symptomatic deep vein thrombosis. The Botticelli DVT dose-ranging study. J Thromb Haemost. 2008;6(8):1313–1318. doi: 10.1111/j.1538-7836.2008.03054.x. [DOI] [PubMed] [Google Scholar]
- 10.Agnelli G, Buller HR, Cohen A, et al. Apixaban for extended treatment of venous thromboembolism. N Engl J Med. 2013;368(8):699–708. doi: 10.1056/NEJMoa1207541. [DOI] [PubMed] [Google Scholar]
- 11.Frost C, Yu Z, Moore K, et al. Apixaban, an oral direct factor Xa inhibitor: multiple-dose safety, pharmacokinetics, and pharmacodynamics in healthy subjects. J Thromb Haemost. 2007;5(Suppl 2):P-M-664. [Google Scholar]
- 12.Frost C, Wang J, Nepal S, et al. Apixaban, an oral, direct factor Xa inhibitor: single-dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects. Br J Clin Pharmacol. 2013;75(2):476–487. doi: 10.1111/j.1365-2125.2012.04369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L, Zhang D, Raghavan N, et al. In vitro assessment of metabolic drug–drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab Dispos. 2010;38(3):448–458. doi: 10.1124/dmd.109.029694. [DOI] [PubMed] [Google Scholar]
- 14.Frost C, Yu Z, Nepal S, et al. Apixaban, a direct factor Xa inhibitor: single-dose pharmacokinetics and pharmacodynamics of an intravenous formulation. J Clin Pharmacol. 2008;48:1132. doi: 10.1111/j.1365-2125.2012.04369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raghavan N, Frost CE, Yu Z, et al. Apixaban metabolism and pharmacokinetics after oral administration to humans. Drug Metab Dispos. 2009;37(1):74–81. doi: 10.1124/dmd.108.023143. [DOI] [PubMed] [Google Scholar]
- 16.Vakkalagadda B, Frost C, Wang J, et al. Effect of rifampin on the pharmacokinetics of apixaban, an oral direct inhibitor of factor Xa. J Clin Pharmacol. 2009;49:1124. doi: 10.1007/s40256-015-0157-9. [DOI] [PubMed] [Google Scholar]
- 17.Sonnenberg A. The US temporal and geographic variations of diseases related to Helicobacter pylori. Am J Public Health. 1993;83(7):1006–1010. doi: 10.2105/ajph.83.7.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Majumdar SR, Soumerai SB, Farraye FA, et al. Chronic acid-related disorders are common and underinvestigated. Am J Gastroenterol. 2003;98(11):2409–2414. doi: 10.1111/j.1572-0241.2003.07706.x. [DOI] [PubMed] [Google Scholar]
- 19.Echizen H, Ishizaki T. Clinical pharmacokinetics of famotidine. Clin Pharmacokinet. 1991;21(3):178–194. doi: 10.2165/00003088-199121030-00003. [DOI] [PubMed] [Google Scholar]
- 20.Pepcid [package insert] Whitehouse Station, NJ: Merck and Co, Inc; 2010. [Google Scholar]
- 21.Bologna M, Napolitano T, Biordi L, Carlucci G. HPLC reveals famotidine in the urine up to five days after a single 20 mg oral dose. Drugs Exp Clin Res. 1988;14(6):419–421. [PubMed] [Google Scholar]
- 22.Humphries TJ. Famotidine: a notable lack of drug interactions. Scand J Gastroenterol Suppl. 1987;134:55–60. doi: 10.3109/00365528709090142. [DOI] [PubMed] [Google Scholar]
- 23.Klotz U, Arvela P, Rosenkranz B. Famotidine, a new H2-receptor antagonist, does not affect hepatic elimination of diazepam or tubular secretion of procainamide. Eur J Clin Pharmacol. 1985;28(6):671–675. doi: 10.1007/BF00607913. [DOI] [PubMed] [Google Scholar]
- 24.Lin JH, Chremos AN, Chiou R, Yeh KC, Williams R. Comparative effect of famotidine and cimetidine on the pharmacokinetics of theophylline in normal volunteers. Br J Clin Pharmacol. 1987;24(5):669–672. doi: 10.1111/j.1365-2125.1987.tb03228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bourdet DL, Pritchard JB, Thakker DR. Differential substrate and inhibitory activities of ranitidine and famotidine toward human organic cation transporter 1 (hOCT1; SLC22A1), hOCT2 (SLC22A2), and hOCT3 (SLC22A3) J Pharmacol Exp Ther. 2005;315(3):1288–1297. doi: 10.1124/jpet.105.091223. [DOI] [PubMed] [Google Scholar]
- 26.Diletti E, Hauschke D, Steinijans VW. Sample size determination for bioequivalence assessment by means of confidence intervals. Int J Clin Pharmacol Ther Toxicol. 1991;29(1):1–8. [PubMed] [Google Scholar]
- 27.Barrett YC, Wang J, Song Y, et al. A randomised assessment of the pharmacokinetic, pharmacodynamic and safety interaction between apixaban and enoxaparin in healthy subjects. Thromb Haemost. 2012;107(5):916–924. doi: 10.1160/TH11-09-0634. [DOI] [PubMed] [Google Scholar]