

Abstract
We conducted a combined genome-wide association (GWAS) analysis of 7,481 individuals affected with bipolar disorder and 9,250 control individuals within the Psychiatric Genomewide Association Study Consortium Bipolar Disorder group (PGC-BD). We performed a replication study in which we tested 34 independent SNPs in 4,493 independent bipolar disorder cases and 42,542 independent controls and found strong evidence for replication. In the replication sample, 18 of 34 SNPs had P value < 0.05, and 31 of 34 SNPs had signals with the same direction of effect (P = 3.8 × 10−7). In the combined analysis of all 63,766 subjects (11,974 cases and 51,792 controls), genome-wide significant evidence for association was confirmed for CACNA1C and found for a novel gene ODZ4. In a combined analysis of non-overlapping schizophrenia and bipolar GWAS samples we observed strong evidence for association with SNPs in CACNA1C and in the region of NEK4/ITIH1,3,4. Pathway analysis identified a pathway comprised of subunits of calcium channels enriched in the bipolar disorder association intervals. The strength of the replication data implies that increasing samples sizes in bipolar disorder will confirm many additional loci.
Keywords: Alleles; Bipolar Disorder; genetics; Calcium Channels, L-Type; genetics; metabolism; Case-Control Studies; Databases, Genetic; Genetic Loci; Genetic Predisposition to Disease; Genome, Human; Genome-Wide Association Study; Humans; Linkage Disequilibrium; Nuclear Proteins; genetics; metabolism; Polymorphism, Single Nucleotide; Schizophrenia; genetics
Bipolar disorder (BD) is a severe mood disorder affecting greater than 1% of the population[1]. Classical BD is characterized by recurrent manic episodes that often alternate with depression. Its onset is in late adolescence or early adulthood and results in chronic illness with moderate to severe impairment. Although the pathogenesis of BD is not understood, family, twin and adoption studies consistently find relative risks to first-degree relatives of ~8 and concordance of ~40–70% for a monozygotic co-twin[1,2]. BD shares phenotypic similarities with other psychiatric diseases including schizophrenia (SCZ), major depression and schizoaffective disorder. Relatives of BD individuals are at increased risk of psychiatric phenotypes including SCZ, major depression and schizoaffective disorder, suggesting these disorders have a partially shared genetic basis[3,4]. Despite robust evidence for a substantial heritability, single causal mutations have not been identified through linkage or candidate gene association studies[1].
Genome-wide association studies (GWAS) for BD have been performed with multiple partially overlapping case and control samples[5–11]. In a small study, Baum et al. reported genome-wide significant (defined here as P < 5×10−8) association to diacylglycerol kinase eta (DGKH)[5]. Subsequently, Ferreira et al.[8] identified genome-wide significant association in the region of the gene ankyrin 3 (ANK3) and Cichon et al.[12] recently reported neurocan (NCAN); other studies did not report genome-wide significant loci[5,9,10,13]. A critical need for psychiatric genetics is to identify consistently associated loci. Towards that end, the Psychiatric Genome-wide Association Study Consortium (PGC) was established in 2007 to facilitate combination of primary genotype data from studies with overlapping samples and to subsequently allow analyses both within and across the following disorders: autism, attention-deficit hyperactivity disorder, BD, major depressive disorder and SCZ[14,15]. Here, the Bipolar Disorder Working Group of the PGC reports results from our primary association study of combined data in BD from 16,731 samples, and a replication sample of 47,035 individuals.
We received primary genotype and phenotype data for all samples (Table 1; Supplementary Information and Table S1). Results from sets of samples have been reported singly[6,7,9–11] and in combinations[8,9,12] in 7 publications with varying levels of overlap of case and control samples. Data were divided into the 11 case and control groupings shown in Table 1 and each individual was assigned to only one group, with the assignment chosen to maximize power of the combined analysis (See Supplementary Information S2 & S3 for details). The final dataset was comprised of 7,481 unique cases and 9,250 unique controls. Cases had the following diagnoses: BD type 1 (n=6,289; 84%), BD type 2 (n=824; 11%), schizoaffective disorder bipolar type (n=263; 4%), and 104 individuals with other bipolar diagnoses (BD NOS, 1%, Table S1). 46,234 SNPs were directly genotyped by all 11 groups and 1,016,924 SNPs were genotyped by 2–11 groups. Based on reference haplotypes from the HapMap phase 2 CEU sample, genotypes were imputed using BEAGLE[16]. We analyzed imputed SNP dosages from 2,415,422 autosomal SNPs with a minor allele frequency (MAF) ≥ 1% and imputation quality score r2 > 0.3. We performed logistic regression of case status on imputed SNP dosage, including as covariates 5 multidimensional scaling components (based on linkage disequilibrium (LD) pruned genotype data, Figure S1) and indicator variables for each sample grouping using PLINK[17]. We observed a genomic control[18] value of λ=1.148. Consistent with previous work suggesting a highly polygenic architecture for SCZ and BD[19], this estimate will likely reflect a mixture of signals arising from a large number of true risk variants of weak effect as well as some degree of residual confounding. Nonetheless, below we designate an association as “genome-wide significant” only if the genomic-control P-value (Pgc) is below 5 × 10−8. Where reported, nominal P-values are labeled Praw. Results for the primary analyses can be found in the supplementary data (Figure S2 (QQ plot); Figure S3 (Manhattan Plot); Figure S4 (Region Plots)), Table S2 lists regions containing an associated SNP with Pgc < 5 × 10−5.
Table 1.
Description of individual samples
| Sample | Ancestry | Case (n)a | Control (n)b | Platformc | Publicationd |
|---|---|---|---|---|---|
| BOMA- Bipolar Study, University of Bonn and CIMH Mannheim | German | 675 | 1297 | 550 | [7]–[10] |
| Genetic Association Information Network (GAIN)/Bipolar Genome Study (BiGS) | European-American | 542 | 649 | 6.0 | [7]–[10] |
| GlaxoSmithKline (GSK) | British/Canadian/Scottish | 890 | 902 | 550 | [9] |
| Pritzker Neuropsychiatric Disorders Research Consortium | European-American | 1130 | 718 | 550 | [9] |
| Systematic Treatment Enhancement Program for Bipolar Disorder (STEP1) | European-American | 922 | 645 | 500K | [7],[8] |
| Systematic Treatment Enhancement Program for Bipolar Disorder (STEP2) | European-American | 659 | 192 | 5.0 | [8] |
| Thematically Organized Psychosis (TOP) Study | Norwegian | 203 | 349 | 6.0 | [11] |
| Trinity College Dublin | Irish | 150 | 797 | 6.0 | [8] |
| University College London (UCL) | British | 457 | 495 | 500K | [7],[8] |
| University of Edinburgh | Scottish | 282 | 275 | 6.0 | [8] |
| Wellcome Trust Case-Control Consortium (WTCCC) | British | 1571 | 2931 | 500K | [6],[8],[9] |
| TOTAL | 7481 | 9250 |
Cases include BD1, BD2, SAB, BD-NOS (see Table S1).
Most controls were not screened for psychiatric disease. A subset of 33% however were, see supplement.
Platforms are 6.0 = Affymetrix Genome-Wide Human SNP Array 6.0; 5.0 = Affymetrix Genome-Wide Human SNP Array 5.0; 500K = Affymetrix GeneChip Human Mapping 500K Array; 550 = Illumina HumanHap 550.
Primary publication reporting individual sample level genotypes for BD listed. See supplement for fuller description of publications and table S1 for sample origins in primary GWAS analyses.
Table 2 lists four regions from our primary GWAS analysis that contain SNPs with Praw < 5 × 10−8; two regions reach Pgc ≤ 5 × 10−8 (see Figure S4 for plots of the regions). Association was detected in ankyrin 3 (ANK3) on chromosome 10q21 for the imputed SNP rs10994397 (Pgc = 7.1 × 10−9, odds ratio (OR) = 1.35). The second SNP, rs9371601, was located in synaptic nuclear envelope protein 1 (SYNE1) on chromosome 6q25 (Praw = 4.3 × 10−8, OR = 1.15). Intergenic SNP rs7296288 (Pgc = 8.4 × 10−8; OR = 1.15) is found in a region of LD of ~100 kb on chromosome 12q13 that contains 7 genes. SNP rs12576775 (Pgc = 2.1 × 10−7, OR = 1.18) is found at chromosome 11q14 in ODZ4, a human homologue of a Drosophila pair-rule gene odz. Generally consistent signals were observed across studies, with no single study driving the overall association results (Figure S5). Meta-analysis of the 11 samples under both fixed- and random-effects models yielded results similar to the combined analysis (Table S3 and S4).
Table 2.
Primary GWAS association results for four most significant regions
| SNP | Chra | Positiona | Nearest Gene | A1b/A2 | A1 freqc | ORd 95% CI | Praw | Pgc |
|---|---|---|---|---|---|---|---|---|
| rs10994397 | 10 | 61949130 | ANK3 | T/C | 0.06 | 1.35 (1.48–1.23) | 5.5 × 10−10 | 7.1 × 10−9 |
| rs9371601 | 6 | 152832266 | SYNE1 | T/G | 0.36 | 1.15 (1.21–1.10) | 4.3 × 10−9 | 4.3 × 10−8 |
| rs7296288 | 12 | 47766235 | Many | C/A | 0.48 | 1.15 (1.20–1.09) | 9.4 × 10−9 | 8.4 × 10−8 |
| rs12576775 | 11 | 78754841 | ODZ4 | G/A | 0.18 | 1.18 (1.25–1.11) | 2.7 × 10−8 | 2.1 × 10−7 |
Chromosome;
SNP basepair position on Build 36;
Allele frequency in the total sample;
OR is predicted towards allele A1
We next sought to replicate these findings in independent samples (Table S5). 38 SNPs were selected that had Pgc < 5 × 10−5 and were not in LD with each other (Table 3). Of these, four SNPs were not considered to be completely independent signals and are not used for further analyses. (For completeness, data for these 4 SNPs are listed and denoted by an asterisk in Table 3, supplementary Section 6 for details). We received unpublished data from investigators on a further 4,493 cases and 42,542 controls for the top 34 independent SNPs. Significantly more SNPs replicated at all levels than would be expected by chance (Table 3). Four of 34 had Prep values < 0.01, 18 of 34 SNPs had Prep values < 0.05 and 31 of 34 had signal in the same direction of effect (binomial test, P = 3.8 × 10−7). Within the replication samples, two SNPs remained significant following correction for multiple testing. The first, rs4765913, is found on chromosome 12 in CACNA1C, the alpha subunit of the L-type voltage-gated calcium channel (Prep = 1.6 × 10−4, OR = 1.13). The second, rs10896135 is in a 17 exon, 98kb open reading frame C11orf80 (Prep = 0.0015, OR = 0.91). Nominally significant Prep values were also obtained in another calcium channel subunit, CACNB3. Only 2 of the 4 SNPs in Table 2 had Prep values < 0.05; the genome-wide significant SNPs from the primary analysis, rs10994397 and rs9371601 did not (Prep = 0.11 and 0.10, respectively). Finally, we performed a fixed-effects meta-analysis, as described in the supplementary information our primary PGC and Prep data and established genome-wide significant evidence for association with rs4765913 in CACNA1C (P = 1.82 × 10−9, OR= 1.14) and rs12576775 in ODZ4 (P = 2.77 × 10−8; OR = 0.89). As in the primary analyses, generally consistent signals were observed across replication studies and meta-analysis of the replication data also did not reveal significant heterogeneity between the samples (Tables S6 and S7).
Table 3.
Bipolar association results for primary GWAS, replication and combined samples for the most significant SNP from regions with Pgc < 5 × 10−5
| SNP | CHRb | POSc | A1 | A2 | PRIMARY GWAS | REPLICATIONa | COMBINED GWAS and REPLICATION | GENES IN LD REGION | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pgc | ORd | P1-sided | OR | P | OR | ||||||
| rs4765913 | 12 | 2290157 | A | T | 6.50 × 10−6 | 1.15 | 1.6 × 10−4 | 1.13 | 1.82 × 10−9 | 1.14 | CACNA1C |
| rs10896135 | 11 | 66307578 | C | G | 8.46 × 10−6 | 0.88 | 1.47 × 10−3 | 0.91 | 2.77 × 10−8 | 0.89 | ZDHHC24,YIF1A,TMEM151A, SYT12,SPTBN2,SLC29A2,SF3B2, RIN1,RCE1,RBM4B,RBM4,RBM14, RAB1B,PELI3,PC,PACS1,NPAS4, MRPL11,LRFN4,KLC2,GAL3ST3, DPP3,CTSF,CNIH2,CD248,CCS, CCDC87,C11orf86,C11orf80, BRMS1,BBS1,B3GNT1,ACTN3 |
| rs2070615* | 12 | 47504438 | A | G | 4.00 × 10−5 | 0.90 | 2.52 × 10−3 | 0.93 | 2.48 × 10−7 | 0.91 | RND1,DDX23,CACNB3 |
| rs12576775 | 11 | 78754841 | A | G | 2.09 × 10−7 | 0.85 | 7.59 × 10−3 | 0.92 | 6.32 × 10−9 | 0.88 | ODZ4 |
| rs2175420* | 11 | 78801531 | C | T | 2.90 × 10−5 | 0.87 | 7.80 × 10−3 | 0.92 | 6.55 × 10−7 | 0.89 | ODZ4 |
| rs3845817 | 2 | 65612029 | C | T | 1.65 × 10−5 | 0.90 | 8.98 × 10−3 | 0.94 | 4.68 × 10−7 | 0.91 | |
| rs2176528 | 2 | 194580428 | C | G | 3.98 × 10−5 | 1.15 | 1.04 × 10−2 | 1.09 | 1.12 × 10−6 | 1.12 | |
| rs4660531 | 1 | 41612409 | G | T | 3.16 × 10−5 | 0.89 | 1.11 × 10−2 | 0.93 | 1.02 × 10−6 | 0.91 | |
| rs7578035 | 2 | 98749324 | G | T | 1.83 × 10−5 | 1.12 | 1.29 × 10−2 | 1.06 | 7.93 × 10−7 | 1.09 | TXNDC9,TSGA10,REV1,MRPL30, MITD1,MGAT4A,LYG1,LYG2,LIPT1, EIF5B,C2orf55,C2orf15 |
| rs2287921 | 19 | 53920084 | C | T | 1.68 × 10−5 | 1.12 | 1.37 × 10−2 | 1.06 | 8.99 × 10−7 | 1.10 | SPHK2,SEC1,RPL18,RASIP1, NTN5,MAMSTR,IZUMO1,FUT2, FUT1,FGF21,FAM83E,DBP,CA11 |
| rs11168751* | 12 | 47505405 | C | G | 1.80 × 10−5 | 0.84 | 1.43 × 10−2 | 0.90 | 7.08 × 10−7 | 0.86 | CACNB3 |
| rs7296288 | 12 | 47766235 | A | C | 8.39 × 10−8 | 0.87 | 1.50 × 10−2 | 0.94 | 8.06 × 10−9 | 0.90 | TUBA1B,TUBA1A,RHEBL1, PRKAG1,MLL2,LMBR1L,DHH,DDN |
| rs7827290 | 8 | 142369497 | G | T | 3.54 × 10−5 | 1.13 | 1.67 × 10−2 | 1.06 | 3.03 × 10−6 | 1.10 | LOC731779,GPR20 |
| rs12730292 | 1 | 79027350 | C | G | 2.37 × 10−5 | 1.12 | 1.71 × 10−2 | 1.06 | 1.59 × 10−6 | 1.10 | |
| rs12912251 | 15 | 36773660 | G | T | 9.57 × 10−6 | 1.13 | 2.04 × 10−2 | 1.06 | 9.63 × 10−7 | 1.10 | C15orf53 |
| rs4332037 | 7 | 1917335 | C | T | 1.78 × 10−5 | 0.87 | 3.00 × 10−2 | 0.93 | 2.44 × 10−6 | 0.90 | MAD1L1 |
| rs6550435 | 3 | 36839493 | G | T | 1.97 × 10−5 | 1.12 | 3.26 × 10−2 | 1.05 | 3.26 × 10−6 | 1.09 | LBA1 |
| rs17395886 | 4 | 162498835 | A | C | 2.18 × 10−5 | 0.86 | 3.51 × 10−2 | 0.93 | 3.78 × 10−6 | 0.89 | FSTL5 |
| rs6746896 | 2 | 96774676 | A | G | 2.33 × 10−6 | 1.14 | 3.86 × 10−2 | 1.05 | 6.59 × 10−7 | 1.10 | LMAN2L,FER1L5,CNNM4 |
| rs736408 | 3 | 52810394 | C | T | 1.22 × 10−6 | 1.14 | 4.65 × 10−2 | 1.05 | 6.03 × 10−7 | 1.10 | WDR82,TWF2,TNNC1,TMEM110, TLR9,STAB1,SPCS1,SNORD69, SNORD19,SNORD19B,SFMBT1, SEMA3G,RFT1,PRKCD,PPM1M, PHF7,PBRM1,NT5DC2,NISCH, NEK4,MUSTN1,LOC440957,ITIH1, ITIH3,ITIH4,GNL3,GLYCTK, GLT8D1,DNAH1,BAP1,ALAS1 |
| rs11162405 | 1 | 78242248 | A | G | 2.54 × 10−5 | 0.90 | 4.76 × 10−2 | 0.96 | 7.11 × 10−6 | 0.92 | ZZZ3,USP33,NEXN,MGC27382, GIPC2,FUBP1,FAM73A,DNAJB4, AK5 |
| rs9804190 | 10 | 61509837 | C | T | 3.06 × 10−5 | 1.17 | 9.63 × 10−2 | 1.04 | 6.32 × 10−5 | 1.10 | ANK3 |
| rs9371601 | 6 | 152832266 | G | T | 4.27 × 10−8 | 0.87 | 0.103 | 0.97 | 1.52 × 10−7 | 0.91 | SYNE1 |
| rs3774609 | 3 | 53807943 | G | T | 1.14 × 10−5 | 0.89 | 0.107 | 0.97 | 1.63 × 10−5 | 0.92 | CHDH,CACNA1D |
| rs10994397 | 10 | 61949130 | C | T | 7.08 × 10−9 | 0.74 | 0.116 | 0.94 | 6.14 × 10−8 | 0.82 | ANK3 |
| rs4668059 | 2 | 168874528 | C | T | 4.45 × 10−5 | 1.18 | 0.158 | 1.04 | 7.05 × 10−5 | 1.12 | STK39 |
| rs16966413 | 15 | 36267191 | A | G | 4.74 × 10−5 | 0.84 | 0.160 | 0.95 | 5.10 × 10−5 | 0.88 | SPRED1 |
| rs6102917 | 20 | 40652833 | C | G | 3.88 × 10−5 | 1.44 | 0.165 | 1.11 | 4.22 × 10−5 | 1.31 | PTPRT |
| rs11085829 | 19 | 13035312 | A | G | 4.03 × 10−6 | 0.87 | 0.175 | 0.97 | 3.37 × 10−5 | 0.92 | NFIX |
| rs875326 | 1 | 173556022 | C | T | 2.51 × 10−5 | 1.15 | 0.183 | 1.03 | 5.75 × 10−5 | 1.10 | TNR |
| rs13245097* | 7 | 2307581 | C | T | 3.81 × 10−5 | 1.13 | 0.196 | 1.02 | 0.0001992 | 1.08 | SNX8,NUDT1,MAD1L1,FTSJ2 |
| rs780148 | 10 | 80605089 | C | G | 4.66 × 10−5 | 1.12 | 0.230 | 1.03 | 7.59 × 10−5 | 1.09 | ZMIZ1 |
| rs2281587 | 10 | 105367339 | C | T | 1.96 × 10−5 | 1.12 | 0.372 | 1.01 | 0.000238 | 1.07 | SH3PXD2A,NEURL |
| rs10776799 | 1 | 115674570 | G | T | 4.84 × 10−5 | 1.15 | 0.434 | 1.01 | 0.0009391 | 1.08 | NGF |
| rs263906 | 1 | 101750922 | C | T | 2.42 × 10−5 | 1.13 | 0.440 | 1.01 | 0.0002859 | 1.08 | |
| rs10028075 | 4 | 87186854 | C | T | 8.96 × 10−6 | 0.89 | 1.00 | 1.02 | 0.001651 | 0.95 | MAPK10 |
| rs3968 | 9 | 4931997 | C | G | 2.09 × 10−5 | 1.17 | 1.00 | 0.92 | 0.07571 | 1.04 | |
| rs8006348 | 14 | 50595223 | A | G | 4.91 × 10−5 | 0.89 | 1.00 | 1.05 | 0.01855 | 0.95 | TRIM9 |
Replication case and control sample details can be found in the supplement; 4% of the controls were screened for psychiatric disorder
Chromosome;
SNP basepair position on Build 36;
OR is predicted towards allele A1
To interpret why two of the significant associations in the primary analysis appear to fail to replicate, it is important to quantify the role of the “winner’s curse” on estimates of power to replicate individual signals. Given a polygenic model, power will be very low to detect any one variant at genome-wide significant levels, but there will be many chances to “get lucky” with at least one variant. Those that are discovered will have relatively inflated effect estimates. A simple simulation of the distribution of ORs around several “true” ORs (conditioning on a genome-wide significant P value of 5 × 10−8, fixed minor allele frequency (0.20), and our sample size (Table S8)) demonstrates a distinct inflation of the estimated OR leading to a marked overestimate of the power to replicate an individual result. For example, for a true genotypic relative risk of 1.05 the mean estimated OR is 1.17, conditioning on having P < 5 × 10−8. Thus, although the nominal power for replication is 100% for the inflated OR, the true power to replicate at P < 0.05 is only 30%. Thus any single failure to replicate is by itself less informative. This simulation is consistent with the positive signal we observed in the independent replication where many more than expected show nominal replication with all but one in the original, expected, direction of effect.
We performed an analysis to look for enrichment of Gene Ontology (GO) terms among genes in the association intervals containing the same top 34 independent SNPs used in the replication analysis (Pgc < 5 × 10−5) from Table 3 using a permutation-based approach that controlled for potential biases due to SNP density, gene density, and gene size and found enrichment in GO:0015270, dihydropyridine-sensitive calcium channel activity. This GO category contains 8 genes, 3 of which (CACNA1C, CACNA1D and CACNB3) are present among the 34 independent association-intervals tested (P = 0.00002); the probability of observing an empirical P value this small, given all the targets tested, is P = 0.021 (See Supplementary section 7). Overall, these analyses suggest that the set of intervals ranked highly in our GWAS do not represent a random set with respect to annotated gene function. This analysis focused only on the most significant loci, consistent with the other results presented in this manuscript. It is likely that a study based on a larger number of loci, defined by a more liberal P value cutoff, would indicate other promising areas for biological investigation.
We performed a conditional analysis that included the 34 independent SNPs listed in Table 3. In three of the 34 regions with Pgc 5 < 10−5, we identified SNPs within 1 MB of the most strongly associated SNP that continued to show evidence of association (conditional Pgc <10−4). We next performed region specific conditional analysis in these regions and observed conditional association at 3p21.1 (rs736408, conditional Pgc = 8.1 × 10−7), 10q21.2 (rs9804190, conditional Pgc = 7.3 × 10−5) and 15q14 (rs16966413, conditional Pgc = 7.3 × 10−5) (Figure S6). On chromosomes 3 and 15, the SNP most strongly associated after conditioning was > 500kb from the conditioning SNP with multiple genes in the intervening interval. On chromosome 10 we observed additional less strongly associated conditionally independent SNPs located upstream of the 5′ end of ANK3, in an intron of ANK3, and at the 3′ end of the longest transcript (704kb). In each of these three regions, the association signals remaining after conditioning could arise from multiple causal variants, from a single rare causal variant that is incomplete LD with the tested SNPs or represent false positive associations. The presence of additional SNPs with evidence for association in three of the regions of interest, including ANK3 (10q21.2) (previously reported by Schulze et al.[13] in partially overlapping samples), might increase the likelihood that these loci are causal. The 3p21.1 and 15q.14 regions also each showed evidence for association (P < .05) in the replication sample for one of the SNPs.
Finally, to provide direct and independent evidence for a highly polygenic basis for BD – as implied by a polygenic component shared between BD and SCZ, International Schizophrenia Consortium (2009) – we repeated the analysis performed by the ISC in these samples, with BD discovery samples. We observed a significant enrichment of putatively-associated BD “score alleles” in target sample cases compared to controls for all discovery P value thresholds tested (see Supplementary Section 9; Table S9).
A parallel study has been performed by the PGC investigators for SCZ. Given the known overlap in risk factors between BD and SCZ, we asked if a combined analysis of PGC-BD and PGC-SCZ (eliminating overlapping control samples, see Supplementary S10 section) would show stronger evidence of association than the original BD GWAS analysis for 5 of the most strongly associated SNPs from the primary GWAS and meta-analysis, supplemented by the additional genome-wide significant region (i.e. CACNA1C) in our replication analyses. In the combined BD and SCZ analysis of GWAS samples two SNPs showed stronger association compared to the BD GWAS analysis, rs4765913 in CACNA1C ( SCZ Praw = 7.0 × 10−9 compared to BD Praw=1.35 × 10−6) and rs736408 in a multigene region containing NEK4/ITIH1,3,4 (SCZ Praw =8.4 × 10−9 compared to BD Praw =2.00 × 10−7) (Table S10).
In the current analysis of BD we observed primary association signals that reached genome-wide significance (Pgc < 5 × 10−8) in the primary analyses in the region of ANK3 and SYNE1 and two signals near genome-wide significant on chromosome 12 and in the region of ODZ4. While in our independent replication sample we did not find additional support for ANK3 or SYNE1, this is consistent with a potential overestimation of the original ORs and should not be taken to disprove the involvement of these genes. Data from additional samples will be needed to resolve this question.
The most striking finding is the overall abundance of replication signals observed. Among our top 34 signals, the number of nominal associations in the same direction of effect is highly unlikely to be a chance observation. That the enrichment of replication results is almost entirely in the direction of the original observations strongly implies that many, if not most of the signals will ultimately turn out to be true associations with BD. Such results are expected under a highly polygenic model, where there are few or no variants of large effect. BD has a heritability estimated at higher than 80%. As is typical in studies of complex disorders, our findings explain only a small fraction of this heritability. Our data are consistent with the presence of many common susceptibility variants of relatively weak[19] effect, potentially operating together with rarer variants with a range of effect sizes[20]. Although this is the largest GWAS study of BD to date, our sample size remains modest in comparison with some other recent meta-analyses of common, complex diseases and is therefore likely to be underpowered to detect the majority of risk variants. Variation among the eleven studies in patient ascertainment, assessment and population could also potentially reduce power to detect loci with relatively specific phenotypic effects. Alternative analytic approaches that consider a broader approach to phenotype, both within and across psychiatric disorders, are underway in the PGC.
In order to understand the implications of these results for disease pathogenesis, we focused on one approach based on the joint analysis of variation at biologically meaningful sets of polymorphisms (e.g. specific genes, gene families or biological pathways). The connections drawn by INRICH between calcium channel subunits are not novel, but are consistent with a prior literature regarding the role of ion channels in BD, the mood stabilizing effects of ion channel modulating drugs, and the specific treatment literature suggesting direct efficacy of L-type calcium channel blockers in the treatment of BD[21]. We observed significant enrichment of CACNA1C and CACNA1D which are the major L-type alpha subunits found in the brain and their specific association with BD suggests a value to designing calcium channel antagonists that are selective for these subunits. Magnetic resonance imaging studies have implicated CACNA1C SNP rs1006737 with several alterations in structural[22], and functional imaging[23–25]. Several groups have previously implicated CACNA1C in other adult psychiatric disorders, in particular, SCZ and major depression[26–29]. L-type calcium channels also regulate changes in gene regulation responsible for many aspects of neuronal plasticity and have more recently been shown to have direct effects on transcription[29]. Taken together these lines of evidence should lead to renewed biological investigation of calcium channels in the pathogenesis of BD and other psychiatric diseases. ODZ4, located on chromosome 11, is a member of a family of cell surface proteins, the teneurins, related to the Drosophila pair-rule gene ten-m/odz. These genes are likely involved in cell surface signaling and neuronal pathfinding.
Three of our top 5 regions have non-coding RNAs present within the associated region (none are found in the remaining regions in Table S2). MicroRNAs are small RNA molecules known to regulate gene expression. Mir708, a member of a conserved mammalian microRNA family, is located in the first intron of ODZ4. Three small nucleolar RNAs, SNORD69 and SNORD19, and SNORD19B are located on chromosome 3p21.1 and belong to the C/D family of snoRNAs involved in processing and modification of ribosome assembly. Finally, a 121 base non-coding RNA with homology to the 5S-rRNA is also located within the SYNE1 association region. The role of microRNAs in neurodevelopmental disorders is increasingly apparent in Rett’s syndrome, Fragile × and SCZ. Our study represents the first connections to BD.
Our combined analyses with SCZ illuminates the growing appreciation of shared genetic epidemiology[30] and shared polygenic contribution to risk [31]. This adds to the evidence that best supported loci have an effect across the traditional bipolar/schizophrenia diagnostic divide.
In conclusion, we have obtained strong evidence for replication of multiple signals in BD. In particular, we support prior findings in CACNA1C, and now identify ODZ4 as associated with BD. The strongly positive replication results imply that data from additional samples, both from GWAS and sequencing, will identify more of the genetic architecture of BD. When the biological concomitants of the association signals have been characterized they are likely to provide important novel insights into the pathogenesis of BD.
Supplementary Material
Figure 1.
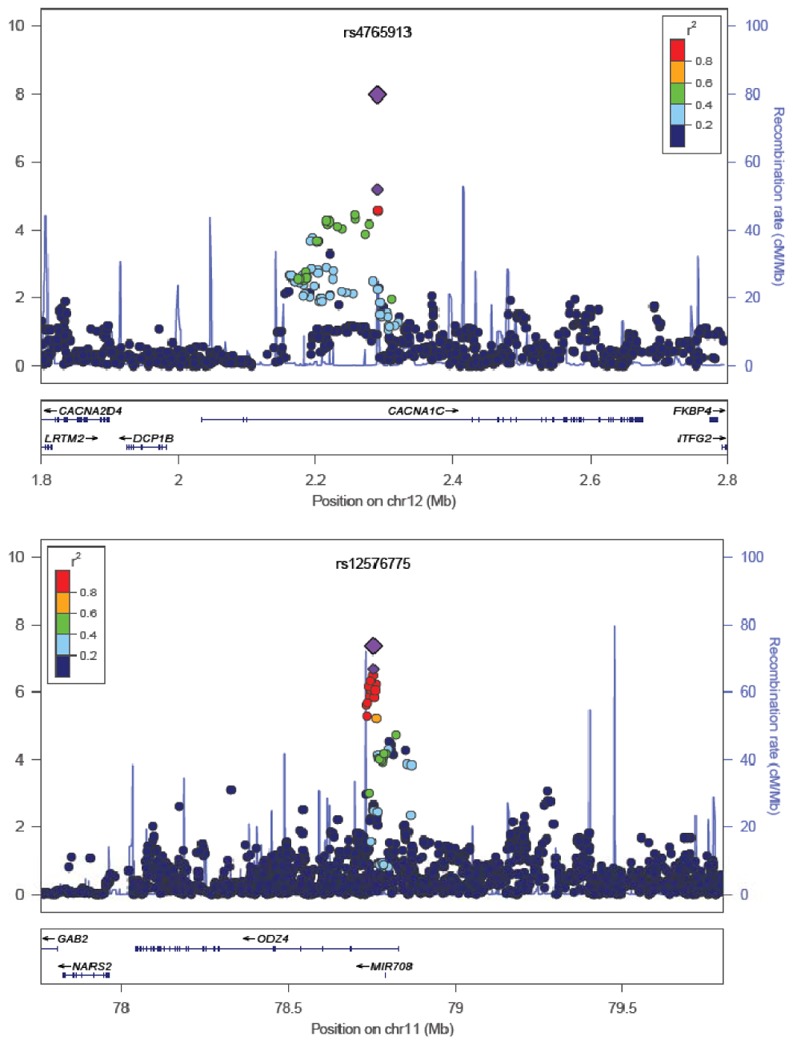
Results are shown as –log10(P value) for genotyped and imputed SNPs. The most associated SNP in the primary analysis is shown as the small purple triangle. The most associated SNP in the combined analysis is shown as the large purple triangle. The color of the remaining markers reflects r2 with the most associated SNP. The recombination rate from CEU HapMap (second y axis) is plotted in light blue.
Acknowledgments
We would like to recognize the contribution of thousands of subjects without whom this work would not be possible. Thomas Lehner (NIMH) was instrumental in initiating and planning the overall project. Daniella Posthuma and the Dutch Genetic Cluster Computer provided invaluable computational resources. We also that the PGC schizophrenia group for allowing us perform the combined analyses of 6 loci prior to publication. This work was supported by many grants from NIH (MH078151, MH081804, MH059567 supplement, MH59553, MH080372, 1U54RR025204). Other sources of support include: the Genetic Association Information Network (GAIN), the NIMH Intramural Research Program, the Tzedakah Foundation, the American Philosophical Society, the Stardust foundation, the National Library of Medicine, the Stanley Foundation for Medical Research, and the Wellcome Trust, the Pritzker Neuropsychiatric Disorders Research Fund L.L.C., GlaxoSmithKline, as well as grants for individual studies (see supplemental acknowledgements). TOP Study was supported by grants from the Research Council of Norway (167153/V50, 163070/V50, 175345/V50) and South-East Norway Health Authority (123-2004), and EU (ENBREC).
Protocols and assessment procedures were approved by the relevant ethical review mechanisms for each study. All participants provided written informed consent prior to participation in the primary study and consent allowed the samples to be used within the current combined analyses. Genotype data from this manuscript for 10,257 samples can be obtained from the Center on Collaborative Genetic Studies of Mental Disorders in accordance with NIMH data release policies (http://zork.wustl.edu/nimh/). Genotype data from the WTCCC sample can be obtained from https://www.wtccc.org.uk/info/access_to_data_samples.shtml. Genotype data from the BOMA-Bipolar Study can be obtained by contacting S. Cichon directly (sven.cichon@uni-bonn.de).
Footnotes
COMPETING FINANCIAL STATEMENT
We have no competing financial interests.
DATA RELEASE POLICY
Data will be released through the NIMH Genetics Initiative Repository.
References
- 1.Craddock N, Sklar P. Genetics of bipolar disorder: successful start to a long journey. Trends Genet. 2009;25:99–105. doi: 10.1016/j.tig.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Cardno AG, et al. Heritability estimates for psychotic disorders: the Maudsley twin psychosis series. Arch Gen Psychiatry. 1999;56:162–8. doi: 10.1001/archpsyc.56.2.162. [DOI] [PubMed] [Google Scholar]
- 3.Craddock N, Jones I. Genetics of bipolar disorder. J Med Genet. 1999;36:585–94. doi: 10.1136/jmg.36.8.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Craddock N, O’Donovan MC, Owen MJ. The genetics of schizophrenia and bipolar disorder: dissecting psychosis. J Med Genet. 2005;42:193–204. doi: 10.1136/jmg.2005.030718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baum AE, et al. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry. 2007 doi: 10.1038/sj.mp.4002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.WTCCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sklar P, et al. Whole-genome association study of bipolar disorder. Mol Psychiatry. 2008;13:558–69. doi: 10.1038/sj.mp.4002151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferreira MA, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet. 2008 doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott LJ, et al. Genome-wide association and meta-analysis of bipolar disorder in individuals of European ancestry. Proc Natl Acad Sci U S A. 2009;106:7501–6. doi: 10.1073/pnas.0813386106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith EN, et al. Genome-wide association study of bipolar disorder in European American and African American individuals. Mol Psychiatry. 2009;14:755–63. doi: 10.1038/mp.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Djurovic S, et al. A genome side association study of bipolar disorder in Norwegian individuals, followed by replication in Icelandic sample. J Affect Disord. 2010 doi: 10.1016/j.jad.2010.04.007. in press. [DOI] [PubMed] [Google Scholar]
- 12.Cichon S. Genome-wide association study identifies genetic variation in neurocan as a susceptibility factor for bipolar disorder. Am J Hum Genet. doi: 10.1016/j.ajhg.2011.01.017. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schulze TG, et al. Two variants in Ankyrin 3 (ANK3) are independent genetic risk factors for bipolar disorder. Mol Psychiatry. 2009;14:487–91. doi: 10.1038/mp.2008.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.PGC. A framework for interpreting genome-wide association studies of psychiatric disorders. Mol Psychiatry. 2009;14:10–7. doi: 10.1038/mp.2008.126. [DOI] [PubMed] [Google Scholar]
- 15.Cichon S, et al. Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am J Psychiatry. 2009;166:540–56. doi: 10.1176/appi.ajp.2008.08091354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–97. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purcell S, et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 19.ISC. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–52. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang D, et al. Singleton deletions throughout the genome increase risk of bipolar disorder. Mol Psychiatry. 2009;14:376–80. doi: 10.1038/mp.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levy NA, Janicak PG. Calcium channel antagonists for the treatment of bipolar disorder. Bipolar Disord. 2000;2:108–19. doi: 10.1034/j.1399-5618.2000.020204.x. [DOI] [PubMed] [Google Scholar]
- 22.Kempton MJ, et al. Effects of the CACNA1C risk allele for bipolar disorder on cerebral gray matter volume in healthy individuals. Am J Psychiatry. 2009;166:1413–4. doi: 10.1176/appi.ajp.2009.09050680. [DOI] [PubMed] [Google Scholar]
- 23.Krug A, et al. Effect of CACNA1C rs1006737 on neural correlates of verbal fluency in healthy individuals. Neuroimage. 49:1831–6. doi: 10.1016/j.neuroimage.2009.09.028. [DOI] [PubMed] [Google Scholar]
- 24.Bigos KL, et al. Genetic variation in CACNA1C affects brain circuitries related to mental illness. Arch Gen Psychiatry. 67:939–45. doi: 10.1001/archgenpsychiatry.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erk S, et al. Brain function in carriers of a genome-wide supported bipolar disorder variant. Arch Gen Psychiatry. 67:803–11. doi: 10.1001/archgenpsychiatry.2010.94. [DOI] [PubMed] [Google Scholar]
- 26.Nyegaard M, et al. CACNA1C (rs1006737) is associated with schizophrenia. Mol Psychiatry. 15:119–21. doi: 10.1038/mp.2009.69. [DOI] [PubMed] [Google Scholar]
- 27.Curtis D, et al. Case-case genome-wide association analysis shows markers differentially associated with schizophrenia and bipolar disorder and implicates calcium channel genes. Psychiatr Genet. 21:1–4. doi: 10.1097/YPG.0b013e3283413382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Green EK, et al. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Mol Psychiatry. 2009 doi: 10.1038/mp.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gomez-Ospina N, Tsuruta F, Barreto-Chang O, Hu L, Dolmetsch R. The C terminus of the L-type voltage-gated calcium channel Ca(V)1.2 encodes a transcription factor. Cell. 2006;127:591–606. doi: 10.1016/j.cell.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lichtenstein P, et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet. 2009;373:234–9. doi: 10.1016/S0140-6736(09)60072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.International Schizophrenia Consortium. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–52. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.