

Abstract
We sought to evaluate the relationship between cell division and protein expression when using commercial poly(ethylenimine) (PEI)-based polyplexes. The membrane dye PKH26 was used to assess cell division, and cyan fluorescent protein (CFP) was used to monitor protein expression. When analyzed at the whole population level, a greater number of cells divided than expressed protein, regardless of the level of protein expression observed, giving apparent consistency with the hypothesis that protein expression requires cells to pass through mitosis in order for the transgene to overcome the nuclear membrane. However, when the polyplex-exposed population was evaluated for the amount of division in the protein-expressing subpopulation, it was observed that substantial amounts of expression had occurred in the absence of division. Indeed, in HeLa S3 cells, this represented the majority of expressing cells. Of interest, the doubling time for both cell lines was slowed by ~2-fold upon exposure to polyplexes. This change was not altered by the origin of the plasmid DNA (pDNA) transgene promoter (cytomegalovirus (CMV) or elongation factor-1 alpha (EF1α)). Gene expression arrays in polyplex-exposed HeLa S3 cells showed upregulation of cell cycle arrest genes and downregulation of genes related to mitosis. Chemokine, interleukin, and toll-like receptor genes were also upregulated, suggesting activation of proinflammatory pathways. In summary, we find evidence that a cell division-independent expression pathway exists, and that polyplex exposure slows cell division and increases inflammatory response.
Keywords: Polyplexes, poly(ethyleneimine), PEI, transfection, gene expression, protein expression, doubling time, promoter, gene array, HeLa S3, 293A
Introduction
The role of cell division in gene expression is important to understand in order to guide the development of better nonviral gene delivery materials. Microinjection experiments have repeatedly confirmed that transgenes must reach the nucleus to generate a gene product,1–5 indicating that the nuclear membrane is a major barrier to gene expression. Some studies have shown that nuclear uptake of plasmid DNA (pDNA) is strongly dependent on cell division4,6–11 with seminal work having been done by Mario Capecchi more than 30 years ago.1 Other research has shown that cell division is not required for nuclear uptake of pDNA12–16 with most studies exploiting nuclear localization signals (NLS) for transport through nuclear pore complexes.17 Indeed, Zanta et al. have shown that a single NLS is able to translocate pDNA to the nucleus.18
Much of this previous research employed microinjection or synchronization methods. Cooper has raised concerns that chemically synchronized cells do not reflect specific cell ages that are representative of the normal cell cycle.19 Additionally, microarray analysis of gene expression patterns has cast doubt that a conventional double thymidine block is able to synchronize cells.20 The drawbacks to microinjection experiments are that relatively low numbers of cells can be analyzed (usually on the order of tens to hundreds), the average volume injected into each cell can vary substantially,21 and material intended for the nucleus can be deposited into the cytoplasm. The limitations of synchronization and microinjection techniques indicate a need for a complementary method that can analyze the relationship of cell division and gene expression.
We designed a flow cytometry experiment to test the relationship of protein expression and cell division. This method utilizes large numbers of cells without perturbing the cell cycle with physical or chemical methods. The lipophilic dye PKH26 was used to assess division because it evenly stains the cell membrane and is divided approximately equally between daughter cells upon mitosis.22–24 Protein expression was monitored by fluorescence of cyan fluorescent protein (CFP). Polyplexes were formed between pDNA and jetPEI™, a potent poly(ethylenimine) (PEI)-derivative transfection reagent, and delivered to HeLa S3 and 293A cells. As an early clone of the parent HeLa cell line,25 HeLa S3 cells were used because they are established and commonly used. 293A cells were used because they produce high levels of transgene expression as the parent line was transformed with sheared human adenovirus type 5 DNA.26 Our experiment was designed to test whether or not cell division was required for protein expression.
We find that the number of polyplex-exposed cells that has divided is consistently greater than that expressing protein. This result provides apparent consistency with a model where cells divide in the course of gene expression because enough division has occurred to account for the entire expressing population. However, when we analyzed the amount of division in only the protein-expressing cells, we obtained evidence for expression occurring in the absence of cell division. This result substantiates a division-independent pathway. In the course of these experiments, we also discovered that exposure to polyplexes slowed the doubling time of both HeLa S3 and 293A cells by ~1.2 to 2.5 times. Gene expression arrays suggest that the cells are arrested in the G1 phase of the cell cycle and that polyplex exposure induces innate inflammatory gene expression. Together, these results demonstrate the need for development of nonviral gene delivery particles that mitigate the induction of inflammatory responses and alteration of the cell cycle progression.
Experimental
Cell Culture
HeLa S3 (human epithelial; Cat. No. CCL-2.2™) and COS-7 (monkey fibroblast; Cat. No. CRL-1651™) cells were purchased from ATCC® (Manassas, VA). 293A cells (human epithelial; Cat. No. R705-07) were purchased from Life Technologies (Carlsbad, CA). HeLa S3 cells are a derivative of the parent HeLa line (Cat. No. CCL-2™; ATCC®), and 293A cells are a subclone of HEK 293 cells (Cat. No. CRL-1573™; ATCC®). Each line tested negative for mycoplasma contamination (Cat. No. 6601; Takara Bio; Kyoto, Japan), was expanded, and then cryopreserved in LN2. HeLa S3 cells were cultured in F-12K medium (Cat. No. 30-2004; ATCC®). COS-7 and 293A cells were cultured in Dulbecco’s modified Eagle’s medium (D-MEM) with high glucose (Cat. No. 11995; Life Technologies). The media were supplemented with 10% fetal bovine serum (Cat. No. SH30910.03; Thermo Fisher Scientific; Waltham, MA) and 100 units/mL penicillin and 100 μg/mL streptomycin (Cat. No. 15140; Life Technologies). The 293A media was additionally supplemented with 100 μM MEM non-essential amino acids solution (Cat. No. 11140; Life Technologies). Cells were maintained at 37 °C with 5% CO2 in a humidified atmosphere and subcultured by trypsinization (Cat. No. 25200; Life Technologies).
PKH26 Staining
Cells were stained with PKH26 (Cat. No. PKH26GL; Sigma-Aldrich; Saint Louis, MO) according to the manufacturer’s protocol with final staining concentrations of 2 μM PKH26 and 1 × 107 cells/mL. After staining, 293A cell viability as measured by exclusion of trypan blue stain (Cat. No. 15250; Life Technologies) and percent recovery averaged 91 ± 3% and 72 ± 16%, respectively. Control cells that were not stained with PKH26 were still subjected to the staining reaction, albeit with no PKH26 dye present, and showed similar viability (94 ± 3%) and recovery (84 ± 17%) as the stained cells. Similar percentages were also obtained for stained and control HeLa S3 cells. Cells were seeded at 80,000 cells/well in 12-well tissue culture-treated plates (Cat. No. 353043; Becton, Dickinson and Company; Franklin Lakes, NJ) in 800 μL complete medium, and incubated at 37 °C with 5% CO2 for 5 to 6 h prior to transfection (see SI for the rationale for the short incubation time).
DNA Preparation
The pDNAs used were 4.1 kb CFP-encoding pDNA (pAmCyan1-C1; Cat. No. 632441; Clontech; Mountain View, CA), 5.1 kb blank pDNA that encoded for no fluorescent reporter protein (gWIZ™-blank; Cat. No. 5008; Aldevron; Fargo, ND), and 5.8 kb green fluorescent protein (GFP)-encoding pDNA (gWIZ™-GFP; Cat. No. 5006; Aldevron). These pDNAs were all driven by the CMV immediate-early promoter (PCMV IE). A fourth pDNA, 5.5 kb in size and GFP-encoding, was custom synthesized by Aldevron to be the same as gWIZ™-GFP except that it contained the human elongation factor-1 alpha (EF1α) nonviral transgene promoter from pEF1α-AcGFP1-C1 (Cat. No. 631974; Clontech) instead of the CMV promoter. All components of the gWIZ™-GFP-EF1α pDNA were nonviral in origin. All pDNAs were amplified after transformation of DH5α™ Escherichia coli cells (Cat. No. 18265; Life Technologies) according to the manufacturer’s protocol, and LB Broth (Cat. No. 12780; Life Technologies) supplemented with 50 μg/mL kanamycin (Cat. No. K0254; Sigma-Aldrich) was used as the medium. The pDNAs were purified from the cells with a Qiagen EndoFree® Plasmid Mega Kit (Cat. No. 12381; Venlo, Netherlands) according to the manufacturer’s instructions. A fifth DNA sample was sheared salmon sperm DNA (ssDNA; Cat. No. AM9680; Applied Biosystems; Foster City, CA) which was also taken through the Qiagen EndoFree® purification for consistency with the pDNA samples. All DNA samples were nonpyrogenic according to a Kinetic-QCL™ assay (BioWhittaker, Inc.; Walkersville, MD) and tested negative for RAW-Blue™ cell (Cat. No. raw-sp; InvivoGen; San Diego, CA) activation according to a QUANTI-Blue™ enzymatic assay (Cat. No. rep-qb1; InvivoGen).
Polyplex Formation
Polyplexes were formed in DNase/RNase-free water (Cat. No. 10977; Life Technologies) between jetPEI™ (Cat. No. 89129-940; VWR International; West Chester, PA) and each of the five DNA samples at an N/P (+/−) ratio of 10 where N represents the nitrogen residues in the cationic polymer and P represents the phosphate residues in the DNA backbone. An equal volume (20 μL) of jetPEI™ in water was added to 20 μL of 40 μg/mL DNA in water, and the resultant solution was incubated at room temperature for ~20 to 30 min. Cells plated in 12-well plates were transfected with 40 μL polyplex solution/well containing 0.8 μg DNA. For samples exposed to jetPEI™ only, the same amount of jetPEI™ per 40 μL of total solution was used, replacing the DNA with an equal volume of water.
Cell Transfection
Prior to transfection, the complete medium was aspirated, the cells were washed once with phosphate buffered saline (PBS; Cat. No. 10010; Life Technologies), and 800 μL of serum-free medium (SFM) was added back to each well. To transfect the cells, 40 μL of either the polyplex or polymer-only solution, was added to the appropriate wells, and the cells were incubated at 37 °C with 5% CO2. After 3 h, the SFM was aspirated and replaced with 800 μL complete medium. The cells were again incubated at 37 °C with 5% CO2 until the selected time point, whereupon the cells were harvested for analysis by flow cytometry. A given time point includes the 3 h incubation in SFM. A 24 h transfection or time point, for example, indicates cells that were exposed to nanoparticles in SFM for 3 h and then incubated 21 h further in complete medium prior to harvesting.
Flow Cytometry
Cells were harvested for analysis by flow cytometry by trypsinization with 200 μL trypsin/well after rinsing once with PBS. Complete medium (1 mL) was added to each well to inhibit the trypsin, and resultant suspensions were centrifuged for 5 min at 835 × g. Cell pellets were resuspended in 1 mL PBS, and 7-aminoactinomycin D (7AAD; Cat. No. A1310; Life Technologies) was added to a final concentration of 1 μg/mL as a viability marker. 7AAD single dye controls were made by resuspending the cell pellet in 500 μL PBS, adding an equal volume of 200 proof ethanol (Cat. No. 2716; Decon Labs; King of Prussia, PA), and subsequently adding 7AAD to a final concentration of 5 μg/mL. All samples were incubated on ice for 30 min, centrifuged for 5 min at 835 × g, and resuspended in PBS. The cells were analyzed with an Epics XL-MCL Flow Cytometer (Beckman Coulter; Brea, CA), collecting 10,000 events per sample. We analyzed three technical replicates per sample, and experiments were repeated at least three independent times.
Because three (or more) colors cannot be easily manually compensated due to their interdependence,27 data analyses were performed using Weasel software (Version 2.7.4; Walter and Eliza Hall Institute; Melbourne, Australia) and a custom code written in MATLAB (Version 7.8.0.347; The MathWorks; Natick, MA) that follows standard compensation protocols.28–31 Median (as opposed to mean) fluorescence intensity is used throughout this manuscript because it is relatively insensitive to outliers in the data. Curves of best exponential fit were determined using Microsoft® Excel® for Mac (Redmond, WA). The exponential fits were constrained to go through the PKH26 only data point at 0 h because the cells for all the different treatments were stained in a single reaction. Cells positive for 7AAD were gated out of the analyses but never represented more than 2–3% of any given sample. Previous data from our group has also shown that these transfection conditions are not cytotoxic according to XTT and LDH assays.32
Total RNA Extraction and PCR Arrays
HeLa S3 cells were plated in triplicate as described above except that cells were not stained with PKH26. (Control experiments showed that PKH26-staining did not affect the rate of proliferation—see Figure S1.) The transfection experiment was performed in 6-well tissue culture-treated plates (Cat. No. 353046; Becton, Dickinson and Company) with all reagents and cell numbers scaled up according to the growth area of the wells. Again, cells were exposed to jetPEI™ only or to polyplexes made between jetPEI™ and either gWIZ™-GFP or gWIZ™-GFP-EF1α pDNA. At 4 and 24 h following transfection, total RNA from each sample was isolated using the Qiagen RNeasy Mini Kit (Cat. No. 74104). Cell lysates were homogenized using Qiagen QIAshredder spin columns (Cat. No. 79654) and on-column DNase digestions were performed with an RNase-Free DNase Set (Cat. No. 79254; Qiagen). The experiment was repeated three independent times yielding three biological replicates for each sample (the one exception is described in the Figure S7 caption), and all biological replicates were subjected to quality control procedures and analysis by PCR arrays at the same time.
Quality control for each sample was assessed by measuring RNA concentration and purity with a UV spectrophotometer (NanoDrop 2000; Thermo Fisher Scientific), and by measuring ribosomal RNA integrity with an Agilent 2100 BioAnalyzer (Agilent Technologies; Santa Clara, CA). The RT2 First Strand Kit (Cat. No. 330401; Qiagen) was used according to the manufacturer’s protocol (500 ng total RNA per sample) to synthesize the complementary DNA (cDNA). The cDNA samples were analyzed for changes in gene expression as compared to the untreated control by both Human Cell Cycle (Cat. No. PAHS-020E) and Inflammatory Response and Autoimmunity (Cat. No. PAHS-077E) RT2 Profiler™ PCR arrays from Qiagen. We used an ABI 7900HT Fast Real-Time PCR System (Life Technologies; Carlsbad, CA) with a 384-well block module, the manufacturer’s recommended thermal cycling conditions, and the Qiagen RT2 SYBR® Green qPCR Mastermix (Cat. No. PA-112). Each plate contained 4 × 96 genes with each set of 96 containing 84 pathway-related genes plus genomic DNA contamination, reverse transcription, and positive PCR controls, and 5 housekeeping genes (β2 microglobulin, hypoxanthine phosphoribosyltransferase 1, ribosomal protein L13a, glyceraldehyde 3-phosphate dehydrogenase, and β-actin), all of which were used for normalization. The data were analyzed using the 2−ΔΔ CT method and genes that exhibited greater than or equal to 2-fold up- or downregulation were considered different from the control. Statistical significance was determined using false discovery rate, where a p-value of 0.05 indicates at most 5% false positives.33,34
Results
Relationship of Cell Division and Protein Expression
A two-color flow cytometry experiment was designed to quantify the relationship between cell division and protein expression for PEI-based polyplexes. The membrane dye PKH26 was used to assess division and fluorescence of CFP was used to monitor protein expression. PKH26-stained HeLa S3 and 293A cells were transfected for 3 h with polyplexes formed between CFP-encoding pDNA and jetPEI™. The dilution of PKH26 in treated and untreated samples was observed by flow cytometry over 2 days following transfection (Figure 1; see Figure S2 for representative 293A data).
Figure 1.

PKH26 intensity (arbitrary units) of representative HeLa S3 cells treated with polyplexes formed between jetPEI™ and CFP pDNA over 2 days following transfection as compared to control cells (PKH26 only). Cells expressing CFP (□) and those not expressing CFP (□) are subsets of the whole population (▲) that was treated with polyplexes. Cells were transfected for 3 h beginning at time = 0 h. Each point shows the mean ± SD of three technical replicates with curve of best exponential fit. All data are fit with the PKH26 only datum at 0 h.
We compared the number of polyplex-exposed cells that had divided to the number that expressed protein. The number of cells that had divided was measured by comparing the initial PKH26 intensity (at 0 h) to the final PKH26 intensity (at ~48 h). The number of cells expressing CFP was measured by comparing CFP fluorescence intensity to that of the untreated (but still PKH26-stained) control (Figure 2; see Figure S3 for data showing pDNA uptake). Consistently, the number of cells that had divided was greater than the number of cells that expressed CFP (Table 1). This held true regardless of the percent of protein expression observed (38 ± 9% for HeLa S3 and 84 ± 5% for 293A). For example, in HeLa S3 Experiment #1, out of 10,000 measured cells, 6000 ± 800 had divided and 2400 ± 200 expressed CFP. Table 1 shows the results from the full sets of experiments in HeLa S3 and 293A cells.
Figure 2.

Flow cytometry data for representative 293A cells showing expression efficiency for CFP pDNA (CMV) polyplexes. The gate was drawn based on the control cells, and the inset table shows the average percent of expression efficiency for all three protein-encoding polyplexes across both cell lines at ~48 h following transfection. The chart also shows that cells exposed to CFP pDNA (CMV) polyplexes (red) have higher PKH26 intensity than control cells (gray) at ~48 h following transfection.
Table 1.
Breakdown of the number of cells for each experiment that have divided and/or expressed CFP ~48 h following transfection.
| Cell Line | Expt # | All Cells | Cells Expressing CFPa | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| # of Cells Analyzed | # Dividedb | # Expressing CFPc | # Dividedd | ||||||
|
| |||||||||
| Avge | SD | Avge | SD | Avge | SD | Avge | SD | ||
| HeLa S3 | 1 | 10000 | 0 | 6000 | 800 | 2400 | 200 | 1100 | 100 |
| 2f | - | - | - | - | - | - | - | - | |
| 3 | 10000 | 0 | 4400 | 300 | 4300 | 100 | 900 | 100 | |
| 4 | 10000 | 0 | 8200 | 200 | 2400 | 100 | 1300 | 100 | |
| 5 | 10000 | 0 | 6400 | 100 | 4800 | 200 | 700 | 200 | |
|
| |||||||||
| 293A | 1 | 10000 | 0 | 9800 | 700 | 8100 | 100 | 7500 | 500 |
| 2 | 10000 | 0 | 8500 | 200 | 7700 | 0 | 5300 | 200 | |
| 3 | 10000 | 0 | 9000 | 2100 | 7900 | 100 | 6300 | 1600 | |
| 4f | - | - | - | - | - | - | - | - | |
| 5 | 10000 | 0 | 9200 | 1500 | 7400 | 0 | 6000 | 1100 | |
| 6 | 10000 | 0 | 9500 | 900 | 8300 | 100 | 7600 | 700 | |
| 7g | 7000 | 2700 | 6100 | 700 | 5600 | 100 | 4400 | 400 | |
Cells expressing CFP is a subset of all cells.
Calculated based on the measured PKH26 intensity of all cells and the number of cells analyzed.
Measured based on the amount of CFP fluorescence over the untreated control.
Calculated based on the measured PKH26 intensity of cells expressing CFP and the number of all cells expressing CFP.
Avg is an average of three technical replicates.
HeLa S3 Expt #2 was not included because the percent CFP expression was a statistical outlier. 293A Expt #4 was not included because the doubling time of polyplex-exposed cells was a stastical outlier.
Occassionally we were unable to collect 10,000 cells for all three technical replicates.
The polyplex-exposed population was subdivided into cells that expressed CFP and cells that did not express CFP. By comparing the initial PKH26 intensity to the final PKH26 intensity, we determined the number of cells that had divided in the population expressing CFP (Table 1). For 293A cells, the majority of cells that expressed CFP had also divided (69% to 93% of expressing cells) whereas for HeLa S3 cells, the majority of cells that expressed protein had not divided (44% to 86% of expressing cells). Schematics of representative HeLa S3 and 293A experiments showing the relative numbers of divided versus not divided and expressing versus nonexpressing cells are shown in Figure S4.
Effect of Cationic Polymer- and Polyplex-Exposure on Cell Doubling Times
HeLa S3 and 293A cells exposed to polyplexes formed with any of the five types of DNA divided ~1.2 to 2.5 times more slowly than control cells, but cells exposed to only the cationic polymer, jetPEI™, divided at a similar rate as control cells (Figure 3). For both cell lines, these effects were consistently observed over multiple independent experiments, each sample being tested at least three independent times. The doubling times of the samples were extracted from curves of best exponential fit, the averages of which are shown in Figure 3b and Table S1.
Figure 3.
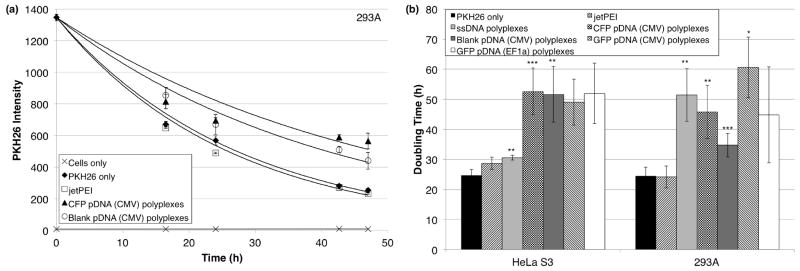
As measured by PKH26 intensity, cells treated with polyplexes formed between jetPEI™ and any of five types of DNA divide more slowly than control cells (PKH26 only) over 2 days following transfection. Cells treated with polymer alone (jetPEI™ only) are not different than the control. (a) Representative data for 293A cells. The cells were transfected for 3 h beginning at time = 0 h. Each point shows the mean ± SD of three technical replicates with curve of best exponential fit except for the cells only sample, which has a linear line of best fit. All data are fit with the PKH26 only datum at 0 h except for the cells only sample. (b) Summary of five and seven independent experiments with HeLa S3 and 293A cells, respectively. All samples were tested in three or more independent experiments even with statistical outliers having been removed. For HeLa S3 cells, F(6, 20) = 15.6, p < 0.05, ω= 0.87. For 293A cells, F(6, 29) = 16.9, p < 0.05, ω = 0.85. Differences were determined according to the post hoc Games-Howell test, with equal variance not assumed (p < 0.05 for the Levene statistic). * p < 0.10, ** p < 0.05, *** p < 0.01.
Representative data showing the effect of blank pDNA polyplexes on COS-7 cells also demonstrates the slowed doubling time result. Figures S5a and S5b show representative data for the dilution of PKH26 in HeLa S3 and COS-7 cells over time, respectively. Figure S5c shows that exposure to the pDNA alone does not affect doubling time, and Figure S5d shows the average doubling times (from Figure 3b) of untreated and CFP polyplex-treated HeLa S3 cells in terms of PKH26 intensity over time, illustrating that even when the error on the doubling times is considered, polyplex-exposed cells exhibit a substantially slowed doubling time.
Senescence-Associated β-Galactosidase Assays
The semiquantitative senescence-associated β-galactosidase (SA-β-gal) assay detects the level of the lysosomal hydrolase β-galactosidase that is increased in senescent cells over quiescent or normally proliferating cells. At pH 6.0, the β-galactosidase cleaves X-gal in senescent cells, leading to formation of a perinuclear blue-green precipitate that is observable by light microscopy.35,36 In both polymer- and polyplex-treated HeLa S3 and 293A cells, senescence was not observed 48 h post-transfection over that of background levels observed in normally proliferating cells (Figure S6).
Gene Expression in HeLa S3 Cells Related to the Cell Cycle and Inflammatory Response and Autoimmunity by Targeted PCR Arrays
The changes in expression of 84 cell cycle-related genes and 84 inflammatory response and autoimmunity-related genes were observed in polymer- and polyplex-exposed cells. The goal of the array study was to probe possible mechanisms by which polyplex exposure slowed HeLa S3 cell division, and to investigate differential responses based on the viral versus nonviral nature of the transgene promoter. Overall, the changes in gene expression were similar for both kinds of polyplexes (i.e., differences due to the viral vs nonviral nature of the transgene promoter were not observed). For the majority of affected genes at 24 h following transfection on both types of arrays, the polyplex-exposed samples were similarly up- or downregulated and the polymer-only sample was unchanged as compared to the untreated control.
The genes upregulated by the largest magnitude on the cell cycle array were CDKN1A, CHEK2, DIRAS3, GADD45A, HERC5, and SERTAD1 (Table 2). These genes were upregulated 24 h following transfection in both polyplex-exposed samples but were unchanged in the polymer-only sample, with the exception of GADD45A which was upregulated 4-fold in the polymer-only sample. Four of these genes (CDKN1A, CHEK2, DIRAS3, and GADD45A) have been shown to function in cell cycle arrest or inhibition pathways, generally in response to stress stimuli such as DNA damage. The genes downregulated by the largest magnitude on the cell cycle array were CCNB1, CCNF, CDK5R1, CDKN3, KPNA2, MAD2L1, MRE11A, and TFDP1 (Table 2). At 24 h following transfection, these genes were generally downregulated between 2- and 3-fold in both polyplex-exposed samples but were unchanged in the polymer-only sample. These genes are all related to normal proliferation pathways except for CDKN3, which encodes for a cyclin-dependent kinase inhibitor.37 Figures 4a and 4b show scatterplots comparing the gene regulation of the jetPEI™ and GFP pDNA (CMV) polyplex treatments to the untreated control at 24 h. Figure S7 shows scatterplots of the 4 h treatments as well as the 24 h treatment with GFP pDNA (EF1α) polyplexes, and Table S2 shows the full cell cycle array results.
Table 2.
Changes in expression of selected genes related to the cell cycle pathway in jetPEI™ or polyplex-exposed HeLa S3 cells.
| GeneBank | Symbol | Descriptiona | Fold Change in Gene Expressionb
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| jetPEI ™ | GFP (CMV) polyplexes | GFP (EF1α) polyplexes | |||||||
|
| |||||||||
| 4h | 24h | 4h | 24h | 4h | 24h | ||||
| Upregulated | NM_000389 | CDKN1A | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | 2.5 | 1.4 | 2.2 | 2.1 | 2.5 | 2.6 |
| NM_007194 | CHEK2 | CHK2 checkpoint homolog (S. pombe) | −1.2 | −1.3 | 1.0 | 2.2 | 1.0 | 2.4 | |
| NM_004675 | DIRAS3 | DIRAS family, GTP-binding RAS-like 3 | 1.1 | −2.8 | 2.7 | 4.2 | 3.5 | 5.5 | |
| NM_001924 | GADD45A | Growth arrest and DNA-damage-inducible, alpha | 2.5 | 4.0 | 1.6 | 3.6 | 1.5 | 3.8 | |
| NM_016323 | HERC5 | Hect domain and RLD 5 | −1.1 | 1.4 | 5.3 | 25.8 | 4.8 | 27.9 | |
| NM_013376 | SERTAD1 | SERTA domain containing 1 | 4.1 | 1.7 | 3.5 | 2.8 | 4.1 | 2.8 | |
|
| |||||||||
| Downregulated | NM_031966 | CCNB1 | Cyclin B1 | −1.3 | −1.1 | −1.3 | −2.1 | −1.2 | −2.1 |
| NM_001761 | CCNF | Cyclin F | −2.8 | −1.1 | −2.3 | −2.0 | −2.8 | −2.0 | |
| NM_003885 | CDK5R1 | Cyclin-dependent kinase 5, regulatory subunit 1 (p35) | 1.6 | −1.1 | 2.0 | −2.2 | 2.2 | −1.6 | |
| NM_005192 | CDKN3 | Cyclin-dependent kinase inhibitor 3 | −1.3 | −1.1 | −1.3 | −2.3 | −1.2 | −2.3 | |
| NM_002266 | KPNA2 | Karyopherin alpha 2 (RAG cohort 1, importin alpha 1) | −1.2 | −1.5 | −1.0 | −2.3 | −1.1 | −2.1 | |
| NM_002358 | MAD2L1 | MAD2 mitotic arrest deficient-like 1 (yeast) | −1.1 | −1.2 | −1.1 | −2.2 | 1.0 | −2.3 | |
| NM_005590 | MRE11A | MRE11 meiotic recombination 11 homolog A (S. cerevisiae) | −1.6 | −1.5 | −1.3 | −2.6 | −1.1 | −3.1 | |
| NM_007111 | TFDP1 | Transcription factor Dp-1 | −1.2 | −1.2 | −1.3 | −2.1 | −1.2 | −2.2 | |
The gene descriptions are reproduced from the array product information from Qiagen.
Green and red shading indicate fold-changes of ≥ 2 and ≤ 2, respectively, with p ≤ 0.05.
Figure 4.

Scatterplots showing the relative number of up- (▲) and downregulated (■) genes for the jetPEI™ and GFP pDNA (CMV) polyplex treatments compared to the untreated control at 24 h following transfection. (a) and (b) Cell cycle array. (c) and (d) Inflammatory response and autoimmunity array. The centerline indicates a fold change (2−ΔΔCt) of 1, and the upper and lower lines indicate fold changes of 2 in gene expression. Circles indicate fold changes ≤ 2. Comparing to the untreated control, filled symbols indicate regulation that is significantly different (p ≤ 0.05), and open symbols indicate regulation that is not significantly different. Only the significantly up- or downregulated genes shown in Tables 2 and 3 are labeled in this figure.
We observed substantial upregulation of chemokine, interleukin, and toll-like receptor (TLR) genes on the inflammatory response and autoimmunity array (Table 3). The chemokine ligand genes (CCL22, CCL3, CCL5, and CXCL10) in the polyplex-exposed samples exhibited the highest magnitude upregulation, up to 1000-fold over the negative control. TNF and a number of interleukin genes (e.g., IL1A, IL1B, IL1R1, IL1RN, IL23A, and IL6), a class of cytokines that act as key regulators of the inflammatory response, were also highly upregulated in polyplex-exposed samples in comparison to the polymer-only treatment. Some genes encoding TLRs (TLR1, TLR3, TLR4, and TLR5), membrane-spanning proteins that activate inflammatory responses,38 were additionally strongly affected by the polyplex treatments. Table S3 shows the full inflammatory response and autoimmunity array results. Figures 4c and 4d show selected scatterplots for the 24 h time point comparing the gene regulation of the jetPEI™ and GFP pDNA (CMV) polyplex treatments to the untreated control. Figure S7 shows scatterplots of the 4 h treatments as well as the 24 h treatment with GFP pDNA (EF1α) polyplexes.
Table 3.
Changes in expression of selected genes related to inflammatory response and autoimmunity in jetPEI™ or polyplex-exposed HeLa S3 cells.
| GeneBank | Symbol | Descriptiona | Fold Change in Gene Expressionb
|
|||||
|---|---|---|---|---|---|---|---|---|
| jetPEI ™ | GFP (CMV) polyplexes | GFP (EF1α) polyplexes | ||||||
|
| ||||||||
| 4h | 24h | 4h | 24h | 4h | 24h | |||
| NM_002990 | CCL22 | Chemokine (C-C motif) ligand 22 | 3.6 | 6.8 | 5.4 | 272.8 | 4.3 | 213.1 |
| NM_002983 | CCL3 | Chemokine (C-C motif) ligand 3 | −1.2 | 46.5 | −1.1 | 1084.1 | 1.9 | 989.4 |
| NM_002985 | CCL5 | Chemokine (C-C motif) ligand 5 | 1.1 | 2.4 | 4.5 | 109.6 | 4.1 | 116.2 |
| NM_001295 | CCR1 | Chemokine (C-C motif) receptor 1 | 1.4 | 1.1 | 1.8 | 24.2 | −2.0 | 20.1 |
| NM_001565 | CXCL10 | Chemokine (C-X-C motif) ligand 10 | −2.4 | 2.6 | 2.3 | 553.0 | 2.1 | 652.0 |
|
| ||||||||
| NM_000575 | IL1A | Interleukin 1, alpha | −2.1 | 3.3 | −1.0 | 9.0 | 1.1 | 7.3 |
| NM_000576 | IL1B | Interleukin 1, beta | −1.7 | 2.6 | −1.2 | 7.3 | −1.3 | 6.3 |
| NM_000877 | IL1R1 | Interleukin 1 receptor, type I | −6.8 | 1.0 | −3.4 | 3.6 | −5.8 | 3.1 |
| NM_000577 | IL1RN | Interleukin 1 receptor antagonist | 1.4 | 2.0 | −1.1 | 4.6 | 1.0 | 6.0 |
| NM_016584 | IL23A | Interleukin 23, alpha subunit p19 | −1.2 | 1.1 | 1.2 | 7.2 | −1.6 | 6.1 |
| NM_000600 | IL6 | Interleukin 6 (interferon, beta 2) | 1.4 | 1.6 | 7.5 | 11.8 | 7.1 | 12.4 |
|
| ||||||||
| NM_003263 | TLR1 | Toll-like receptor 1 | −5.8 | 1.2 | −1.6 | 3.4 | −2.4 | 2.8 |
| NM_003265 | TLR3 | Toll-like receptor 3 | −2.2 | 1.1 | −1.9 | 11.5 | −1.7 | 10.8 |
| NM_138554 | TLR4 | Toll-like receptor 4 | 1.1 | 1.6 | −1.2 | 3.9 | −1.3 | 4.3 |
| NM_003268 | TLR5 | Toll-like receptor 5 | 1.2 | −6.5 | 1.5 | 5.8 | 1.5 | 9.6 |
|
| ||||||||
| NM_000594 | TNF | Tumor necrosis factor (TNF superfamily, member 2) | 13.0 | 1.2 | 18.6 | 14.9 | 14.6 | 14.1 |
The gene descriptions are reproduced from the array product information from Qiagen.
Green and red shading indicate fold-changes of ≥ 2 and ≤ 2, respectively, with p ≤ .05.
Discussion
Protein Expression Can Occur Without Cell Division
We developed a new approach to assess the relationship of cell division and protein expression. The results show that the number of divided cells is consistently higher than the number of protein-expressing cells, in apparent support of a model where division is required for gene expression. However, when we delineated the number of protein-expressing cells that had divided, it became clear that a division-independent protein expression pathway was operative, regardless of the level of protein expression.
For 293A cells, the majority of cells that expressed protein had divided. Although this appears to support a division-dependent pathway for protein expression, we observed that 7% to 31% of protein-expressing cells had not divided. In HeLa S3 cells, 44 to 86% of protein-expressing cells had not divided, again showing that protein expression can occur without division, and consistent with a previous study that showed a lack of cell cycle dependence in HeLa cells for transfections with linear PEI.39 Grandinetti et al. have also shown that linear PEI can induce nuclear membrane permeability, providing a possible route for nucleic acid entry into the nucleus.40
Our study improves understanding of transfection pathways by demonstrating that the apparent expression pathway is consistent with previous studies showing that cell division is required, but only when one cannot look at the different cell subpopulations. However, when using the quantitative methods employed here, which allow the division of protein-expressing cells to be analyzed, we find that protein expression can occur without division. Indeed, the apparent consistency for the overall cell population with a division-dependent pathway may arise from a coincidence of similar rates of protein expression and cell division.
Polyplex Exposure Slows Cell Doubling Time
This study also improves current understanding of transfection by showing that transfection and subsequent protein expression slow cell division, which studies that employ nondividing cells could not detect. We employed a number of controls to determine the cause of the slowed division, such as a “blank” pDNA of comparable size to the CFP-encoding pDNA. The blank pDNA contains the same cytomegalovirus (CMV) transgene promoter but does not encode for any fluorescent reporter protein. The slowed division of HeLa S3 and 293A cells is not attributable to toxicity of the generated fluorescent protein because when cells are exposed to blank pDNA polyplexes, division is slowed to a similar extent.
We also formed polyplexes with two pDNAs that encode for GFP, one containing a viral transgene promoter (CMV) and the other of completely nonviral origin, containing the EF1α transgene promoter. These pDNAs were used because it has previously been shown that transfection can lead to upregulation of some cytokines (e.g., TNFα, IFNγ, and IL-10) that are able to differentially affect the activity of viral promoters such as CMV in comparison to nonviral promoters such as EF1α.41–46 We had hypothesized that a pDNA lacking any viral origin would be unaffected by the cytokine upregulation, possibly yielding a normal doubling time. However, regardless of the type of DNA used in the polyplexes, division was consistently slowed.
We also find that the slowed division is not related to a cationic charge density effect of the free polymer, because cells exposed to only free polymer divide at a similar rate as control cells. The amount of free polymer that cells are exposed to in the polymer-only treatments is the same as the total amount of polymer used in the polyplex formulations. The amount of excess free polymer in the polyplex formulations is necessarily less than that in the free polymer only case because the DNA in the polyplex formulations electrostatically binds some of the polymer.
In summary, we find that the generated protein product, the origin of the pDNA transgene promoter, and the cationic charge concentration do not cause the slowed cell division. As another control, we made polyplexes with sheared salmon sperm DNA (ssDNA), which is nonfunctional in terms of expression. Dynamic light scattering measurements of the hydrodynamic diameter of polyplexes formed with the pDNAs or the ssDNA show no appreciable differences (data not shown). Interestingly, exposure to polyplexes made with ssDNA slowed the doubling times of both cell lines, suggesting that a combination of the size and charge of the polyplexes was required to slow the doubling time. Particle size and surface properties have previously been found to affect cellular uptake and responses.47,48 The slowed division appears to be a result of toxicity of the polyplexes on the cell, and this was further explored using gene expression arrays.
Cells Exposed to Polyplexes Do Not Enter Cellular Senescence
We investigated senescence, a postmitotic state where cells are no longer able to divide, as a possible mechanism by which the doubling times of polyplex-exposed HeLa S3 and 293A cells were slowed. Replicative senescence was not induced in any sample over the basal level observed in the control. Bringold and Serrano49 have previously shown that in vitro upregulation of cyclin-dependent kinase inhibitors p16INK4a, p21Cip1, and p27Kip1 can cause senescence; the PCR arrays described here show no change in gene expression for p16INK4a or p27Kip1 in any sample at either time point. CDKN1A, which encodes for p21Cip1, was upregulated 2- to 3-fold at 24 h in the cells exposed to both types of polyplexes, but the gene encoding for p53 (another protein involved in the p21Cip1 pathway) was unaffected. Additionally, that p21Cip1 is required for activating senescence is debated.50 Together, these data indicate that entering senescence is not a biological effect of exposure to the types of polyplexes used in this study.
Cell Cycle and Inflammatory Response PCR Arrays Suggest Pathways for Slowed HeLa S3 Cell Division
To further probe possible mechanisms by which cell division was being slowed, gene arrays were run on total RNA was isolated from HeLa S3 cells that had been exposed to either jetPEI™ only, or to polyplexes formed between jetPEI™ and either gWIZ™-GFP or gWIZ™-GFP-EF1α pDNA. The isolated RNA was analyzed by both cell cycle and inflammatory response and autoimmunity PCR arrays for changes in gene expression relative to untreated controls. The inflammatory response and autoimmunity array was used because a preliminary study (data not shown) using the cell cycle array revealed that the hit of greatest magnitude fold change, HERC5, had some relationship in the literature to genes related to inflammatory response.
The cell cycle array shows that the upregulated genes are generally related to stress and cell cycle arrest, and the downregulated genes are generally related to normal pathways of proliferation. SERTAD1 and HERC5 are exceptions to this result that are also described here. The downregulated genes on the cell cycle array, specifically CCNB1, CCNF, and MAD2L1, suggest that normal progression through mitosis is being inhibited. CCNB1 encodes for cyclin B1, a G2/M-specific regulatory protein that, once activated, is involved in several of the early events of mitosis;51 CCNB1 is downregulated 2-fold in both polyplex-exposed samples at 24 h. CCNF is another gene that is similarly downregulated and encodes for a G2/M-specific cyclin.52 MAD2L1 functions as a mitotic checkpoint gene as it encodes for a protein that delays anaphase until the chromosomes have been properly aligned.53 The downregulation of these three genes suggest that normal progression through mitosis is being inhibited. Interestingly, KPNA2 encodes for a protein that has been shown to interact with NLSs and may regulate transport of proteins across the nuclear membrane;54 downregulation of KPNA2 could represent a defense mechanism against foreign genetic material entering the nucleus.
The upregulation of CDKN1A, CHEK2, and DIRAS3 in polyplex- but not polymer-exposed cells at 24 h specifically suggests arrest in the G1 phase of the cell cycle. First, CDKN1 encodes for the p21Cip1/Waf1 protein in mammalian cells and is upregulated 2- to 3-fold in polyplex-exposed cells. In response to DNA damage, p21Cip1/Waf1 has been shown to inhibit G1/S- and S-Cdks and therefore block cell cycle progression in the G1 phase,55 and overexpression of p21Cip1/Waf1 has been shown to decrease mammalian cell proliferation.56 Second, though it has also been linked to G2 phase arrest,57 CHEK2 has been shown to function as a DNA damage checkpoint by stabilization of tumor suppressor protein p53, which leads to growth arrest at the G1/S regulation point.58 Third, expression of the ARHI protein, encoded for by DIRAS3 which is upregulated 4- to 6-fold in polyplex-exposed samples, has been associated with decreased proliferation in some cancerous cell types.59 In this study, Rosen et al. reported that the ARHI acted by inducing p21Cip1/Waf1 and downregulating Cyclin D1 activity; Cyclin D1 activates G1-specific Cdks, suggesting that expression of ARHI can inhibit the cell cycle at the G1 phase. Together, the upregulation of these three genes indicates that a key mechanism of slowed doubling time in polyplex-exposed cells may be arrest in the G1 phase of the cell cycle, allowing the cells time to attempt to repair the “damage” done by the polyplexes before starting DNA replication in the S phase.
The upregulation of GADD45A in polyplex-exposed samples at 24 h is a biologically interesting result in terms of cell cycle inhibition, although the polymer-only sample shows similar upregulation. GADD45A can be induced by DNA damage and cell growth arrest,60 suggesting a mechanism by which growth may be arrested in the G2/M cell cycle phase. Tumor suppressor protein p53 transcriptionally regulates production of GADD45A protein,60,61 however, no significant changes in TP53 were observed here at 24 h, consistent with p53 regulation primarily occurring through post-transcriptional mechanisms.62 Subsequent research has shown that GADD45A protein interacts with the well-known cell cycle inhibitor p21Cip1 that is also transcriptionally regulated by p53,63 but that GADD45A protein is also able to arrest cell cycle progression independently of p21 by inhibiting Cdk1/Cyclin B1 kinase activity,64,65 which normally regulates cell cycle progression from G2 to M.51 Additionally, CCNB1, which encodes for Cyclin B1, is downregulated 2-fold in the polyplex-exposed samples at 24 h. Because similar upregulation is observed in polymer-exposed samples at 24 h, the upregulation of GADD45A in polyplex-exposed samples may be irrelevant to the observed slowed division. Alternatively, it may be the case that upregulation of GADD45A is a contributing factor to the observed slowed division via inhibition of progression through the M phase and subsequent division, yet that it is not sufficient to cause the slowed division alone.
SERTAD1 is an upregulated gene in polyplex-exposed samples on the cell cycle array that does not represent a cell cycle inhibition pathway. SERTAD1 encodes for the Cdk4-binding protein p34SEI-1, also known as TRIP-BR1, and is able to antagonize the function of the Cdk inhibitor protein p16INK4a. p16INK4a is normally able to target monomeric Cdks 4 and 6 and inhibit binding to cyclins in response to DNA damage or other environmental inhibitory signals.66 A subsequent study of p34SEI-1 revealed that it was able to activate Cdk4 in a concentration-dependent manner.67 The 3-fold upregulation of SERTAD1 in polyplex-exposed samples at 24 h may indicate that the cells are attempting to recover functional growth mechanisms, though recovery of a normal doubling time after 24 h was not observed.
HERC5 is upregulated by the largest magnitude of any gene on the cell cycle array, with polyplex-exposed samples showing 26- to 28-fold upregulation at 24 h. HERC5 upregulation is indicative of an inflammatory response as it has previously been induced by the proinflammatory cytokines tumor necrosis factor α (TNFα) and interleukin 1β (IL1β).68 Indeed, TNF and IL1B are also strongly upregulated at 24 h in both polyplex-exposed samples, consistent with the characterization of HERC5 as a late inflammatory response gene.68 TNFα was previously shown to be upregulated in keratinocytes in response to polyplex exposure,69 and activation of TNFα and IL1β has been shown to be effected through TLRs,38 which are also strongly affected here by the polyplex and polymer treatments. Interestingly, HERC5 has recently been shown to inhibit the replication of viral particles by blocking assembly,70 and Gao et al.71 observed a 5-fold decrease in HERC5 by the same cell cycle array when human epidermal keratinocytes were treated with a fullerene derivative that induces cellular senescence. HERC5 encodes for a ubiquitin ligase member of the HERC protein family called CEB172. This protein has been established as an active E3 enzyme required for posttranslational conjugation of ISG15, a ubiquitin-like protein, to target proteins, with ISG15 upregulation induced by interferons α and β during antiviral responses to infection.73 More recently, interferon regulatory factor 3 (IRF3) was shown to be a target protein of ISGylation by HERC5 protein;74 IRF3 is a tightly regulated transcription factor that has been shown to mediate antiviral responses.75
Upregulation of various chemokine ligand and receptor genes at 24 h also indicates a significant inflammatory response to the polyplexes and, in some cases, even to the polymer alone. Chemokines play a key role in inflammatory response by chemically attracting lymphocytes to the endothelial cells secreting the chemokines. Here, a number of chemokine ligand genes were strongly upregulated in the polyplex-exposed samples, with fold changes ranging from 110- to 1084-fold above the untreated control. CCR1, which encodes for a chemokine receptor, was also upregulated 20- to 24-fold in polyplex-exposed samples; some chemokine receptors have previously been shown to be upregulated in murine kidney tissue following polyplex administration at the same N/P ratio (10) used in this study.76 The chemokine ligand 10 gene (CXCL10) was also upregulated 553- to 652-fold in polyplex-exposed samples but unchanged in the polymer-only treatments.
Interleukins are another class of cytokines whose gene expression was strongly affected at 24 h by the polyplexes, including IL6, IL1R1, IL1RN, and IL23A. Upregulation of these interleukins was observed for both polyplex-exposed samples, whereas polymer-treated samples were either unchanged or minimally upregulated (~2-fold upregulation for IL1RN). Upregulation of interleukins IL10 and IL4 in murine spleen cells in response to polyplex treatment was previously observed.69 These gene expression data, coupled with the chemokine ligand and receptor responses, suggests that the strong inflammatory response displayed in the polyplex-treated cells is a contributing factor to the observed slowed division.
It is well appreciated that the presence of free DNA in different cellular spaces, notably the cytosol that is normally void of DNA, has been shown to induce antiviral responses.77–80 Even so, the purported benefits of nonviral over viral gene delivery systems almost always include that nonviral systems are less immunogenic than viral systems,81–87 with the chief drawback of nonviral systems being a lack of comparable transfection and expression efficiency. Indeed, nonviral carriers have been used to specifically deliver anti-inflammatory drugs.88 Our data demonstrate that exposure to nonviral gene delivery nanoparticles induces an inflammatory response in terms of gene expression, though the severity of this response in comparison to a response induced by viral gene delivery agents is not established here. It is possible that the inflammatory responses identified here are primarily related to cytosolic free pDNA that has been released from the polycationic polymeric carrier. For this reason it may be most beneficial for in vivo and clinical applications to pursue polymeric vectors that minimally separate from the pDNA prior to nuclear entry.89–91
Conclusion
In summary, when viewed at the whole population level, this study employing PEI/pDNA polyplexes in HeLa S3 and 293A cells appears to support a model where the pathway for protein expression requires cell division. However, when the division of only protein-expressing cells is considered, there is evidence that protein expression occurs without division. These experiments provide a complementary data set using independent methods to previous studies that showed that cell division is not required for protein expression.12–16 Additionally, we have shown that exposure to polyplexes can cause up to a 2-fold decrease in the doubling time of HeLa S3 and 293A cells. Analysis of changes in gene expression suggests that polyplex exposure slows cell division by upregulation of pathways that arrest the cell cycle in G1, and downregulation of pathways related to mitosis. Differences in proliferation rates upon exposure to polyplexes have not been apparent via other techniques and may affect data interpretation. The implications of nanoparticle exposure causing slowed cell division and inflammatory response should be considered in research that employs nanoscale drug or gene delivery vectors.
Supplementary Material
Acknowledgments
We thank Rahul Rattan and Drs. Thommey P. Thomas, Andrzej Myc, Anna U. Bielinska, and Lisa E. Prevette for helpful discussions and advice. We also thank University of Michigan’s Microarray Core Group for conducting quality control assays on the total RNA samples as well as for performing the PCR arrays, Gayle E. Carroll for doing the Kinetic-QCL™ assay, and Catherine H. Mullen for performing the QUANTI-Blue™ assay. This project has been funded in part with Federal funds from the National Institutes of Health, National Institute of Biomedical Imaging and Bioengineering under award EB005028.
Abbreviations
- 7AAD
7-aminoactinomycin D
- cDNA
complementary DNA
- CFP
cyan fluorescent protein
- CMV
cytomegalovirus
- EF1α
elongation factor-1 alpha
- GFP
green fluorescent protein
- NLS
nuclear localization signal
- PBS
phosphate buffered saline
- pDNA
plasmid DNA
- PEI
poly(ethylenimine)
- SA-β-gal
senescence-associated β-galactosidase
- SFM
serum-free medium
- ssDNA
salmon sperm DNA
Footnotes
Effect of PKH26 staining on cell proliferation rate; PKH26 intensity of CFP-expressing and nonexpressing 293A cells; pDNA uptake in 293A and HeLa S3 cells; representative schemes showing the approximate breakdown of divided versus not divided and expressing versus not expressing cells; PKH26 intensity of HeLa S3 cells exposed to CFP pDNA (CMV) and blank pDNA (CMV) polyplexes; average PKH26 intensity of untreated and CFP pDNA (CMV) polyplex-treated HeLa S3 cells over time; cellular senescence assay results; scatterplots showing gene up- and downregulation; average cell doubling times; complete tables of gene array results. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Capecchi MR. High efficiency transformation by direct microinjection of DNA into cultured mammalian cells. Cell. 1980;22:479–488. doi: 10.1016/0092-8674(80)90358-x. [DOI] [PubMed] [Google Scholar]
- 2.Dean DA, Dean BS, Muller S, Smith LC. Sequence Requirements for Plasmid Nuclear Import. Exp Cell Res. 1999;253:713–722. doi: 10.1006/excr.1999.4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zabner J, Fasbender AJ, Moninger T, Poellinger KA, Welsh MJ. Cellular and Molecular Barriers to Gene Transfer by a Cationic Lipid. J Biol Chem. 1995;270:18997–19007. doi: 10.1074/jbc.270.32.18997. [DOI] [PubMed] [Google Scholar]
- 4.Dean DA, Strong DD, Zimmer WE. Nuclear entry of nonviral vectors. Gene Ther. 2005;12:881–890. doi: 10.1038/sj.gt.3302534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Escriou V, Ciolina C, Helbling-Leclerc A, Wils P, Scherman D. Cationic lipid-mediated gene transfer: Analysis of cellular uptake and nuclear import of plasmid DNA. Cell Biol Toxicol. 1998;14:95–104. doi: 10.1023/a:1007425803756. [DOI] [PubMed] [Google Scholar]
- 6.Lam AP, Dean DA. Progress and prospects: nuclear import of nonviral vectors. Gene Ther. 2010;17:439–447. doi: 10.1038/gt.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vaughan EE, DeGiulio JV, Dean DA. Intracellular trafficking of plasmids for gene therapy: Mechanisms of cytoplasmic movement and nuclear import. Curr Gene Ther. 2006;6:671–681. doi: 10.2174/156652306779010688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brunner S, Sauer T, Carotta S, Cotten M, Saltik M, Wagner E. Cell cycle dependence of gene transfer by lipoplex, polyplex and recombinant adenovirus. Gene Ther. 2000;7:401–407. doi: 10.1038/sj.gt.3301102. [DOI] [PubMed] [Google Scholar]
- 9.Grosse S, Thévenot G, Monsigny M, Fajac I. Which mechanism for nuclear import of plasmid DNA complexed with polyethylenimine derivatives? J Gene Med. 2006;8:845–851. doi: 10.1002/jgm.915. [DOI] [PubMed] [Google Scholar]
- 10.Mortimer I, Tam P, MacLachlan I, Graham RW, Saravolac EG, Joshi PB. Cationic lipid-mediated transfection of cells in culture requires mitotic activity. Gene Ther. 1999;6:403–411. doi: 10.1038/sj.gt.3300837. [DOI] [PubMed] [Google Scholar]
- 11.Tseng WC, Haselton FR, Giorgio TD. Mitosis enhances transgene expression of plasmid delivered by cationic liposomes. Biochimica et Biophysica Acta. 1999;1445:53–64. doi: 10.1016/s0167-4781(99)00039-1. [DOI] [PubMed] [Google Scholar]
- 12.Dean DA. Import of Plasmid DNA into the Nucleus Is Sequence Specific. Exp Cell Res. 1997;230:293–302. doi: 10.1006/excr.1996.3427. [DOI] [PubMed] [Google Scholar]
- 13.Dowty ME, Williams P, Zhang G, Hagstrom JE, Wolff JA. Plasmid DNA entry into postmitotic nuclei of primary rat myotubes. Proc Natl Acad Sci US A. 1995;92:4572–4576. doi: 10.1073/pnas.92.10.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ludtke JJ, Sebestyen MG, Wolff JA. The Effect of Cell Division on the Cellular Dynamics of Microinjected DNA and Dextran. Mol Ther. 2002;5:579–588. doi: 10.1006/mthe.2002.0581. [DOI] [PubMed] [Google Scholar]
- 15.Pollard H, Remy JS, Loussouarn G, Demolombe S, Behr JP, Escande D. Polyethylenimine but Not Cationic Lipids Promotes Transgene Delivery to the Nucleus in Mammalian Cells. J Biol Chem. 1998;273:7507–7511. doi: 10.1074/jbc.273.13.7507. [DOI] [PubMed] [Google Scholar]
- 16.Wilson GL, Dean BS, Wang G, Dean DA. Nuclear Import of Plasmid DNA in Digitonin-permeabilized Cells Requires Both Cytoplasmic Factors and Specific DNA Sequences. J Biol Chem. 1999;274:22025–22032. doi: 10.1074/jbc.274.31.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bergen JM, Pun SH. Peptide-Enhanced Nucleic Acid Delivery. MRS Bull. 2005;30:663–667. [Google Scholar]
- 18.Zanta MA, Belguise-Valladier P, Behr JP. Gene delivery: A single nuclear localization signal peptide is sufficient to carry DNA to the cell nucleus. Proc Natl Acad Sci US A. 1999;96:91–96. doi: 10.1073/pnas.96.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooper S. Rethinking synchronization of mammalian cells for cell cycle analysis. Cell Mol Life Sci. 2003;60:1099–1106. doi: 10.1007/s00018-003-2253-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shedden K, Cooper S. Analysis of cell-cycle-specific gene expression in human cells as determined by microarrays and double-thymidine block synchronization. Proc Natl Acad Sci US A. 2002;99:4379–4384. doi: 10.1073/pnas.062569899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minaschek G, Bereiter-Hahn J, Bertholdt G. Quantitation of the volume of liquid injected into cells by means of pressure. Exp Cell Res. 1989;183:434–442. doi: 10.1016/0014-4827(89)90402-3. [DOI] [PubMed] [Google Scholar]
- 22.Horan PK, Slezak SE. Stable cell membrane labelling. Nature. 1989;340:167–168. doi: 10.1038/340167a0. [DOI] [PubMed] [Google Scholar]
- 23.Horan PK, Jensen BD, Slezak SE. Viable Cell Labelling. 4,783,401. US Patent. 1988 Nov 8;
- 24.Wallace PK, Tario JD, Fisher JL, Wallace SS, Ernstoff MS, Muirhead KA. Tracking antigen-driven responses by flow cytometry: Monitoring proliferation by dye dilution. Cytometry, Part A. 2008;73A:1019–1034. doi: 10.1002/cyto.a.20619. [DOI] [PubMed] [Google Scholar]
- 25.Puck TT, Marcus PI, Cieciura SJ. Clonal Growth of Mammalian Cells In Vitro. J Exp Med. 1956;103:273–283. doi: 10.1084/jem.103.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a Human Cell Line Transformed by DNA from Human Adenovirus Type 5. J Gen Virol. 1977;36:59–72. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 27.Roederer M, De Rosa S, Gerstein R, Anderson M, Bigos M, Stovel R, Nozaki T, Parks D, Herzenberg L, Herzenberg L. 8 Color, 10-parameter flow cytometry to elucidate complex leukocyte heterogeneity. Cytometry. 1997;29:328–339. doi: 10.1002/(sici)1097-0320(19971201)29:4<328::aid-cyto10>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 28.Roederer M. Current Protocols in Cytometry; Vol. 1. John Wiley & Sons; Hoboken, NJ: 2002. Compensation in Flow Cytometry; pp. 1.14.1–1.14.20. [DOI] [PubMed] [Google Scholar]
- 29.Roederer M. Spectral compensation for flow cytometry: Visualization artifacts, limitations, and caveats. Cytometry. 2001;45:194–205. doi: 10.1002/1097-0320(20011101)45:3<194::aid-cyto1163>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 30.Baumgarth N, Roederer M. A practical approach to multicolor flow cytometry for immunophenotyping. J Immunol Methods. 2000;243:77–97. doi: 10.1016/s0022-1759(00)00229-5. [DOI] [PubMed] [Google Scholar]
- 31.Tung JW, Parks DR, Moore WA, Herzenberg LA, Herzenberg LA. New approaches to fluorescence compensation and visualization of FACS data. Clin Immunol. 2004;110:277–283. doi: 10.1016/j.clim.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 32.Prevette LE, Mullen DG, Banaszak Holl MM. Polycation-Induced Cell Membrane Permeability Does Not Enhance Cellular Uptake or Expression Efficiency of Delivered DNA. Mol Pharmaceutics. 2010;7:870–883. doi: 10.1021/mp100027g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met. 1995;57:289–300. [Google Scholar]
- 34.Smyth GK. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat Appl Genet Mol Biol. 2004;3:1–25. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 35.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci US A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bandyopadhyay D, Gatza C, Donehower LA, Medrano EE. Current Protocols in Cell Biology; Vol. 18. John Wiley & Sons; Hoboken, NJ: 2005. Analysis of Cellular Senescence in Culture In Vivo: The Senescence-Associated β-Galactosidase Assay; pp. 18.9.1–18.9.9. [DOI] [PubMed] [Google Scholar]
- 37.Gyuris J, Golemis E, Chertkov H, Brent R. Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- 38.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–80. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 39.Brunner S, Furtbauer E, Sauer T, Kursa M, Wagner E. Overcoming the Nuclear Barrier: Cell Cycle Independent Nonviral Gene Transfer with Linear Polyethylenimine or Electroporation. Mol Ther. 2002;5:80–86. doi: 10.1006/mthe.2001.0509. [DOI] [PubMed] [Google Scholar]
- 40.Grandinetti G, Smith AE, Reineke TM. Membrane and Nuclear Permeabilization by Polymeric pDNA Vehicles: Efficient Method for Gene Delivery or Mechanism of Cytotoxicity? Mol Pharmaceutics. 2011;9:523–538. doi: 10.1021/mp200368p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Audouy SAL, de Leij LFMH, Hoekstra D, Molema G. In Vivo Characteristics of Cationic Liposomes as Delivery Vectors for Gene Therapy. Pharm Res. 2002;19:1599–1605. doi: 10.1023/a:1020989709019. [DOI] [PubMed] [Google Scholar]
- 42.Gribaudo G, Ravaglia S, Gaboli M, Gariglio M, Cavallo R, Landolfo S. Interferon-α Inhibits the Murine Cytomegalovirus Immediate-Early Gene Expression by Down-Regulating NF-κ B Activity. Virology. 1995;211:251–260. doi: 10.1006/viro.1995.1398. [DOI] [PubMed] [Google Scholar]
- 43.Harms JS, Splitter GA. Interferon-γ inhibits transgene expression driven by SV40 or CMV promoters but augments expression driven by the mammalian MHC I promoter. Hum Gene Ther. 1995;6:1291–1297. doi: 10.1089/hum.1995.6.10-1291. [DOI] [PubMed] [Google Scholar]
- 44.Qin L, Ding Y, Pahud DR, Chang E, Imperiale MJ, Bromberg JS. Promoter attenuation in gene therapy: Interferon-γ and tumor necrosis factor-α inhibit transgene expression. Hum Gene Ther. 1997;8:2019–2029. doi: 10.1089/hum.1997.8.17-2019. [DOI] [PubMed] [Google Scholar]
- 45.Ritter T, Brandt C, Prösch S, Vergopoulos A, Vogt K, Kolls J, Volk HD. Stimulatory and inhibitory action of cytokines on the regulation of hCMV-IE promoter activity in human endothelial cells. Cytokine. 2000;12:1163–1170. doi: 10.1006/cyto.2000.0689. [DOI] [PubMed] [Google Scholar]
- 46.Teschendorf C, Warrington KH, Siemann DW, Muzyczka N. Comparison of the EF-1 alpha and the CMV promoter for engineering stable tumor cell lines using recombinant adeno-associated virus. Anticancer Res. 2002;22:3325–30. [PubMed] [Google Scholar]
- 47.Kim JA, Aberg C, Salvati A, Dawson KA. Role of cell cycle on the cellular uptake and dilution of nanoparticles in a cell population. Nat Nanotechnol. 2012;7:62–68. doi: 10.1038/nnano.2011.191. [DOI] [PubMed] [Google Scholar]
- 48.Jiang W, Kim BYS, Rutka JT, Chan WCW. Nanoparticle-mediated cellular response is size-dependent. Nat Nanotechnol. 2008;3:145–150. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 49.Bringold F, Serrano M. Tumor suppressors and oncogenes in cellular senescence. Exp Gerontol. 2000;35:317–329. doi: 10.1016/s0531-5565(00)00083-8. [DOI] [PubMed] [Google Scholar]
- 50.Pantoja C, Serrano M. Murine fibroblasts lacking p21 undergo senescence and are resistant to transformation by oncogenic Ras. Oncogene. 1999;18:4974–4982. doi: 10.1038/sj.onc.1202880. [DOI] [PubMed] [Google Scholar]
- 51.Morgan DO. The cell cycle: principles of control; New Science Press; London: 2007. [Google Scholar]
- 52.Kong M, Barnes EA, Ollendorff V, Donoghue DJ. Cyclin F regulates the nuclear localization of cyclin B1 through a cyclin-cyclin interaction. EMBO J. 2000;19:1378–1388. doi: 10.1093/emboj/19.6.1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Y, Benezra R. Identification of a Human Mitotic Checkpoint Gene: hsMAD2. Science. 1996;274:246–248. doi: 10.1126/science.274.5285.246. [DOI] [PubMed] [Google Scholar]
- 54.Braem CV, Kas K, Meyen E, Debiec-Rychter M, Van de Ven WJM, Voz ML. Identification of a Karyopherin α2 Recognition Site in PLAG1, Which Functions As a Nuclear Localization Signal. J Biol Chem. 2002;277:19673–19678. doi: 10.1074/jbc.M112112200. [DOI] [PubMed] [Google Scholar]
- 55.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 56.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 57.Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to Cell Cycle Regulation by the Chk2 Protein Kinase. Science. 1998;282:1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- 58.Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/hCds1 functions as a DNA damage checkpoint in G1 by stabilizing p53. Genes Dev. 2000;14:278–288. [PMC free article] [PubMed] [Google Scholar]
- 59.Rosen DG, Wang L, Jain AN, Lu KH, Luo RZ, Yu Y, Liu J, Bast RC. Expression of the Tumor Suppressor Gene ARHI in Epithelial Ovarian Cancer Is Associated with Increased Expression of p21WAF1/CIP1 and Prolonged Progression-Free Survival. Clin Cancer Res. 2004;10:6559–6566. doi: 10.1158/1078-0432.CCR-04-0698. [DOI] [PubMed] [Google Scholar]
- 60.Kastan MB, Zhan Q, El-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 61.Zhan Q, Bae I, Kastan MB, Fornace AJ. The p53-dependent γ-Ray Response of GADD45. Cancer Res. 1994;54:2755–2760. [PubMed] [Google Scholar]
- 62.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 Protein in the Cellular Response to DNA Damage. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- 63.Kearsey JM, Coates PJ, Prescott AR, Warbrick E, Hall PA. Gadd45 is a nuclear cell cycle regulated protein which interacts with p21Cip1. Oncogene. 1995;11:1675–1683. [PubMed] [Google Scholar]
- 64.Zhan Q, Antinore MJ, Wang XW, Carrier F, Smith ML, Harris CC, Fornace AJ. Association with Cdc2 and inhibition of Cdc2/Cyclin B1 kinase activity by the p53-regulated protein Gadd45. Oncogene. 1999;18:2892–2900. doi: 10.1038/sj.onc.1202667. [DOI] [PubMed] [Google Scholar]
- 65.Zhao H, Jin S, Antinore MJ, Lung FDT, Fan F, Blanck P, Roller P, Fornace AJ, Zhan Q. The Central Region of Gadd45 Is Required for Its Interaction with p21/WAF1. Exp Cell Res. 2000;258:92–100. doi: 10.1006/excr.2000.4906. [DOI] [PubMed] [Google Scholar]
- 66.Sugimoto M, Nakamura T, Ohtani N, Hampson L, Hampson IN, Shimamoto A, Furuichi Y, Okumura K, Niwa S, Taya Y, Hara E. Regulation of CDK4 activity by a novel CDK4-binding protein, p34SEI-1. Genes Dev. 1999;13:3027–3033. doi: 10.1101/gad.13.22.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li J, Melvin WS, Tsai MD, Muscarella P. The Nuclear Protein p34SEI-1 Regulates the Kinase Activity of Cyclin-Dependent Kinase 4 in a Concentration-Dependent Manner. Biochemistry. 2004;43:4394–4399. doi: 10.1021/bi035601s. [DOI] [PubMed] [Google Scholar]
- 68.Kroismayr R, Baranyi U, Stehlik C, Dorfleutner A, Binder BR, Lipp J. HERC5, a HECT E3 ubiquitin ligase tightly regulated in LPS activated endothelial cells. J Cell Sci. 2004;117:4749–4756. doi: 10.1242/jcs.01338. [DOI] [PubMed] [Google Scholar]
- 69.Kawase A, Isaji K, Yamaoka A, Kobayashi N, Nishikawa M, Takakura Y. Enhanced antigen-specific antibody production following polyplex-based DNA vaccination via the intradermal route in mice. Vaccine. 2006;24:5535–5545. doi: 10.1016/j.vaccine.2006.04.056. [DOI] [PubMed] [Google Scholar]
- 70.Woods MW, Kelly JN, Hattlmann CJ, Tong JGK, Xu LS, Coleman MD, Quest GR, Smiley JR, Barr SD. Human HERC5 restricts an early state of HIV-1 assembly by a mechanism correlating with the ISGylation of Gag. Retrovirology. 2011;8:1–17. doi: 10.1186/1742-4690-8-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gao J, Wang HL, Shreve A, Iyer R. Fullerene derivatives induce premature senescence: A new toxicity paradigm or novel biomedical applications. Toxicol Appl Pharmacol. 2010;244:130–143. doi: 10.1016/j.taap.2009.12.025. [DOI] [PubMed] [Google Scholar]
- 72.Mitsui K, Nakanishi M, Ohtsuka S, Norwood TH, Okabayashi K, Miyamoto C, Tanaka K, Yoshimura A, Ohtsubo M. A Novel Human Gene Encoding HECT Domain and RCC1-like Repeats Interacts with Cyclins and Is Potentially Regulated by the Tumor Suppressor Proteins. Biochem Biophys Res Commun. 1999;266:115–122. doi: 10.1006/bbrc.1999.1777. [DOI] [PubMed] [Google Scholar]
- 73.Dastur A, Beaudenon S, Kelley M, Krug RM, Huibregtse JM. Herc5, an Interferon-induced HECT E3 Enzyme, Is Required for Conjugation of ISG15 in Human Cells. J Biol Chem. 2006;281:4334–4338. doi: 10.1074/jbc.M512830200. [DOI] [PubMed] [Google Scholar]
- 74.Shi HX, Yang K, Liu X, Liu XY, Wei B, Shan YF, Zhu LH, Wang C. Positive Regulation of Interferon Regulatory Factor 3 Activation by Herc5 via ISG15 Modification. Mol Cell Biol. 2010;30:2424–2436. doi: 10.1128/MCB.01466-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Collins SE, Noyce RS, Mossman KL. Innate Cellular Response to Virus Particle Entry Requires IRF3 but Not Virus Replication. J Virol. 2004;78:1706–1717. doi: 10.1128/JVI.78.4.1706-1717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jeong GJ, Byun HM, Kim JM, Yoon H, Choi HG, Kim WK, Kim SJ, Oh YK. Biodistribution and tissue expression kinetics of plasmid DNA complexed with polyethylenimines of different molecular weight and structure. J Controlled Release. 2007;118:118–125. doi: 10.1016/j.jconrel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 77.Hornung V, Latz E. Intracellular DNA recognition. Nat Rev Immunol. 2010;10:123–130. doi: 10.1038/nri2690. [DOI] [PubMed] [Google Scholar]
- 78.Park JH, Chang SH, Kim MC, Shin SH, Youn HJ, Kim JK, Jang YS, Kim CW. Up-regulation of the expression of major histocompatibility complex class I antigens by plasmid DNA transfection in non-hematopoietic cells. FEBS Lett. 1998;436:55–60. doi: 10.1016/s0014-5793(98)01097-7. [DOI] [PubMed] [Google Scholar]
- 79.Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, Ludwig H, Sutter G, Suzuki K, Hemmi H, Sato S, Yamamoto M, Uematsu S, Kawai T, Takeuchi O, Akira S. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol. 2006;7:40–48. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- 80.Stetson DB, Medzhitov R. Recognition of Cytosolic DNA Activates an IRF3-Dependent Innate Immune Response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 81.Thomas M, Klibanov AM. Non-viral gene therapy: polycation-mediated DNA delivery. Appl Microbiol Biotechnol. 2003;62:27–34. doi: 10.1007/s00253-003-1321-8. [DOI] [PubMed] [Google Scholar]
- 82.Lechardeur D, Verkman AS, Lukacs GL. Intracellular routing of plasmid DNA during non-viral gene transfer. Adv Drug Delivery Rev. 2005;57:755–767. doi: 10.1016/j.addr.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 83.Medina-Kauwe LK, Xie J, Hamm-Alvarez S. Intracellular trafficking of nonviral vectors. Gene Ther. 2005;12:1734–1751. doi: 10.1038/sj.gt.3302592. [DOI] [PubMed] [Google Scholar]
- 84.Nabel GJ, Nabel EG, Yang ZY, Fox BA, Plautz GE, Gao X, Huang L, Shu S, Gordon D, Chang AE. Direct gene transfer with DNA-liposome complexes in melanoma: expression, biologic activity, and lack of toxicity in humans. Proc Natl Acad Sci US A. 1993;90:11307–11311. doi: 10.1073/pnas.90.23.11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Khalil IA, Kogure K, Akita H, Harashima H. Uptake Pathways and Subsequent Intracellular Trafficking in Nonviral Gene Delivery. Pharmacol Rev. 2006;58:32–45. doi: 10.1124/pr.58.1.8. [DOI] [PubMed] [Google Scholar]
- 86.Glover DJ, Lipps HJ, Jans DA. Towards safe, non-viral therapeutic gene expression in humans. Nat Rev Genet. 2005;6:299–310. doi: 10.1038/nrg1577. [DOI] [PubMed] [Google Scholar]
- 87.Hsu J, Muro S. Nanomedicine and Drug Delivery Strategies for Treatment of Genetic Diseases. In: Plaseska-Karanfilska D, editor. Genetic Disease; InTech; Rijeka, Croatia: 2011. pp. 241–266. [Google Scholar]
- 88.Moon JJ, Huang B, Irvine DJ. Engineering Nano- and Microparticles to Tune Immunity. Adv Mater. 2012;24:3724–3746. doi: 10.1002/adma.201200446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Godbey WT, Wu KK, Mikos AG. Tracking the intracellular path of poly(ethylenimine)/DNA complexes for gene delivery. Proc Natl Acad Sci US A. 1999;96:5177–5181. doi: 10.1073/pnas.96.9.5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Godbey WT, Barry MA, Saggau P, Wu KK, Mikos AG. Poly(ethylenimine)-mediated transfection: A new paradigm for gene delivery. J Biomed Mater Res. 2000;51:321–328. doi: 10.1002/1097-4636(20000905)51:3<321::aid-jbm5>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 91.Bieber T, Meissner W, Kostin S, Niemann A, Elsasser HP. Intracellular route and transcriptional competence of polyethylenimine-DNA complexes. J Controlled Release. 2002;82:441–454. doi: 10.1016/s0168-3659(02)00129-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.