

Abstract
Receptor-mediated endocytosis, involving megalin and cubilin, mediates renal proximal-tubular reabsorption and is decreased in Dent disease because of mutations of the chloride/proton antiporter, chloride channel-5 (CLC-5), resulting in low-molecular-weight proteinuria, hypercalciuria, nephrolithiasis, and renal failure. To facilitate studies of receptor-mediated endocytosis and the role of CLC-5, we established conditionally immortalized proximal-tubular epithelial cell lines (ciPTECs) from three patients with CLC-5 mutations (30:insH, R637X, and del132-241) and a normal male. Confocal microscopy using the tight junction marker zona occludens-1 (ZO-1) and end-binding protein-1 (EB-1), which is specific for the plus end of microtubules demonstrated that the ciPTECs polarized. Receptor-mediated endocytic uptake of fluorescent albumin and transferrin in 30:insH and R637X ciPTECs was significantly decreased, compared with normal ciPTECs, and could be further reduced by competition with 10-fold excess of unlabeled albumin and transferrin, whereas in the del132-241 ciPTEC, receptor-mediated endocytic uptake was abolished. Investigation of endosomal acidification by live-cell imaging of pHluorin-VAMP2 (vesicle-associated membrane protein-2), a pH-sensitive-GFP construct, revealed that the endosomal pH in normal and 30:insH ciPTECs was similar, whereas in del132-241 and R637X ciPTECs, it was significantly more alkaline, indicating defective acidification in these ciPTECs. The addition of bafilomycin-A1, a V-ATPase inhibitor, raised the pH significantly in all ciPTECs, demonstrating that the differences in acidification were not due to alterations in the V-ATPase, but instead to abnormalities of CLC-5. Thus, our studies, which have established human Dent disease ciPTECs that will facilitate studies of mechanisms in renal reabsorption, demonstrate that Dent disease-causing CLC-5 mutations have differing effects on endosomal acidification and receptor-mediated endocytosis that may not be coupled.
Renal proximal tubular epithelial cells (PTECs) are of central importance in facilitating receptor-mediated endocytosis of >80% of plasma proteins, such as albumin, and low-molecular-weight proteins (LMWPs) that are present in the glomerular filtrate (1). These LMWPs include vitamin-binding proteins and hormones (e.g., insulin and parathyroid hormone), which are important for maintaining extracellular fluid homeostasis, as well as vitamin and hormonal metabolism (2). PTEC receptor-mediated endocytosis uses the multiligand receptors, megalin and cubilin, which are located on the apical brush-border membrane, where they interact and function as coreceptors (3). Ligand binding by the coreceptors results in internalization of the receptor–ligand complex into clathrin-coated vesicles, which are anterogradely transported along microtubules to fuse with early endosomes and, thereby, enter the endosomal-lysosomal pathway for further processing, recycling, and degradation of the proteins (3–5). Progress through this pathway requires endosomal luminal acidification that facilitates ligand-receptor dissociation, ligand processing, receptor recycling or degradation, vesicular trafficking, and fusion to late endosomes and lysosomes (5). In PTECs, the endosomal acidification is provided by the electrogenic vacuolar H+ATPase (V-ATPase) and the countercurrent to maintain electroneutrality has been reported to be likely provided by the chloride channel-5, CLC-5, a chloride/proton exchanger (GenBank accession no. NM_001127899.1) (6) (Fig. 1A), which is codistributed with V-ATPase in renal endosomes (7, 8). Moreover, loss-of-function mutations of CLC-5, encoded by the CLCN5 gene located on chromosome Xp11.22, result in Dent disease (Online Mendelian Inheritance in Man [OMIM]:300009) (9), a renal tubular disorder characterized by: low-molecular-weight proteinuria; hypercalciuria with nephrolithiasis and renal failure; and hyperphosphaturia that may lead to hypophosphataemic rickets (2) (Table 1). These clinical features have been attributed to impaired proximal tubular endocytosis, and CLC-5–deficient male mice (Clcn5y/−), which develop some of these manifestations of Dent disease (Table 1), have in vivo defects of proximal tubular endocytosis (10, 11), and in vitro studies have demonstrated Clcn5y/− renal endosomes to have decreased ATP-dependent vesicular acidification (2). Furthermore, CLC-5 deficiency was associated with a generalized trafficking defect in mouse proximal tubules, which had a loss of megalin and cubilin at the brush-border membrane (12) and an impairment of lysosomal formation (13).
Fig. 1.

Establishing conditionally immortalized proximal tubule epithelial cell lines from the urine of patients with Dent disease. (A) Schematic representation of the chloride/proton exchanger, CLC-5, a 746-aa protein that consists of 18 α-helices (A to R) with the NH2 and COOH terminus domains being cytoplasmic (membrane boundaries indicated by broken lines). The C-terminal domain contains two CBS domains. The locations of three mutations identified in male patients with Dent disease (Table 1), from whom ciPTECs were established, 30:insH, del132-241 [predicted to result in a loss of helices C-F and a portion of helix G (indicated in black)], and R637X, predicted to result in a loss of CBS1 and the whole of CBS2 are indicated (arrows). (B) Flow cytometry analysis of ciPTECs from a normal male (control, wild-type) and three Dent disease patients, using a FITC-labeled antibody against CD13 (22). Readings were initially made in unstained cells (Left), and then in CD13-FITC–treated cells (Center). Forward scatter was plotted (on y axis) against the CD13-FITC count (on x axis). The readings from the unstained (red peak) and CD13-FITC stained cells (blue peak) are representative for each ciPTEC (Right). (C) RT-PCR analysis of genes expressed in the renal proximal tubule (ABCC4, ABCB1), loop of Henle (NKCC2 and KCNJ1), distal tubule (CALB1 and TRPV5), and bladder (UPKIIIA). RNA extracted from the four ciPTECs (three Dent disease patients and one control, wild-type) was used. RNA from a normal kidney and a human bladder cell line, and a water blank were used as positive and negative controls, respectively. Calmodulin (CaM) expression was used as a loading control. (D) Western blot analysis of proteins expressed in the renal proximal tubule (AQP1), distal tubule (TRPV5), collecting duct (AQP2), and bladder (UPKIIIA). Cell lysates from each of the four ciPTECs were used. Cell lysates from a normal human kidney and a bladder cell line were used as positive controls, and α-tubulin was used as a loading control. (E) Expression of some genes that encode components of the proximal tubular receptor-mediated endocytic pathway. RT-PCR analysis of the expression of cubilin (CUBN), low-density lipoprotein related protein 2 (LRP2), kinesin family member 3B (KIF3B) and V-ATPase subunits D (ATP6V1D) and A2 (ATP6V0A2) genes using RNA extracted from the four ciPTECs. RNA from a normal human kidney and a water blank was used as a positive control and negative control, respectively. CaM expression was used as a loading control. (F) Confocal microscopy of the four ciPTECs stained with anti–ZO-1 (green) and anti–EB-1 (red). The presence of tight junctions (green) at the apical surface with EB-1–labeled microtubules below (red) demonstrates that the ciPTECs are polarized. (F Upper) XY image. (F Lower) Orthogonal view. (Scale bars: 10 μm.)
Table 1.
Comparison of clinical and cellular phenotypes and mouse models with CLC-5 mutations
| CLC-5 mutation* |
CLC-5 transport† |
Dent disease phenotype‡ |
PTEC phenotype§ |
|||||||||||
| Origin of Dent Disease | Type | Location | Cl− | H+ | Pr | Ca | Nc | Rf | Ri | RME | MGi | FPE | EAc | |
| Patient¶ | Normal | — | — | ++ | ++ | − | − | − | − | − | ++ | ++ | ++ | ++ |
| 1 | 30:insH | A | + | ++ | + | + | − | − | − | + | + | ++ | ++ | |
| 2 | del132-241 | C-G | (−) | (−) | + | + | + | + | + | − | − | + | + | |
| 3 | R637X | CBS1 | − | − | + | + | + | − | ? | + | + | ++ | + | |
| Mouse model | A | KO | E | (−) | (−) | + | − | − | ? | ↑ UPi | + | − | + | + |
| B | KO | C-F | (−) | (−) | + | + | + | ? | ↑ALP | + | + | + | ? | |
| C | KD | L-M loop | ? | ? | ? | + | ? | ? | ? | ? | ? | ? | ? | |
| D | KI-E211A | F | ++ | − | + | + | ? | ? | ? | + | + | + | ++ | |
CLC-5 mutation details and location provided with reference to helix or CBS domain (Fig. 1A).
CLC-5 transport activity assessed by heterologous expression of CLC-5s in HEK293 cells. Cl− conductance and H+ efflux were measured. ++, normal, +, impaired; −, absent; (−) likely absent, but not measured; ?, not known.
ALP, alkaline phosphatase; Ca, hypercalciuria; Nc, nephrocalcinosis; Pr, low-molecular-weight proteinuria; Rf, renal failure; Ri, rickets; glycosuria and phosphaturia were not reported in any of the patients; UPi, hyperphosphaturia; +, present; −, absent; ?, not known.
EAc, endosomal acidification; FPE, fluid-phase endocytosis; MGi, megalin internalization; RME, receptor-mediated endocytosis. ++, normal, +, impaired; −, absent; ?, not known.
The role of CLC-5 in Cl− conductance and endosomal acidification has been investigated in vitro by expression of wild-type and mutant CLC-5s in Xenopus oocytes (9, 14) and human embryonic kidney (HEK) cells (15), and in vivo by generating mice that harbor a CLC-5 mutation (E211A) that converts the CLC-5 (chloride/proton exchanger) into a pure Cl− conductor (16). Thus, in vitro studies have demonstrated that Dent disease causing CLC-5 mutants markedly reduce or abolish Cl− conductance and that some, but not all, CLC-5 mutants may also impair endosomal acidification (17). Thus, the in vitro expression studies of Dent disease causing CLC-5 mutants in HEK cells have shown that: CLC-5 mutants S270R, located in the H-I loop, G513E and R516W (in helix O), I524K and E527D (in helix P), and R637X [in cystathionine-β-synthase (CBS) domain-1 in the C terminus] resulted in a lack of Cl− currents, largely due to retention and degradation of the mutant CLC-5s in the endoplasmic reticulum (ER); whereas CLC-5 mutants consisting of a His insertion at codon 30 (30:insH), G57V, and R280P (in helix A, B, and I, respectively), had markedly reduced Cl− currents but did not impair endosomal acidification (Fig. 1A, Fig. S1, and Table 1) (17). Thus, some Dent disease causing CLC-5 mutations may not impair endosomal acidification, while resulting in defective proximal tubular endocytosis, suggesting endocytosis and endosomal trafficking may not be coupled to endosomal acidification. Indeed, recent in vivo studies of a mouse harboring the CLC-5 mutant E211A, in helix F (Table 1), have demonstrated that proximal tubular endocytosis and endosomal acidification is uncoupled. The E211A CLC-5 mutant, which has not been identified in a patient with Dent disease, alters the “gating” glutamate of CLC-5 chloride/proton exchanger to generate an uncoupled Cl− conductor. The E211A CLC-5 mutant was found neither to alter Cl− currents nor to impair endosomal acidification, but resulted in defects of receptor-mediated and fluid-phase endocytosis (16). Thus, these findings demonstrate that the impairment of endocytosis observed in patients with Dent disease due to CLC-5 mutations may not result from reduced endosomal acidification.
Further investigation of the effects of Dent disease causing CLC-5 mutations on the relationship between endocytosis and endosomal acidification is hampered because CLC-5 is predominantly expressed in renal epithelia, and such cells from patients with Dent disease are not available. However, PTECs may be isolated from fresh urine samples (18, 19), and to facilitate investigations of the effects of CLC-5 mutations on endocytosis and endosomal acidification, we embarked on studies to establish human conditionally immortalized PTECs (ciPTECs) from patients with Dent disease due to CLC-5 mutations (Table 1), with the aim of using these ciPTECs to further study the effects of Dent disease causing CLC-5 mutants on endocytosis and endosomal acidification.
Results
Establishment of Dent Disease ciPTECs.
Three males with known CLCN5 mutations, comprising 30:insH, deletion of codons 132–241 (del132-241) and R637X (Table 1) that affected different functional domains of CLC-5 (Fig. 1A) were identified (9, 14, 20, 21), first-void morning urine samples were obtained, and primary cultures were established (19). After conditional immortalization by infecting with SV40-T and human telomerase reverse transcriptase (hTERT), the presence of the CLCN5 mutation was confirmed by DNA sequence analysis of each cell line and in matched leukocyte DNA samples from each patient. Cultures of immortalized urinary cells may contain different types of urinary tract cells, including proximal tubular cells (18). To determine the content of proximal tubular cells in these cultures, flow cytometry using fluorescein-isothiocyanate (FITC)-labeled antibody against cluster of differentiation (CD)13, which is aminopeptidase-N and an established marker for the proximal tubule brush border (22), was performed. This analysis revealed that these cultures contained either <40% or >95% of CD13-FITC stained cells (Fig. S2). To obtain a homogenous population of >95% proximal tubular cells, fluorescence-activated cell sorting (FACS) was performed to isolate CD13-positive cells, which were then cloned and reassessed by flow cytometry (Fig. 1B). Comparison of cells stained for CD13-FITC with unstained (control) cells from the same patient (Fig. 1B) detected CD13-FITC in >95% of the cells, which was similar to that observed in previously established ciPTECs from a normal male (Fig. 1B) (19). Thus, ciPTECs from three patients with Dent disease were established.
Characterization of the ciPTECs from the patients with Dent disease and a normal (wild-type) male by RT-PCR using extracted total RNA showed the following: the presence of the proximal tubular expressed genes for ATP-binding cassette family members multidrug resistance protein 4 (ABCC4) and P-glycoprotein (ABCB1), and an absence of genes that are expressed in: the thick ascending limb, Na+-K+-Cl− cotransporter (NKCC2) and K+ inwardly-rectifying channel, subfamily J, member 1 (KCNJ1); distal tubule, Calbindin-1 (CALB1) and Transient receptor potential cation channel, subfamily V, member 5 (TRPV5); and bladder, Uroplakin IIIA (UPKIIIA). The appropriate renal or bladder expression of these genes was shown in total RNA from human kidney and bladder, respectively (Fig. 1C). Similarly, Western blot analysis revealed that the ciPTECs expressed the proximal tubular water channel, aquaporin-1 (AQP1), but not the collecting duct specific water channel, aquaporin-2 (AQP2), the distal tubular TRPV5, or the bladder UPKIIIA (Fig. 1D). Thus, the ciPTECs from the patients with Dent disease and the normal male were demonstrated only to express markers of proximal tubule cells. A critical function of PTECs is reabsorption of solutes from the glomerular filtrate, by the receptor-mediated endocytic pathway, whose components include the multiligand receptors megalin, also referred to as LRP2, and cubilin (CUBN) and intracellular facilitators such as kinesin family member 3B (KIF3B), and V-ATPase subunits A2 and D (ATP6V0A2 and ATP6V1D). RT-PCR using total RNA obtained from the ciPTECs revealed expression of these receptor-mediated endocytic pathway components (Fig. 1E) as well as CLCN5 (Table S1), thereby indicating that the ciPTECs may be able to reabsorb molecules such as albumin and transferrin by endocytosis. However, Western blot analysis revealed that CLC-5 was markedly degraded, absent, or truncated in the mutant 30:insH, del132-241, and R637X ciPTECs, respectively (Fig. S3).
Polarization of ciPTECs.
Proximal tubular cells, which are polarized, facilitate endocytosis of proteins at the apical brush border membrane. The apical endocytosed proteins are then transferred, which is most probably mediated by anterograde microtubule transport, to lysosomes for degradation (3–5). The anterograde transport is facilitated by orientation of the microtubules such that their minus ends are toward the apical side and their plus ends, which have the microtubule associated protein end-binding protein-1 (EB-1), are extending through the cell to the basolateral side (4, 23). Confocal microscopy and analysis of orthogonal sections from Z-stack images obtained of the four ciPTECs stained with antibodies to zona occludens-1 (ZO-1) and EB-1 showed the presence of ZO-1 containing tight junctions (green) at the apical side, and the location of EB-1–labeled microtubules (red) below the tight junctions (Fig. 1F), thereby demonstrating that the growing ends of microtubules are predominantly toward the basolateral surface of the polarized ciPTECs. Thus, these results reveal that all four ciPTECs form polarized cells (Fig. 1F), consistent with their derivation from proximal tubular cells (Fig. 1 B–E).
Dent Disease ciPTECs Have Impaired Uptake of Albumin and Transferrin.
Proximal tubular cells reabsorb >80% of the filtered protein load, including albumin and transferrin, by receptor-mediated endocytosis that involves the megalin–cubilin receptor complex and is impaired in patients with Dent disease (24). Albumin is a ligand for both megalin and cubilin, whereas transferrin is a ligand for cubilin only. We therefore investigated the ability of the polarized ciPTECs (Fig. 1F) that express megalin and cubilin (Fig. 1E) to absorb albumin and transferrin. All of the Dent disease ciPTECs, compared with the wild-type ciPTECs, were found to have significant reductions in albumin uptake (Fig. 2A); thus, the 30:insH, del132-241, and R637X ciPTECs had mean ± SEM fluorescent albumin uptakes of 57.0 ± 4.6%, 23.3 ± 4.0%, and 64.9 ± 6.1%, respectively, compared with the wild-type (control) ciPTECs (100.0 ± 8%, n = 10, P < 0.02). However, the Dent disease ciPTECs showed different responses in the uptake of fluorescently labeled albumin when challenged with competition by the presence of excess unlabeled albumin or transferrin in the medium. Thus, in control ciPTECs, fluorescent albumin uptake in the presence of excess unlabeled albumin or transferrin was significantly reduced to 38.4 ± 2.6%, or 43.7 ± 4.1%, compared with the uptake with no competition, respectively (n = 6, P < 0.02), and the 30:insH and R637X ciPTECs also showed significantly reduced albumin uptake in the presence of excess unlabeled albumin (30:insH: 13.1 ± 2.1%, P < 0.02, and R637X: 37.6 ± 5.8%, P < 0.05, n = 6) or excess unlabeled transferrin (30:insH: 17.2 ± 4.2%, P < 0.02 and R637X: 39.6 ± 4.4%, P < 0.02, n = 6). In contrast, the del132-241 ciPTECs did not demonstrate a further reduction in fluorescent albumin uptake (31.3 ± 3.2% and 27.4 ± 5.2%, in the presence of excess unlabeled albumin or transferrin, respectively). These different responses in albumin uptake correlated with differences in cell surface expression and internalization of megalin by the ciPTECs (Fig. 2B). Thus, in control ciPTECs, megalin was predominantly expressed at the cell surface at 2 min after exposure to albumin, but at 5 min, internalized megalin was observed and, at 15 min, most of the megalin was in the cytoplasm. However, in the mutant 30:insH and R637X ciPTECs, which initially had similar megalin cell surface expression to control ciPTECs, megalin internalization at 15 min was reduced and in the del132-241 ciPTECs, megalin was located predominantly in the cytoplasm at all time points (Fig. 2B). Thus, these data indicate that the CLC-5 mutation in del132-241 results in a complete disruption of receptor-mediated endocytosis, whereas the 30:insH and R637X cause a partial loss of receptor-mediated endocytosis.
Fig. 2.
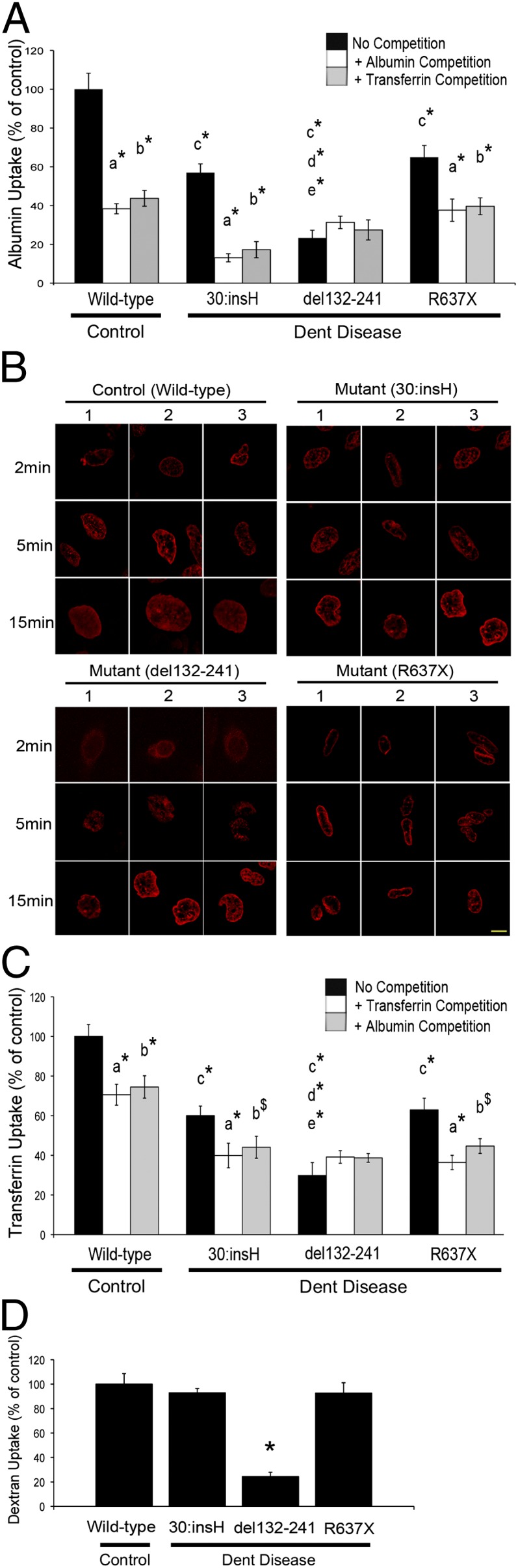
Endocytosis of albumin, transferrin and dextran, and megalin internalization in control and Dent disease ciPTECs. Albumin, transferrin, and dextran uptake by the three ciPTECs from the Dent disease patients and the ciPTEC from a control normal male (wild-type) were measured, and expressed relative to that of the control ciPTEC. (A) Albumin uptake was measured in the absence (no competition, black bars), or presence of excess unlabeled albumin (albumin competition, white bars) or excess unlabeled transferrin (transferrin competition, gray bars). Mean values ± SEM (n = 10) are shown. P values (*P < 0.02) are for: a, no competition vs. albumin competition; b, no competition vs. transferrin competition; c, control ciPTEC vs. 30:insH, del132-241, or R637X ciPTEC; d, 30:insH ciPTEC vs. del132-241 ciPTEC; and e, del132-241 ciPTEC vs. R637X ciPTEC. (B) Megalin internalization in control and Dent disease ciPTECs following exposure to unlabeled albumin for 2, 5, and 15 min. The ciPTECs were immunostained with an antibody against megalin (red), and three representative confocal microscopy images from different cells (labeled 1–3) are shown. (Scale bars: 10 μm.) (C) Transferrin uptake was measured in the absence (no competition, black bars) or presence of excess unlabeled transferrin (transferrin competition, white bars), or in the presence of excess unlabeled albumin (albumin competition, gray bars). Mean values ± SEM (n = 6) are shown. P values ($P < 0.05 and *P < 0.02) are for: a, no competition vs. transferrin competition; b, no competition vs. albumin competition; c, control ciPTEC vs. 30:insH, del132-241, or R637X ciPTEC; d, 30:insH ciPTEC vs. del132-241 ciPTEC; and e, del132-241 ciPTEC vs. R637X ciPTEC. (D) Dextran uptake is shown as mean values ± SEM (n = 44), *P < 0.02. All P values were calculated by Student’s unpaired, two-tailed t test.
The studies of fluorescently labeled transferrin uptake revealed similar abnormalities (Fig. 2C) to those observed for fluorescent-labeled albumin uptake (Fig. 2A). Thus, all of the Dent disease ciPTECs, compared with the wild-type ciPTECs, were found to have significant reductions in transferrin uptake (Fig. 2C); the 30:insH, del132-241, and R637X ciPTECs had mean ± SEM fluorescent transferrin uptake of 60.1 ± 4.8%, 29.8 ± 6.5%, and 62.9 ± 5.9%, respectively, compared with the control ciPTECs (100.0 ± 6%) (n = 8, P < 0.02). However, the Dent disease ciPTECs showed differences in uptake in response to competition with excess unlabeled albumin or transferrin. Thus, in control ciPTECs in the presence of excess unlabeled albumin or transferrin, fluorescent transferrin uptake was significantly reduced to 70.5 ± 5.3% or 60.0 ± 4.8%, respectively, compared with uptake without competition (n = 6, P < 0.02); the 30:insH and R637X ciPTECs also showed significantly reduced transferrin uptake in the presence of excess unlabeled albumin (30:insH: 44.1 ± 5.5%, P < 0.02 and R637X: 44.7 ± 3.7%, P < 0.05, n = 6) or excess unlabeled transferrin (30:insH: 39.9 ± 6.2%, P < 0.02 and R637X: 36.4 ± 3.6%, P < 0.02, n = 6). In contrast, competition with excess unlabeled albumin or transferrin had no effect on fluorescent transferrin uptake in del132-241 cells (38.7 ± 2.1% and 39.1 ± 3.2%, respectively, n = 6) similar to the effects observed with fluorescent albumin.
The combined results demonstrate that the ciPTECs with the del132-241 mutation had a complete disruption of receptor-mediated endocytosis, whereas the ciPTECs with the 30:insH and R637X mutations had active but reduced receptor-mediated endocytosis, compared with that in normal ciPTECs. Investigation of fluid-phase endocytosis using FITC-labeled dextran revealed that the del132-241 ciPTECs, but not the 30:insH and R637X ciPTECs, had significantly reduced dextran uptake compared with control ciPTECs (24.4 ± 1%, n = 44, P < 0.02) (Fig. 2D). The addition of excess unlabeled albumin during the FITC-labeled dextran uptake assays did not affect dextran uptake by the control or Dent disease ciPTECs, thereby confirming that dextran uptake was not via the receptor-mediated endocytic pathway involving the megalin–cubilin complex.
Endosomal Acidification Defects in Dent Disease ciPTECs.
CLC-5 is postulated to have a role in acidification of the endosome by providing a parallel chloride conductance to the V-ATPase (5). Previous studies using heterologous expression systems have shown that CLC-5 mutations have different effects on endosomal acidification (17). Expression of wild-type CLC-5 and CLC-5 mutants 30:insH and R637X in HEK293 cells revealed that proton transport by the CLC-5 mutant R637X was significantly decreased compared with wild-type CLC-5, whereas that by the CLC-5 mutant 30:insH was similar to wild-type CLC-5 (Fig. S4). To investigate whether this proton transport affected endosomal acidification in the Dent disease ciPTECs, live-cell imaging using the ratiometric GFP variant pHluorin, expressing a VAMP2 tag to target it to endosomes (17), was performed (Fig. 3A). The mean endosomal pH in control ciPTECs was 5.69 ± 0.23. In 30:insH cells, endosomal pH was not significantly different from that recorded in control ciPTECs (5.43 ± 0.22). Endosomal pH in del132-241 and R637X ciPTECs was significantly increased to 6.36 ± 0.19 and 6.63 ± 0.27, respectively (P < 0.05) (Fig. 3B), thereby demonstrating defective endosomal acidification. The addition of bafilomycin-A1, an inhibitor of the V-ATPase (15), raised the pH significantly in all ciPTECs, demonstrating that the differences in acidification were likely due to differences between wild-type and mutant CLC-5, and not changes in V-ATPase. Thus, the combined results of these studies show that the CLC-5 mutant 30:insH is associated with reduced Cl− conductance but normal H+ efflux and normal endosomal acidification, compared with wild-type CLC-5; and the del132-241 and R637X CLC-5 mutants result in loss of Cl− conductance and H+ efflux with impaired endosomal CLC-5 acidification (Table 1, Figs. 2 and 3, and Figs. S1 and S4).
Fig. 3.

Endosomal acidification in ciPTECs. Endosomal pH was determined by using the ratiometric GFP variant pHluorin-VAMP2, by comparison with a pH standard curve constructed using surface-exposed GPI-tagged pHluorin in buffers with different pHs. (A) Live-cell imaging using confocal microscopy of the four ciPTECs [three from Dent disease patients and one normal control (wild-type) male]. Images with 488 nm and 760 nm excitation were collected, the fluorescence intensity of individual vesicles was measured, and the ratios of 760 nm/488 nm were calculated. (Scale bars: 10 μm.) (B) Endosomal pH values were determined for each of the four ciPTECs that were either untreated (black bars) or treated with bafilomycin-A1 (white bars). Data are expressed as mean ± SEM for untreated Dent disease ciPTECs versus control ciPTEC ($P < 0.05 and #P < 0.02); and for ciPTECs treated with bafilomycin versus the respective untreated ciPTECs (*P < 0.02). P values were calculated by Student’s unpaired, two-tailed t test. The number of vesicles and cells from which recordings were collected are indicated below each bar. Horizontal dashed line at pH 7 represents the pH of the extracellular solution.
Discussion
Our studies, which establish human renal proximal tubular cells that harbor endogenous CLC-5 mutations, provide human cell models for Dent disease, a disorder manifesting with defects in receptor-mediated endocytosis and endosomal acidification. Thus, these ciPTECs provide valuable resources for the investigation of the receptor-mediated endocytic pathway, the mechanisms and roles of CLC-5 in endosomal acidification, and the assessment of compounds that may be of potential benefit in treating Dent disease and other human renal proximal tubular disorders due to defects in receptor-mediated endocytosis and endosomal acidification. Such applications are illustrated by use of ciPTECs derived from patients with nephropathic cystinosis (OMIM:219800) due to mutations in the lysosomal transporter cystinosin (25) for studies of cell metabolism in the disease (25).
Our investigations of the three human ciPTECs from Dent disease patients who had CLC-5 mutations involving different domains (Fig. 1) reveal that each CLC-5 mutation resulted in a different cellular phenotype (Table 1 and Figs. 2 and 3). Thus, the R637X ciPTECs had impaired receptor-mediated endocytosis, defective endosomal acidification, but normal fluid-phase endocytosis; the 30:insH ciPTECs had impaired receptor-mediated endocytosis, but normal endosomal acidification and normal fluid-phase endocytosis; whereas the del132-241 ciPTECs had a lack of receptor-mediated endocytosis, decreased fluid-phase endocytosis, and defective endosomal acidification. These findings indicate a correlation between the CLCN5 genotype and cellular phenotype, which is consistent with the findings in mouse models with different Clcn5 mutations (Table 1), in which detailed studies have revealed the presence of genotype-phenotype correlations (10, 11, 16, 26). For example, mice deleted for CLC-5 helices C-F were reported to have defective receptor-mediated and fluid-phase endocytosis and impaired endosomal acidification (10) (Table 1), and these defects resemble the defects observed in del132-241 ciPTECs, which would have deletion of helices C-G; whereas Clcn5y/211A mice had defects in receptor-mediated and fluid-phase endocytosis without impairment of endosomal acidification (16) (Table 1), resembling the phenotypic defects observed in the 30:insH ciPTECs. Our observations in the ciPTECs of a possible genotype-cellular phenotype correlation contrasts to the reported lack of a genotype-phenotype correlation in Dent disease patients (2, 27). However, the phenotypic features reported in studies of Dent disease patients, which are usually retrospective and from multiple centers, require cautious interpretation, because the data: could not be collected using uniformly standard conditions but instead was ascertained at different ages and stages of the disease; often lacked detailed assessment of receptor-mediated endocytosis; and could not have undertaken measurements of fluid-phase endocytosis or endosomal acidification. However, our results from these ciPTECs indicate that a detailed and prospective study of Dent disease patients examining for such genotype-phenotype correlation is required. In addition, these ciPTECs will be valuable for studying the mechanisms whereby CLC-5 mutations cause Dent disease as well as for assessing therapeutic compounds, which are not available for Dent disease.
Our investigations of the ciPTECs with CLC-5 mutations indicate that receptor-mediated endocytosis is not always coupled to endosomal acidification, which is consistent with the observations of in vitro studies that have expressed the CLC-5 mutants—G57V, S270R, G513E, R516W, I524K, and E527D—in HEK cells (17), and in vivo studies of Clcn5y/211A mice (16). An examination of the locations of these CLC-5 mutants and their effects on receptor-mediated endocytosis and endosomal acidification helps to provide insights into the structure–function relationships of the CLC-5 domains. Thus, the G57V mutant was reported to be associated with reduced Cl− currents without a defect in endosomal acidification, whereas the E527D mutant was associated with abolition of Cl− currents and impairment of endosomal acidification, and the mutants S270R, G513E, R516W, and I524K were retained in the ER and degraded and, hence, with absent Cl− currents and a lack of endosomal acidification (17).
The 30:insH mutation, which is located in helix A, had similar abnormalities to the G57V mutant, also in helix A (Fig. 1A), in being associated with reduced but not absent Cl− currents (Fig. S1), and resulting in a loss of receptor-mediated endocytosis (Fig. 2) that occurred without a defect in endosomal acidification (Fig. 3) or proton transport (Fig. S4 and Table 1) (17). These abnormalities also parallel the E211A knock-in mouse where a defect in endocytosis is present, although acidification is intact (16). The R637X mutation, which is located in CBS1, had similar abnormalities to the E527D mutant, located in helix P (Fig. 1A) in being associated with abolition of Cl− currents (Fig. S1) and proton transport (Fig. S4) and resulting in defective endosomal acification (Fig. 3), and impaired receptor-mediated endocytosis (Fig. 2) (Table 1). Finally, the del132-241 mutation (which involves helices C-G) (Fig. 1A) had similarities to the S270R (in H-I loop), G513E (in helix O), R516W (in helix O), and I524K (in helix P) (17) and mice deleted for CLC-5 helices C-F (10), in resulting in an absence of CLC-5 protein (Fig. S3) with mislocalization of megalin (Fig. 2B). Thus, the cellular phenotype of the del132-241 ciPTEC may be explained by an absence of CLC-5 protein and impaired megalin internalization.
In conclusion, our studies have established human cell models (ciPTECs) for the proximal tubular defects observed in Dent disease. These ciPTECs provide valuable resources for the investigation of receptor-mediated endocytosis and the mechanisms of endosomal acidification.
Materials and Methods
Detailed methods can be found in SI Materials and Methods.
Patients, Cell Cultures, and Characterization of ciPTECs.
Informed consent was obtained from the individuals, using protocols approved by the National Research Ethics Service (NRES) Committee (London; Protocol number MREC/02/2/93). Primary cell cultures were established from first-void urine samples, and immortalized and proximal tubule cells were isolated by FACS using mouse–anti-human CD13-FITC antibody (Dako), as described (18, 19, 28). Sorted cells were subcloned and treated with CD13-FITC before investigation by flow cytometry (19). DNA sequence analysis of the CLCN5 coding region exons and intron-exon boundaries, and Western blot and RT-PCR analysis of nephron segment specific genes and proteins was performed, as described (4, 9). For Z-stack imaging, cells were fixed and coimmunostained with anti-ZO-1 and anti-microtubule associated protein, RB/EB family, member-1 (MAPRE1) (EB-1), and secondary antibodies anti-rabbit Alexa Fluor 488 and anti-mouse Alexa Fluor 594, respectively (4).
Functional Assays.
Electrophysiological studies, and albumin and transferrin endocytosis assays, were performed as described (4, 29). Dextran uptake was measured by exposing cells to FITC-conjugated dextran followed by solubilization and recording of FITC fluorescence by using a microplate reader. Endosomal pH was determined by using a modification of described methods (17, 30). Following generation of a standard curve (17), endosomal pH was determined by using ciPTECs transfected with pHluorin-VAMP2, in the presence or absence of bafilomycin-A1, and imaging pHluorin intensity with 488 nm (diode laser) and 760 nm (argon two-photon laser), to obtain 760 nm/488 nm ratios that were compared with the standard curve (17).
Supplementary Material
Acknowledgments
This paper is dedicated to Oliver Wrong (1925–2012) for his contribution to this manuscript and his pioneering work in Dent disease. We thank M. O’Hare and J. Galvanovskis for technical assistance. This work was funded by UK Medical Research Council (MRC) Grants G9825289 and G1000467 (to C.M.G., S.E.P., B.H., and R.V.T.); Kidney Research UK (S.E.P. and R.V.T.); European Union Sixth Framework Programme-European Renal Gene Project Grant 05085 (to S.E.P. and R.V.T.); and Fund for Scientific Research, Flanders Fundamental Clinical Investigatorship 1801110N (to E.N.L.). C.M.G. was an MRC doctoral student.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. A.R. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1302063110/-/DCSupplemental.
References
- 1.Madsen KM, Nielsen S, Tisher CC. The Kidney. Philadelphia: Saunders; 2008. pp. 39–50. [Google Scholar]
- 2.Devuyst O, Thakker RV. Dent’s disease. Orphanet J Rare Dis. 2010;5:28. doi: 10.1186/1750-1172-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Christensen EI, Birn H. Megalin and cubilin: Multifunctional endocytic receptors. Nat Rev Mol Cell Biol. 2002;3(4):256–266. doi: 10.1038/nrm778. [DOI] [PubMed] [Google Scholar]
- 4.Reed AA, et al. CLC-5 and KIF3B interact to facilitate CLC-5 plasma membrane expression, endocytosis, and microtubular transport: Relevance to pathophysiology of Dent’s disease. Am J Physiol Renal Physiol. 2010;298(2):F365–F380. doi: 10.1152/ajprenal.00038.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jentsch TJ. Chloride and the endosomal-lysosomal pathway: Emerging roles of CLC chloride transporters. J Physiol. 2007;578(Pt 3):633–640. doi: 10.1113/jphysiol.2006.124719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Picollo A, Pusch M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature. 2005;436(7049):420–423. doi: 10.1038/nature03720. [DOI] [PubMed] [Google Scholar]
- 7.Günther W, Lüchow A, Cluzeaud F, Vandewalle A, Jentsch TJ. ClC-5, the chloride channel mutated in Dent’s disease, colocalizes with the proton pump in endocytotically active kidney cells. Proc Natl Acad Sci USA. 1998;95(14):8075–8080. doi: 10.1073/pnas.95.14.8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Devuyst O, Christie PT, Courtoy PJ, Beauwens R, Thakker RV. Intra-renal and subcellular distribution of the human chloride channel, CLC-5, reveals a pathophysiological basis for Dent’s disease. Hum Mol Genet. 1999;8(2):247–257. doi: 10.1093/hmg/8.2.247. [DOI] [PubMed] [Google Scholar]
- 9.Lloyd SE, et al. A common molecular basis for three inherited kidney stone diseases. Nature. 1996;379(6564):445–449. doi: 10.1038/379445a0. [DOI] [PubMed] [Google Scholar]
- 10.Piwon N, Günther W, Schwake M, Bösl MR, Jentsch TJ. ClC-5 Cl- -channel disruption impairs endocytosis in a mouse model for Dent’s disease. Nature. 2000;408(6810):369–373. doi: 10.1038/35042597. [DOI] [PubMed] [Google Scholar]
- 11.Wang SS, et al. Mice lacking renal chloride channel, CLC-5, are a model for Dent’s disease, a nephrolithiasis disorder associated with defective receptor-mediated endocytosis. Hum Mol Genet. 2000;9(20):2937–2945. doi: 10.1093/hmg/9.20.2937. [DOI] [PubMed] [Google Scholar]
- 12.Christensen EI, et al. Loss of chloride channel ClC-5 impairs endocytosis by defective trafficking of megalin and cubilin in kidney proximal tubules. Proc Natl Acad Sci USA. 2003;100(14):8472–8477. doi: 10.1073/pnas.1432873100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nielsen R, et al. Endocytosis provides a major alternative pathway for lysosomal biogenesis in kidney proximal tubular cells. Proc Natl Acad Sci USA. 2007;104(13):5407–5412. doi: 10.1073/pnas.0700330104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lloyd SE, et al. Characterisation of renal chloride channel, CLCN5, mutations in hypercalciuric nephrolithiasis (kidney stones) disorders. Hum Mol Genet. 1997;6(8):1233–1239. doi: 10.1093/hmg/6.8.1233. [DOI] [PubMed] [Google Scholar]
- 15.Smith AJ, Lippiat JD. Direct endosomal acidification by the outwardly rectifying CLC-5 Cl(-)/H(+) exchanger. J Physiol. 2010;588(Pt 12):2033–2045. doi: 10.1113/jphysiol.2010.188540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novarino G, Weinert S, Rickheit G, Jentsch TJ. Endosomal chloride-proton exchange rather than chloride conductance is crucial for renal endocytosis. Science. 2010;328(5984):1398–1401. doi: 10.1126/science.1188070. [DOI] [PubMed] [Google Scholar]
- 17.Smith AJ, Reed AA, Loh NY, Thakker RV, Lippiat JD. Characterization of Dent’s disease mutations of CLC-5 reveals a correlation between functional and cell biological consequences and protein structure. Am J Physiol Renal Physiol. 2009;296(2):F390–F397. doi: 10.1152/ajprenal.90526.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Racusen LC, Fivush BA, Andersson H, Gahl WA. Culture of renal tubular cells from the urine of patients with nephropathic cystinosis. J Am Soc Nephrol. 1991;1(8):1028–1033. doi: 10.1681/ASN.V181028. [DOI] [PubMed] [Google Scholar]
- 19.Wilmer MJ, et al. Novel conditionally immortalized human proximal tubule cell line expressing functional influx and efflux transporters. Cell Tissue Res. 2010;339(2):449–457. doi: 10.1007/s00441-009-0882-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ludwig M, Utsch B, Monnens LA. Recent advances in understanding the clinical and genetic heterogeneity of Dent’s disease. Nephrol Dial Transplant. 2006;21(10):2708–2717. doi: 10.1093/ndt/gfl346. [DOI] [PubMed] [Google Scholar]
- 21.Takemura T, et al. Identification of two novel mutations in the CLCN5 gene in Japanese patients with familial idiopathic low molecular weight proteinuria (Japanese Dent’s disease) Am J Kidney Dis. 2001;37(1):138–143. doi: 10.1016/s0272-6386(01)80067-6. [DOI] [PubMed] [Google Scholar]
- 22.Kotlo K, Shukla S, Tawar U, Skidgel RA, Danziger RS. Aminopeptidase N reduces basolateral Na+ -K+ -ATPase in proximal tubule cells. Am J Physiol Renal Physiol. 2007;293(4):F1047–F1053. doi: 10.1152/ajprenal.00074.2007. [DOI] [PubMed] [Google Scholar]
- 23.Jaulin F, Xue X, Rodriguez-Boulan E, Kreitzer G. Polarization-dependent selective transport to the apical membrane by KIF5B in MDCK cells. Dev Cell. 2007;13(4):511–522. doi: 10.1016/j.devcel.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Devuyst O. Dent’s disease: Chloride-proton exchange controls proximal tubule endocytosis. Nephrol Dial Transplant. 2010;25(12):3832–3835. doi: 10.1093/ndt/gfq556. [DOI] [PubMed] [Google Scholar]
- 25.Wilmer MJ, et al. Cysteamine restores glutathione redox status in cultured cystinotic proximal tubular epithelial cells. Biochim Biophys Acta. 2011;1812(6):643–651. doi: 10.1016/j.bbadis.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, et al. ClC-5: Role in endocytosis in the proximal tubule. Am J Physiol Renal Physiol. 2005;289(4):F850–F862. doi: 10.1152/ajprenal.00011.2005. [DOI] [PubMed] [Google Scholar]
- 27.Scheinman SJ. X-linked hypercalciuric nephrolithiasis: Clinical syndromes and chloride channel mutations. Kidney Int. 1998;53(1):3–17. doi: 10.1046/j.1523-1755.1998.00718.x. [DOI] [PubMed] [Google Scholar]
- 28.O’Hare MJ, et al. Conditional immortalization of freshly isolated human mammary fibroblasts and endothelial cells. Proc Natl Acad Sci USA. 2001;98(2):646–651. doi: 10.1073/pnas.98.2.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith AJ, Lippiat JD. Voltage-dependent charge movement associated with activation of the CLC-5 2Cl-/1H+ exchanger. FASEB J. 2010;24(10):3696–3705. doi: 10.1096/fj.09-150649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394(6689):192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 31.Silva IV, et al. The ClC-5 knockout mouse model of Dent's disease has renal hypercalciuria and increased bone turnover. J Bone Miner Res. 2003;18(4):615–623. doi: 10.1359/jbmr.2003.18.4.615. [DOI] [PubMed] [Google Scholar]
- 32.Luyckx VA, Leclercq B, Dowland LK, Yu AS. Diet-dependent hypercalciuria in transgenic mice with reduced CLC5 chloride channel expression. Proc Natl Acad Sci USA. 1999;96(21):12174–12179. doi: 10.1073/pnas.96.21.12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.