

Abstract
The Toll/IL-1 receptor (TIR) domains are crucial signaling modules during innate immune responses involving the Toll-like receptors (TLRs) and IL-1 receptor (IL-1R). Myeloid differential factor 88 (MyD88) is a central TIR domain-containing adapter molecule responsible for nearly all TLR-mediated signaling and is targeted by a TIR domain-containing protein C (TcpC) from virulent uropathogenic Escherichia coli, a common human pathogen. The mechanism of such molecular antagonism has remained elusive. We present the crystal structure of the MyD88 TIR domain with distinct loop conformations that underscore the functional specialization of the adapter, receptor, and microbial TIR domains. Our structural analyses shed light on the genetic mutations at these loops as well as the Poc site. We demonstrate that TcpC directly associates with MyD88 and TLR4 through its predicted DD and BB loops to impair the TLR-induced cytokine induction. Furthermore, NMR titration experiments identify the unique CD, DE, and EE loops from MyD88 at the TcpC-interacting surface, suggesting that TcpC specifically engages these MyD88 structural elements for immune suppression. These findings thus provide a molecular basis for the subversion of TLR signaling by the uropathogenic E. coli virulence factor TcpC and furnish a framework for the design of novel therapeutic agents that modulate immune activation.
Keywords: innate immune cells, bacterial pathogens
Toll-like receptors (TLRs) are a family of innate immune receptors that recognize pathogen-associated molecular patterns, such as bacterial cell wall components and microbial nucleic acids. As such, TLR signaling plays critical roles in the initiation and orchestration of both innate and adaptive immune responses (1). Ligand engagement at the TLR extracellular or endosomal domains results in the association of their cytoplasmic Toll/IL-1 receptor (TIR) domains, which in turn recruit downstream TIR domain-containing adapter molecules, such as MyD88 (myeloid differentiation factor 88), MAL (MyD88 adapter-like)/TIRAP (TIR domain-containing adaptor protein), TRIF (TIR domain-containing adapter-inducing IFN-β), and TRIF-related adapter molecule. Recruitment of the TIR domain-containing adapters elicits a signaling cascade that leads to the activation of transcription factors NF-κB and activator protein-1 (AP-1) and the production of cytokines (2, 3).
MyD88 is an essential adapter molecule for the IL-1 receptor and most TLR family members except for TLR3 (3). Its central importance in innate immune responses was underscored by the observations that mice deficient in MyD88 are nonresponsive to LPS challenge (4) and IL-1/IL-18 stimulation (5), and that mutations in human MyD88 render pediatric patients susceptible to pyrogenic bacterial infections (6). Conversely a gain-of-function variant of MyD88, L252P, found in diffuse large B-cell lymphoma, promotes tumor survival through enhanced NF-κB and JAK kinase activation (7). Mechanistically, MyD88 is thought to form homodimers (8) that are recruited by the TLR TIR domains to initiate the assembly of a death domain complex “Myddosome” for downstream signaling (9). Structures of individual TIR domains have been determined for a number of receptors and adapters (10–18), and most TIR domains contain five α helices (A–E) surrounding a central five-stranded β sheet (A–E). Three regions of high sequence conservation are defined as boxes 1–3 motifs (19) and are located at the βA strand, BB loop, and αE helix.
Despite a wealth of reports on TIR domain functions, the molecular mechanism of TIR:TIR domain interactions remains a central unresolved issue. Genetics and mutagenesis studies have identified the conserved Pro residue and the BB loop (box 2) as essential for TLR signaling (10, 20–23). However, the importance of the Pro residue or the BB loop was called into question by others (6, 13, 24–26). Furthermore, modeling and functional studies revealed that residues outside of the BB loop play vital roles in TIR domain associations or TLR signaling, such as the CD, DD, and EE loops (11, 14, 25, 27–30). These observations make conceptualizing a coherent model of the TIR:TIR domain signaling a challenge, which is further complicated by the findings that the modes of receptor:adapter assembly may differ among different TLR receptors (22, 24).
In addition to eukaryotic TIR domain-containing proteins, a number of bacterial proteins have been identified that modulate host immune responses through their TIR domains. These include proteins from uropathogenic Escherichia coli (UPEC) strain CFT073 (TcpC) (31), Brucella melitensis (TcpB) (31), Brucella abortus (TcpB/Btp1) (32, 33), Brucella ovis (TcpB) (32), Salmonella enterica (TlpA) (34), Paracoccus dentrificans (PdTLP) (35) and Yersinia pestis (YpTdp) (36). Many of these microbial proteins have been reported to interact with the host TIR domain proteins and may promote the formation of nonproductive TIR domain complexes and thus prevent immune signaling (31, 35–37). For example, Low et al (35) reported the association of PdTLP with TLR4 and MyD88, although the exact binding interface remains to be characterized. As a result of their ability to suppress immune responses, these microbial TIR domain-containing proteins were proposed as a novel class of virulence factors that subvert host immunity through molecular mimicry (38, 39). The best-characterized bacterial TIR-containing protein is TcpC, which is common in the most virulent UPEC strains from children with acute pyelonephritis. TcpC suppresses TLR-mediated cytokine production upon infection, increases bacterial burden in the urinary tract, and promotes renal tissue damage (31, 37). Likewise, the Brucella protein TcpB has been shown to interact with both TLR signaling adapters MyD88 (31) and MAL (40) and inhibits TLR signaling (32, 33). The clinical relevance of the above bacterial virulence factors makes it imperative to understand the molecular basis of the interactions between the microbial TIR domain-containing proteins and their host targets.
To investigate the role of MyD88 as a central adapter molecule, as well as the mechanisms by which the bacterial virulence factors subvert MyD88-mediated signaling, we have determined the crystal structure of the MyD88 TIR domain and identified a TcpC-binding surface at MyD88. In addition, we show that the predicted TcpC DD and BB loop peptides directly interact with MyD88 and TLR4 and suppress innate immune signaling. Our studies thus provide a molecular mechanism for the interactions of host and pathogen TIR domains and furnish a framework for the design of novel therapeutic agents targeting the TLR and IL-1 receptor (IL-1R) signaling pathways.
Results
MyD88 TIR Domain Loops Adopt Adapter-Specific Conformations.
The crystal structure of the MyD88 TIR domain was determined at 1.45-Å resolution (Table S1), and it reveals a typical TIR domain fold with five parallel β-strands surrounded by helices and loops (Fig. 1A) similar to a reported NMR structure (13). All residues of the TIR domain including the long loops are well resolved by unambiguous electron density map at high resolution (Fig. S1A). A comparison of the MyD88 TIR domain structure with those from TLR2, TLR1, and Paracoccus dentrificans (PdTLP) reveals unique loop conformations for each domain (Fig. 1B). For example, the BB, CD, DD, DE, and EE loops adopt strikingly different conformations (Fig. S2 B–F), and their sequences also seem to be conserved only among the subfamilies of the adapter, receptor, and microbial TIR domains (Fig. S2). Notably, the conserved “box2” Pro residues from TLR2 and PdTLP are 16.5 Å and 22.8 Å apart from that in MyD88, respectively (Fig. S1B). By contrast, the crystal and NMR structures of the MyD88 TIR domain are very similar, with minimal structural changes (Fig. S1G), suggesting that crystallization has no significant effect on the structure of the MyD88 loops. Interestingly, the BB loops of most known TIR domain structures contain a short 310 helix (MyD88 and PdTLP) or α-helix (TLR1, TLR2, TLR10, and IL-1RAPL) preceding the conserved Pro residues (Fig. S2). The functional significance of these short helices is unclear, although previous reports have demonstrated that the MyD88 residue R196 at its 310 helix plays crucial roles in TLR signaling and immune responses (6, 13).
Fig. 1.

Comparison of the TIR domain structures. (A) Cartoon representation of the MyD88 TIR domain crystal structure, labeled with key secondary structure elements. The five parallel β-strands are colored orange and the rest of the structure in cyan. (B) Superposition of structures for the TIR domains from MyD88 (cyan), TLR2 (lime), TLR1 (pale green), and Paracoccus PdTLP (magenta). Regions of major structural differences are marked.
Several TIR:TIR domain interfaces were observed in the MyD88 crystal lattice (Fig. S3A). Two of them bury significant amounts of solvent accessible surface area of 1,200 Å2 (Fig. S3B) and 1,000 Å2 (Fig. S3C), respectively. The former is mediated by αA helix, DD and EE loops from one molecule, and αC’ and αD helices from its symmetry mate (Fig. S3B). Notably, at the center of the interface lies a cluster of hydrophobic interactions between F174, Y257, and Y276 from one TIR domain and L241 from the other. The second crystal lattice interface is centered on residues R196 and D197 from the BB loop of one TIR domain, forming direct or water-mediated hydrogen bonds with the DD and EE loops of the symmetry mate (Fig. S3C). Because some of these TIR domain surfaces have been shown to mediate TIR:TIR domain association and/or TLR signaling (6, 11, 13, 14, 25–29), the above crystal lattice contacts may mimic physiological TIR domain associations.
Pococurante Site Residue I179 Stabilizes the MyD88 BB Loop.
Adjacent to the MyD88 BB loop is the Pococurante (Poc) site mutation I179N that was identified through random germ-line mutagenesis (30). This mutation abrogated MyD88-dependent sensing of most TLR ligands and further diminished TLR signaling by a BB loop mutant. In agreement, an equivalent mutation in TLR4 (V693N) was recently shown to impair signaling (28). The Poc site residue is highly conserved among the TIR domains (Fig. S2). The MyD88 residue I179 buttresses the BB loop through hydrophobic interactions with residues L199 and T202, bracketing the highly conserved “box2” 200PG201 motif (Fig. 2A). To understand the role of the Poc site mutation on the MyD88 TIR domain structure, we carried out molecular dynamics simulations of the wild-type and I179N mutant MyD88 TIR domains (Fig. S4). Analysis of the simulation trajectories illustrates that even though both the wild-type and the mutant domains retained the TIR domain fold throughout the simulation, the I179N mutation significantly enhances the flexibility of the BB loop compared with the wild-type protein. This is demonstrated by its enhanced root mean square fluctuation values, an indication of the atomic position movements (Fig. 2B). A more mobile BB loop may incur higher entropic cost for stable TIR:TIR domain associations mediated by this surface, resulting in diminished signaling ability of the Poc mutant. Because the Poc site mutant may compromise TIR domain-mediated signaling indirectly, a combination of the Poc site and BB loop mutations may result in a more severe phenotype, as demonstrated by Jiang et al. (30).
Fig. 2.

Poc site residue anchors the BB loop. (A) Cartoon representation of the interactions between the MyD88 TIR domain Poc site residue I179 and its BB loop residues. (B) Root mean square fluctuations of the Cα atoms for the wild-type (blue) and I179N mutant (red) MyD88 TIR domains in a 5-ns MD simulation.
TcpC Targets Both MyD88 and TLR4.
MyD88-mediated signaling is suppressed by TcpC that in turn promotes bacterial survival and kidney infection (31). The TIR domain of TcpC was shown to directly interact with that from MyD88 (31). We have shown previously that recombinant TcpC TIR domain suppresses TNFα secretion by wild-type bone marrow-derived macrophages (BMDMs) upon stimulation with TLR ligands (31, 37). Intriguingly, TcpC further diminishes LPS-induced TNF-α secretion by MyD88-deficient BMDMs, implying that it may interact with additional TLR4 pathway components besides MyD88 (31, 37). Using a Strep-tag pull-down assay, we demonstrate that the TcpC TIR domain interacts with both MyD88 and TLR4 (Fig. 3 A and B). To identify the structural elements of the TcpC TIR domain involved in these interactions, we performed similar assays using peptides derived from the predicted TcpC loops (Table S2). These TcpC peptides were identified on the basis of sequence alignment with the only known microbial TIR domain structure of PdTLP (Fig. S2) (16), and we refer to them as the “BB” (198-VIIWYDEQTLEVGDS-212) and “DD” (260-ILPIWHNINAQEVSKY-275) peptides. The last 17 residues of TcpC were shown to facilitate the transport of TcpC across the plasma membrane (31), similar to the HIV TAT sequence. It is hereafter referred to as “TAT” peptide (SAKEIARELAEIAYRRR) and was fused to the C termini of all designed TcpC peptides in the stimulation assay except where stated otherwise.
Fig. 3.

Suppression of TLR signaling by the TcpC TIR domain and its peptides. (A) Pull-down assays of the cellular lysates from the Myc-MyD88 transfected HEK293 cells were performed with the TIR TcpC Strep-Tactin Macroprep beads (+) or empty Strep-Tactin Macroprep beads (-). The beads were washed with 500 mM NaCl, and the remaining proteins were detected by Western blot using an anti-Myc antibody after elution. (B) The same assay was performed as in A using lysates from the Flag-TLR4 transfected cells. The beads were washed with two different NaCl concentrations as indicated before elution. (C and D) BMDMs were stimulated with TLR ligands in the presence of titrated amounts of the TcpC BB loop peptide (0.0026, 0.026, 0.26, or 2.6 µM, C) or the TcpC DD loop peptide (0.0025, 0.025, 0.25, or 2.5 µM, D), and the TNF-α in the culture supernatants was analyzed 3 h after stimulation. BB and DD indicate TNF-α release induced by the highest dose of each peptide in the absence of a TLR ligand. Error bars represent SD of three individual experiments. (E and F) The same pull-down assays were performed as in A and B, except that BB or DD loop peptide bearing a Strep-tag was bound to Strep-Tactin Macroprep beads. Empty Strep-Tactin Macroprep beads (-) were used as negative control. After washing with a buffer containing 500 mM NaCl, bound TLR4 (E) or MyD88 (F) were detected by Western blot after elution.
Using the TNF-α secretion assay, we show that the TcpC BB peptide impaired mainly the LPS-mediated activation of BMDMs (Fig. 3C). The DD peptide inhibited LPS-driven responses, and to some extent also CpG, poly (I:C), and R848-mediated responses of BMDMs (Fig. 3D). In agreement, the pull-down assay revealed that the BB peptide bound to TLR4 but not MyD88 (Fig. 3E), whereas the reverse was true for the DD peptide (Fig. 3F), indicating that the TcpC TIR domain is indeed capable of disrupting signaling by both MyD88 and TLR4. Importantly, the TAT peptide itself did not impair signaling as measured by TNF-α secretion (Fig. S5A) or chemokine KC/CXCL1 production (Fig. S5B). In addition, the scrambled versions of the BB (“BBsc”) and DD (“Disc”) peptides did not significantly inhibit LPS response (Fig. S5B), suggesting that the native BB and DD peptide sequences are critical for their inhibitory functions. Furthermore, removing four residues from the predicted DD loop (“DD-short”: 264-WHNINAQE-271) severely compromised its inhibitory activity (Fig. S5C), demonstrating that these DD loop residues are essential for its function.
Identification of the TcpC-Binding Surface at the MyD88 TIR Domain.
Having identified the functional epitopes at the TcpC TIR domain involved in immune suppression, we sought to map the TcpC-binding surface on MyD88 using NMR titration of 15N-labeled MyD88 TIR domain. The 1H-15N HSQC (hetero-nuclear single quantum coherence) spectra of MyD88 were recorded in the absence or presence of 1:1 molar ratio of unlabeled TcpC, and a number of MyD88 residues showed significant chemical shift changes (Fig. 4A). These include residues M157, S244, K250, R269, F270, T272, V273, C280, and W286 that predominately localize to the CD, DE, and EE loops on one face of the MyD88 TIR domain opposite to its BB loop (Fig. 4B). Similar titration experiments carried out using the DD loop peptide “DD-nostrand” (264-WHNINAQEVSKY-275) showed significant chemical shift changes at residues V204, K250, L268, R269, C280, and W286, which are localized at essentially the same region of the MyD88 CD, DE, and EE loops (Fig. S6A), indicating that the TcpC DD loop is its primary binding surface for the MyD88 TIR domain. The MyD88 CD, DE, and EE loops are not only structurally distinct from those in other TIR domains (Fig. 1B), but their sequences are also very diverse compared with the receptor and microbial TIR domains (Fig. S2), suggesting that TcpC specifically targets this unique surface of the MyD88 TIR domain for immune suppression.
Fig. 4.

Identification of the TcpC-binding site at the MyD88 TIR domain. (A) NMR titration of the 15N-labeled MyD88 TIR domain with TcpC. The 1H-15N chemical shift changes were plotted in the presence of TcpC (black) and buffer (red). The dashed line marks the threshold value (mean + 1.5 SD). (B) Mapping of the TcpC-binding site on the surface of the MyD88 TIR domain. Residues with significant chemical shift changes in A are colored blue and labeled. (C) HEK293 cells were transfected with MyD88-encoding plasmids including the wild-type, C203S, C280S, and C203S plus C280S mutants, as well as titrated amounts of TIR-TcpC ΔTAT, as indicated. Luciferase activities were determined 48 h after transfection. Error bars represent SD of three individual experiments.
To understand the function of these residues in mediating TcpC suppression of MyD88 signaling, we chose to study MyD88 residue C280 at its EE loop that shows the most significant chemical shift changes upon TcpC titration. A solvent-exposed residue C203 at the BB loop on the opposite surface of MyD88 was selected as a control. An NF-κB luciferase assay in HEK293 cells demonstrated that the MyD88 wild-type and C203S, C280S, and C203S/C280S mutants were all capable of stimulating NF-κB activation (Fig. S6B), suggesting that the mutations did not cause intrinsic defects in immune signaling. As expected, introduction of the TcpC TIR domain impaired NF-κB activation by the wild-type MyD88 (Fig. 4C). Similarly, NF-κB activation by the C203S mutant is sensitive to the TcpC inhibition. In contrast, the C280S and C203S/C280S double mutants were no longer inhibited by the TcpC TIR domain (Fig. 4C), demonstrating that the MyD88 residue C280 is an indispensable binding surface for TcpC to exert its inhibitory function, even though this residue is not essential for MyD88 function per se.
Discussion
In this study we present the crystal structure of the MyD88 TIR domain and characterize its interaction with the bacterial virulence factor TcpC. Compared with the receptor and microbial TIR domain structures, the MyD88 crystal structure reveals dramatically different BB, CD, DD, DE, and EE loop conformations, in agreement with their diverse sequences among the adapter, receptor, and microbial TIR domains. Importantly, these loop structures are preserved between the MyD88 crystal and NMR structures, demonstrating their remarkable conformational stability. This suggests that the striking structural differences among the adapter, receptor, and microbial TIR domains are inherent properties of individual proteins that may underpin the functional specialization of the TIR domain subfamilies. Despite their apparent conformational differences, many of the same loops and their connecting secondary structures participate in TIR:TIR domain interfaces across many different crystal lattices: MyD88 (present study), TLR1 (10), TLR2 (10), TLR10 (11), MAL/TIRAP (14, 15), IL-1RAPL (12), PdTLP (16), AtTIR (17), and L6 (18). Even though the specific residue-by-residue interactions at these interfaces differ, the frequent involvement of these loops in crystal lattice contacts suggests that they may indeed mediate biologically relevant TIR:TIR domain interactions, both in host TLR signaling and as a mechanism for microbial antagonism of immune responses. The functional significance of these loops is perhaps best illustrated by their mutations that severely compromise immune signaling. Analogous to the MyD88 DD loop residues K256 and Y257 involved in two different lattice contacts in our crystals, the equivalent MAL residue S180 at its DD loop was shown to be essential for its interaction with TLR2 and human immune responses to infections (41). Similarly, residues R749, F749, L752, and R753 at the DD loop from the TLR2 TIR domain were reported to be critical for TLR1:TLR2 association and signaling (29), and a rare DD loop mutation R753Q in human TLR2 was shown to reduce TLR2 activation and increase the risk of allergic hypersensitivity (42).
The BB loop of TIR domain has long been recognized as crucial for TLR signaling (10, 20, 21). An R196C mutation in the MyD88 BB loop was observed in human patients that compromises immune responses to pyrogenic bacterial infections (6) and diminishes its association with TLR2 (43). It is possible that in addition to modifying the BB loop surface at this location, this mutation may further compromise TIR:TIR domain interactions by either crippling the conformation of the BB loop through aberrant disulfide formation, or chemical modifications of the cysteine that diminish its signaling capacity, analogous to the S nitrosylation of the MyD88 residue C216 (44). As shown in Figs. 1B and 2A, residue R196 is at the 310 helix of the BB loop region that displays the most significant structural differences among the TIR domains. This residue is also located at the center of the BB, DD, and EE loop crystal lattice contacts (Fig. S3C) that may mimic the interaction of MyD88 TIR domain with other signaling domains. One such example was recently reported for an MyD88:MAL docking model with the MyD88 residue R196 in the vicinity of the MAL residue S180 at the DD loop (14), and is consistent with the observation that mutation of the MyD88 residue R196 impaired its interaction with MAL (13). Because R196 is conserved among most TIR domains (Fig. S2), it is likely important for the function of other TIR domains as well: an equivalent R677W mutation at the BB loop of the TLR2 TIR domain was reported to abolish immune responses to Mycobacterium leprae and Mycobacterium tuberculosis (45).
In comparison, the Poc site residue I179 is mostly shielded from the solvent by the BB loop and therefore unlikely to directly participate in TIR:TIR domain interactions. Instead, molecular dynamics simulations reveal that an I179N mutation may enhance the flexibility of the BB loop and perturb its conformation, which in turn destabilize the MyD88 signaling platform assembly. Such an indirect effect may also help reconcile some of the reported TIR domain mutagenesis data that mapped functionally important residues at vastly different TIR domain region. Mutations that compromise TLR signaling may result from diminished homotypic TIR domain dimerization/oligomerization, heterotypic receptor:adapter TIR domain association, or allosteric regulation of both by residues adjacent to the above interfaces, similar to the MyD88 Poc site mutation. Therefore, functionally important TIR domain residues may be directly or indirectly engaged in diverse TIR:TIR domain interfaces in a given receptor signaling assembly.
Chaudhary et al. (40) recently uncovered a new indirect regulatory mechanism of TIR domain function. They showed that the MyD88 death domain and TIR domain interact at a higher affinity than the TIR:TIR domain interaction, which suggested that MyD88 may reside in an autoinhibited “resting” state through intramolecular domain interaction. It is conceivable that some of the TIR domain mutations may modulate such intramolecular domain interactions and thus indirectly regulate the ability of the TIR domain or death domain to associate with their binding partners. Similar autoinhibition mechanisms have been observed for another TIR domain-containing protein L6 from plant (18), as well as other innate immune receptors such as RIG-I (46) and AIM2 (47), and may be a common regulatory mechanism to safeguard against spurious activation of potent immune responses.
Peptides representing functionally important TIR domain loops were shown to interfere with TIR domain-mediated signaling and were used to map important TIR:TIR domain interfaces (22, 23, 48, 49). In this study we identified the BB and DD loops of the TcpC TIR domain and characterized them as important functional epitopes that target TLR4 and MyD88 for immune suppression. The DD peptide inhibited immune response to several TLR ligands, whereas the BB peptide primarily impaired LPS-driven responses. At higher concentrations the BB peptide modestly enhanced response to nucleic acid ligands (Fig. 3C), perhaps through receptors other than TLR4. Similar enhancement of signaling was observed for peptides derived from the TLR4-TIR surface that increased TNF synthesis in response to TLR2 ligands (23), and the MAL TIR domain-derived peptides that enhanced TNF secretion upon LPS stimulation (50). Clearly, further studies are required to investigate the differential activities of the TIR domain-derived peptides.
Our NMR titration mapped the TcpC-binding residues to the CD, DE, and EE loops of the MyD88 TIR domain. How does TcpC binding to MyD88 impair its signaling? The TIR domain αC helix and the following CD loop were reported to be important for TIR domain associations for TLR2 (51), TLR4 (28), TIR10 (11), and IL-1RAcP (12). Similarly, MyD88 residue R288 at the αE helix was reported to be essential for its association with MAL, and the αE helices of the TLR2 and TLR4 TIR domains were shown to mediate their self-associations (23, 30). It is conceivable that the CD or EE loop of MyD88 is at or near the interface between the MyD88 TIR domain and itself or its partner TIR domains, and the TcpC:MyD88 association may prevent the formation of productive TIR domain signaling complexes through steric hindrance (Fig. S7). These molecular interactions between the host and microbial TIR domains thus underline the pathological roles of the TcpC-mediated antagonism of host immune responses. The fact that the CD and EE loops are among the highly divergent structural elements of the TIR domains with the least sequence conservation also provides a possible mechanism for specific targeting of host TIR domain-containing protein by its microbial partners. Future studies will unveil whether the bacterial protein TcpB targets mammalian adapter protein MAL similarly as TcpC does MyD88.
In conclusion, this study reveals the structural basis for the functional specialization of the TIR domain subfamilies, furthers our understandings of the TIR domain-mediated signaling, and provides insights into the mechanisms of antagonism by virulence factors from clinically relevant human pathogens. Furthermore, we identify peptides from the microbial protein TcpC that may serve as useful leads for therapeutics developments to reduce excessive immune activation, similar to the previously reported decoy peptides from the host TIR domain BB loops (23).
Materials and Methods
Protein Expression and Purification.
The human MyD88 residues M157 to P296 was expressed in E. coli and purified to homogeneity using metal ion affinity chromatography and size exclusion chromatography. Recombinant TcpC TIR domain (residues 170–307) was expressed and purified as reported previously (31). Full methods are described in SI Materials and Methods.
Crystallization and Structure Determination.
The MyD88 TIR domain was crystallized using a solution containing 100 mM Tris·HCl (pH 8.0) and 25% (vol/vol) PEG 350 MME. X-ray diffraction data were processed with the HKL2000 program suite (52), and the structure was determined using the deposited NMR structure of the MyD88 TIR domain (Protein Data Bank code 2JS7) as a search model. Refinement was performed using PHENIX (53), and the structures were validated using Molprobity (54) in PHENIX and the Research Collaboratory for Structural Bioinformatics ADIT validation server (55). Figures were produced with the program Pymol (Schrödinger LLC).
Molecular Dynamics Simulation.
The program suite GROMACS 4.5.5 (56) was used for molecular dynamics (MD) simulations with the Biowulf Linux cluster at National Institutes of Health (biowulf.nih.gov). Analysis of the MD simulation trajectories was carried out using the programs g_rms and g_rmsf in the GROMACS package.
Pull-Down Assay.
Purified TcpC TIR domain (TIR-TcpC) or the TcpC peptides carrying a C-terminal Strep-tag II were bound to Strep-Tactin MacroPrep Beads (IBA) and blocked with 300 µg/mL avidin. The beads were incubated with 50–200 mg cleared total cell lysates (“prey”) from HEK293 cells and washed three times with an acetate buffer containing 150 mM or 500 mM NaCl to remove nonspecifically bound proteins. The bound “prey” proteins were then eluted in two consecutive steps with an elution buffer (100 mM sodium citrate, 100 mM lysine, and 3 mM EDTA) at pH 2.8 and neutralized with 2 M Tris·HCl (pH 8.0).
Cell Stimulation Assay.
BMDMs or immortalized BMDMs were stimulated with the TLR ligands poly(I:C) (2.5 µg/mL), ultrapure LPS from E. coli (100 ng/mL), CpG-DNA 1826 (2 µM), or R848 (1 µM) in the absence or presence of titrated amounts of the TcpC peptides. Secreted TNF-α or keratinocyte chemoattractant (KC) was quantitated by ELISA from the culture supernatants 3 h after stimulation. All assays were performed in triplicate.
Luciferase Reporter Assay.
HEK293 cells were transfected using Lipofectamine 2000 (Invitrogen) with NF-κB firefly luciferase (50 ng/mL) and Renilla luciferase reporter constructs (1 ng/mL), as well as plasmids encoding wild-type or mutant MyD88. TIR-TcpC ΔTAT plasmid at 0.2, 2, 20, and 200 ng/mL was cotransfected to test the inhibitory effects of TcpC. Forty-eight hours after transfection the luciferase activities were measured using the dual luciferase reporter assay system (Promega) and a microplate luminometer (Titertek Berthold). All assays were performed in triplicate.
NMR Titration of 15N-Labeled MyD88 TIR Domain.
A 0.35-mM 15N-labeled MyD88 TIR domain sample in PBS buffer (pH 6.5) was titrated with a 3 mM TcpC TIR domain stock solution to 1:1 molar ratio. For titration using the “DD-nostrand” peptide, the labeled MyD88 and synthesized peptide were mixed at 1:1 molar ratio and concentrated to 0.1 mM. The 1H-15N HSQC spectra of MyD88 were collected using a Brüker 800 MHz spectrometer and the normalized 1H-15N chemical shift deviation (δHN) was calculated as 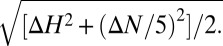 The buffer effect was subtracted using blank buffer titrations.
The buffer effect was subtracted using blank buffer titrations.
Accession Codes.
The atomic coordinates and structural factors for the MyD88 TIR domain have been deposited with the Research Collaboratory for Structural Bioinformatics Protein Data Bank under accession codes 4EO7 and 4DOM.
Supplementary Material
Acknowledgments
We thank the beam line scientists at the Argonne National Laboratory Southeast Regional Collaborative Access Team and the Brookhaven National Laboratory for their support of the X-ray diffraction data collection; Dr. Fred Dyda and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) X-ray facility for their support; Dr. D. Eric Anderson at the mass spectrometry facility of NIDDK for technical support; and David S. Waugh at the National Cancer Institute for the tobacco etch virus protease expression construct. T.S.X. is supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH). T.M. is supported by Deutsche Forschungsgemeinschaft Grant MI471/6-1. N.T. is supported by the Division of Intramural Research, National Heart, Lung and Blood Institute, NIH.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Research Collaboratory for Structural Bioinformatics Protein Data Bank, www.rcsb.org (ID codes 4EO7 and 4DOM).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1215770110/-/DCSupplemental.
References
- 1.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 3.O’Neill LAJ, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7(5):353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 4.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11(1):115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 5.Adachi O, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9(1):143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 6.von Bernuth H, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. 2008;321(5889):691–696. doi: 10.1126/science.1158298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ngo VN, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–119. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burns K, et al. MyD88, an adapter protein involved in interleukin-1 signaling. J Biol Chem. 1998;273(20):12203–12209. doi: 10.1074/jbc.273.20.12203. [DOI] [PubMed] [Google Scholar]
- 9.Lin S-C, Lo Y-C, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465(7300):885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu Y, et al. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature. 2000;408(6808):111–115. doi: 10.1038/35040600. [DOI] [PubMed] [Google Scholar]
- 11.Nyman T, et al. The crystal structure of the human toll-like receptor 10 cytoplasmic domain reveals a putative signaling dimer. J Biol Chem. 2008;283(18):11861–11865. doi: 10.1074/jbc.C800001200. [DOI] [PubMed] [Google Scholar]
- 12.Khan JA, Brint EK, O’Neill LA, Tong L. Crystal structure of the Toll/interleukin-1 receptor domain of human IL-1RAPL. J Biol Chem. 2004;279(30):31664–31670. doi: 10.1074/jbc.M403434200. [DOI] [PubMed] [Google Scholar]
- 13.Ohnishi H, et al. Structural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc Natl Acad Sci USA. 2009;106(25):10260–10265. doi: 10.1073/pnas.0812956106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valkov E, et al. Crystal structure of Toll-like receptor adaptor MAL/TIRAP reveals the molecular basis for signal transduction and disease protection. Proc Natl Acad Sci USA. 2011;108(36):14879–14884. doi: 10.1073/pnas.1104780108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin Z, Lu J, Zhou W, Shen Y. Structural insights into TIR domain specificity of the bridging adaptor Mal in TLR4 signaling. PLoS ONE. 2012;7(4):e34202. doi: 10.1371/journal.pone.0034202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan SL, et al. Molecular mimicry in innate immunity: Crystal structure of a bacterial TIR domain. J Biol Chem. 2009;284(32):21386–21392. doi: 10.1074/jbc.C109.007591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan SL, Mukasa T, Santelli E, Low LY, Pascual J. The crystal structure of a TIR domain from Arabidopsis thaliana reveals a conserved helical region unique to plants. Protein Sci. 2010;19(1):155–161. doi: 10.1002/pro.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernoux M, et al. Structural and functional analysis of a plant resistance protein TIR domain reveals interfaces for self-association, signaling, and autoregulation. Cell Host Microbe. 2011;9(3):200–211. doi: 10.1016/j.chom.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slack JL, et al. Identification of two major sites in the type I interleukin-1 receptor cytoplasmic region responsible for coupling to pro-inflammatory signaling pathways. J Biol Chem. 2000;275(7):4670–4678. doi: 10.1074/jbc.275.7.4670. [DOI] [PubMed] [Google Scholar]
- 20.Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 21.Fitzgerald KA, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413(6851):78–83. doi: 10.1038/35092578. [DOI] [PubMed] [Google Scholar]
- 22.Toshchakov VU, Basu S, Fenton MJ, Vogel SN. Differential involvement of BB loops of toll-IL-1 resistance (TIR) domain-containing adapter proteins in TLR4- versus TLR2-mediated signal transduction. J Immunol. 2005;175(1):494–500. doi: 10.4049/jimmunol.175.1.494. [DOI] [PubMed] [Google Scholar]
- 23.Toshchakov VY, Szmacinski H, Couture LA, Lakowicz JR, Vogel SN. Targeting TLR4 signaling by TLR4 Toll/IL-1 receptor domain-derived decoy peptides: Identification of the TLR4 Toll/IL-1 receptor domain dimerization interface. J Immunol. 2011;186(8):4819–4827. doi: 10.4049/jimmunol.1002424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dunne A, Ejdeback M, Ludidi PL, O’Neill LAJ, Gay NJ. Structural complementarity of Toll/interleukin-1 receptor domains in Toll-like receptors and the adaptors Mal and MyD88. J Biol Chem. 2003;278(42):41443–41451. doi: 10.1074/jbc.M301742200. [DOI] [PubMed] [Google Scholar]
- 25.Li C, Zienkiewicz J, Hawiger J. Interactive sites in the MyD88 Toll/interleukin (IL) 1 receptor domain responsible for coupling to the IL1beta signaling pathway. J Biol Chem. 2005;280(28):26152–26159. doi: 10.1074/jbc.M503262200. [DOI] [PubMed] [Google Scholar]
- 26.Brown V, Brown RA, Ozinsky A, Hesselberth JR, Fields S. Binding specificity of Toll-like receptor cytoplasmic domains. Eur J Immunol. 2006;36(3):742–753. doi: 10.1002/eji.200535158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ronni T, et al. Common interaction surfaces of the toll-like receptor 4 cytoplasmic domain stimulate multiple nuclear targets. Mol Cell Biol. 2003;23(7):2543–2555. doi: 10.1128/MCB.23.7.2543-2555.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bovijn C, et al. Identification of interaction sites for dimerization and adapter recruitment in Toll/interleukin-1 receptor (TIR) domain of Toll-like receptor 4. J Biol Chem. 2012;287(6):4088–4098. doi: 10.1074/jbc.M111.282350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gautam JK, Ashish , Comeau LD, Krueger JK, Smith MF., Jr Structural and functional evidence for the role of the TLR2 DD loop in TLR1/TLR2 heterodimerization and signaling. J Biol Chem. 2006;281(40):30132–30142. doi: 10.1074/jbc.M602057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang Z, et al. Details of Toll-like receptor:adapter interaction revealed by germ-line mutagenesis. Proc Natl Acad Sci USA. 2006;103(29):10961–10966. doi: 10.1073/pnas.0603804103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cirl C, et al. Subversion of Toll-like receptor signaling by a unique family of bacterial Toll/interleukin-1 receptor domain-containing proteins. Nat Med. 2008;14(4):399–406. doi: 10.1038/nm1734. [DOI] [PubMed] [Google Scholar]
- 32.Radhakrishnan GK, Yu Q, Harms JS, Splitter GA. Brucella TIR domain-containing protein mimics properties of the Toll-like receptor adaptor protein TIRAP. J Biol Chem. 2009;284(15):9892–9898. doi: 10.1074/jbc.M805458200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salcedo SP, et al. Brucella control of dendritic cell maturation is dependent on the TIR-containing protein Btp1. PLoS Pathog. 2008;4(2):e21. doi: 10.1371/journal.ppat.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newman RM, Salunkhe P, Godzik A, Reed JC. Identification and characterization of a novel bacterial virulence factor that shares homology with mammalian Toll/interleukin-1 receptor family proteins. Infect Immun. 2006;74(1):594–601. doi: 10.1128/IAI.74.1.594-601.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Low LYL, Mukasa TT, Reed JCJ, Pascual JJ. Characterization of a TIR-like protein from Paracoccus denitrificans. Biochem Biophys Res Commun. 2007;356(2):481–6. doi: 10.1016/j.bbrc.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rana RR, et al. Yersinia pestis TIR-domain protein forms dimers that interact with the human adaptor protein MyD88. Microb Pathog. 2011;51(3):89–95. doi: 10.1016/j.micpath.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 37.Yadav M, et al. Inhibition of TIR domain signaling by TcpC: MyD88-dependent and independent effects on Escherichia coli virulence. PLoS Pathog. 2010;6(9):e1001120. doi: 10.1371/journal.ppat.1001120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cirl C, Miethke T. Microbial Toll/interleukin 1 receptor proteins: A new class of virulence factors. Int J Med Microbiol. 2010;300(6):396–401. doi: 10.1016/j.ijmm.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 39.Xiao TS. Subversion of innate immune signaling through molecular mimicry. J Clin Immunol. 2010;30(5):638–642. doi: 10.1007/s10875-010-9435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chaudhary A, et al. The Brucella TIR-like protein TcpB interacts with the death domain of MyD88. Biochem Biophys Res Commun. 2012;417(1):299–304. doi: 10.1016/j.bbrc.2011.11.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khor CC, et al. A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat Genet. 2007;39(4):523–528. doi: 10.1038/ng1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kormann MSD, et al. Rare TLR2 mutations reduce TLR2 receptor function and can increase atopy risk. Allergy. 2009;64(4):636–642. doi: 10.1111/j.1398-9995.2008.01891.x. [DOI] [PubMed] [Google Scholar]
- 43.Nada M, et al. Molecular analysis of the binding mode of Toll/interleukin-1 receptor (TIR) domain proteins during TLR2 signaling. Mol Immunol. 2012;52(3-4):108–116. doi: 10.1016/j.molimm.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 44.Into T, et al. Regulation of MyD88-dependent signaling events by S nitrosylation retards toll-like receptor signal transduction and initiation of acute-phase immune responses. Mol Cell Biol. 2008;28(4):1338–1347. doi: 10.1128/MCB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bochud P-Y, Hawn TR, Aderem A. Cutting edge: A Toll-like receptor 2 polymorphism that is associated with lepromatous leprosy is unable to mediate mycobacterial signaling. J Immunol. 2003;170(7):3451–3454. doi: 10.4049/jimmunol.170.7.3451. [DOI] [PubMed] [Google Scholar]
- 46.Kowalinski E, et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147(2):423–435. doi: 10.1016/j.cell.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 47.Jin T, et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity. 2012;36(4):561–571. doi: 10.1016/j.immuni.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loiarro M, et al. Pivotal advance: Inhibition of MyD88 dimerization and recruitment of IRAK1 and IRAK4 by a novel peptidomimetic compound. J Leukoc Biol. 2007;82(4):801–810. doi: 10.1189/jlb.1206746. [DOI] [PubMed] [Google Scholar]
- 49.Toshchakov VY, Fenton MJ, Vogel SN. Cutting edge: Differential inhibition of TLR signaling pathways by cell-permeable peptides representing BB loops of TLRs. J Immunol. 2007;178(5):2655–2660. doi: 10.4049/jimmunol.178.5.2655. [DOI] [PubMed] [Google Scholar]
- 50.Couture LA, Piao W, Ru LW, Vogel SN, Toshchakov VY. Targeting Toll-like receptor (TLR) signaling by Toll/interleukin-1 receptor (TIR) domain-containing adapter protein/MyD88 adapter-like (TIRAP/Mal)-derived decoy peptides. J Biol Chem. 2012;287(29):24641–24648. doi: 10.1074/jbc.M112.360925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tao X, Xu Y, Zheng Y, Beg AA, Tong L. An extensively associated dimer in the structure of the C713S mutant of the TIR domain of human TLR2. Biochem Biophys Res Commun. 2002;299(2):216–221. doi: 10.1016/s0006-291x(02)02581-0. [DOI] [PubMed] [Google Scholar]
- 52.Otwinowski Z, Minor W. Processing of X-ray diffraction data. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 53.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang H, et al. Automated and accurate deposition of structures solved by X-ray diffraction to the Protein Data Bank. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 10):1833–1839. doi: 10.1107/S0907444904019419. [DOI] [PubMed] [Google Scholar]
- 56.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.