

Significance
Here we used a visual blue opsin to induce asymmetric signaling in a cell. The opsin recruited endogenous G proteins and allowed immune cell migration to be optically steered with directional precision. Using this approach, cellular and molecular response dynamics were quantitated to facilitate computational modeling of migration. We identified an ultrasensitive switch-like signaling response that explains how immune cells filter background fluctuations in signals and respond decisively to persistent stimuli. This approach can be widely applied to understand G-protein–coupled receptor-stimulated signaling network control of other cell behaviors and potentially to control cell movements in whole animals.
Keywords: optogenetics, ultrasensitivity
Abstract
There is a dearth of approaches to experimentally direct cell migration by continuously varying signal input to a single cell, evoking all possible migratory responses and quantitatively monitoring the cellular and molecular response dynamics. Here we used a visual blue opsin to recruit the endogenous G-protein network that mediates immune cell migration. Specific optical inputs to this optical trigger of signaling helped steer migration in all possible directions with precision. Spectrally selective imaging was used to monitor cell-wide phosphatidylinositol (3,4,5)-triphosphate (PIP3), cytoskeletal, and cellular dynamics. A switch-like PIP3 increase at the cell front and a decrease at the back were identified, underlying the decisive migratory response. Migration was initiated at the rapidly increasing switch stage of PIP3 dynamics. This result explains how a migratory cell filters background fluctuations in the intensity of an extracellular signal but responds by initiating directionally sensitive migration to a persistent signal gradient across the cell. A two-compartment computational model incorporating a localized activator that is antagonistic to a diffusible inhibitor was able to simulate the switch-like PIP3 response. It was also able simulate the slow dissipation of PIP3 on signal termination. The ability to independently apply similar signaling inputs to single cells detected two cell populations with distinct thresholds for migration initiation. Overall the optical approach here can be applied to understand G-protein–coupled receptor network control of other cell behaviors.
A variety of cells sense gradients of chemoattractants and respond by migrating toward increasing concentrations. Migration is central to immune cell function, morphogenesis, cancer cell metastasis, and the life cycle of the social amoeba, Dictyostelium discoideum (1). Cell migration is made up of a characteristic sequence of identifiable cellular events that are governed by G-protein–coupled receptor (GPCR)-driven signaling networks. Although considerable information exists about molecules involved in migration, the challenge is in translating a static map of these molecules into a spatially and temporally dynamic network that orchestrates migratory behavior. Effective methods to probe the basis of network control of migration need to be able to faithfully evoke migratory behavior experimentally and quantitatively monitor response dynamics at the cellular and molecular level. Microfluidic devices and electrical fields have been used to regulate migration and provide insights into the process (2–6). However, there are limitations at present in the ability to direct a series of signaling inputs to a single cell in spatially and temporally complex patterns. Such inputs are essential to continually choreograph the events that constitute the migratory response: initiation, translocation, directional changes, and adaptation. A light-sensitive domain of a plant protein has been inserted into Rac1, a downstream signaling protein, and used to initiate migration (7). We have developed an approach, using members of the family of nonrhodopsin opsins that are light-sensing GPCRs to achieve spatially confined optical activation of signaling activity in a single cell [accompanying paper in PNAS (8)]. Here we use a color opsin from the human retina, to asymmetrically activate the native signaling network in an immune cell in its entirety. Because the protein is a receptor, the extracellular signal stimulated response of the major second messengers and resultant network dynamics can be studied. Furthermore, as the receptor senses the external light signal directly, precise directional control over migration can be exerted optically. The evoked responses recapitulate native migratory cell behavior.
This approach allows network control of migration to be interrogated continually in a single cell. The ability to examine single-cell responses quantitatively using the optical approach allowed us to address some long-standing questions. How does a cell filter background noise but initiate migration initiation decisively? Is there a threshold in the response of signaling molecules at which the cell decisively initiates migration (9)? Is there heterogeneity in network control of migratory behavior among single cells?
A number of models have been described to explain migratory response characteristics (10–12). However, there has been a limitation in methods to test computational models directly. Here, optical control allowed quantitative information on the cellular and molecular dynamics to be acquired during a series of migratory responses. This helped develop a computational model of the G-protein–mediated network that is known to regulate the internal guidance cue for migration, phosphatidylinositol (3,4,5)-triphosphate (PIP3) (1, 13). This mechanistic model accurately simulated the experimental results obtained through optical control.
Results
Asymmetric Optical Activation of Blue Opsin Initiates and Directs Immune Cell Migration Continually.
We used an optical approach to attain spatiotemporal control over single-cell behavior and simultaneously obtain quantitative information about second messenger dynamics in that cell. We examined whether asymmetric signaling in a single cell and resultant migratory behavior can be evoked using a visual opsin to activate endogenous G-protein pathways. Macrophage cell migration is evoked by asymmetric activation of Gi-coupled receptors across the cell (1, 13). We examined the capability of optical activation of human cone photoreceptor blue opsin (bOpsin) to activate the Gi pathway native to mouse macrophage RAW 264.7 cells.
The energy distribution of an optical input followed a Gaussian distribution (Fig. S1A). When the input was placed in front of a cell, a localized light gradient could be created in contrast to a molecular gradient (Fig. 1). Optical inputs were designed by varying the area of exposure, intensity, number of pulses, and intervals between pulses required to activate the opsin (Fig. 1B).
Fig. 1.
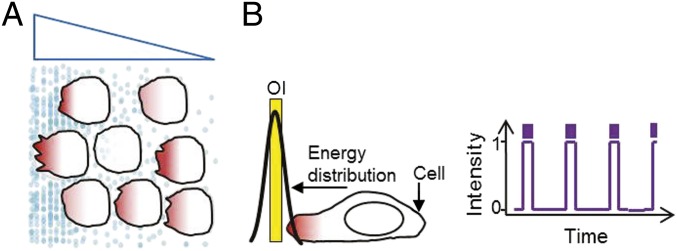
Migration induced by a diffusible gradient and by optical activation. (A) Cells migrate toward the higher concentration of chemoattractant molecules. Blue triangle depicts gradient. (B) Optical input location, area, intensity, number of pulses, and intervals between pulses are designed to evoke spatially asymmetric signaling activity.
We expressed human cone photoreceptor bOpsin in RAW 264.7 cells and examined the response of the cell to confined optical inputs (OIs) at 445 nm. The laser used to create the OI had a power of 5 μW at the image plane. We found that longer wavelengths used to excite fluorescent proteins in the red range mCherry (mCh) (595 nm) do not induce activation of bOpsin whereas medium wavelengths that excite GFP (488 nm excitation) and YFP (515 nm excitation) have minimal potential to do so. This spectral selectivity facilitated the imaging of cellular and molecular changes that occurred during bOpsin activation.
We examined the response of a mouse macrophage RAW 264.7 cell expressing bOpsin-mCh to confined optical inputs. Cells were imaged for mCh (595 nm excitation/630 nm emission) continuously. Fig. 2 and Movie S1 show that cells responded initially to an optical input applied every 5 s (red box) with a protrusion of the cell body in the direction of light. This was followed by lamellipodia formation toward the stimuli. We continually relocated the optical signal to direct the migration. The ability to apply spatially discrete multiple optical inputs allowed independent control of two different cells simultaneously (Movie S1 and Fig. 2: 23:07). The plot shows continuous synchronized response of the cell front and back to the movement of the optical input. Together, these results demonstrated that optical gradients could mimic chemoattractants but with more precise spatial and temporal control at the single-cell level.
Fig. 2.

Optical control of directional cell migration. Confined OIs (red box) pulsing every 5 s continually activate bOpsin-mCh and direct RAW 264.7 cell migration. Representative result (n > 40) is shown. Plot shows tracking of the leading edge (black), the trailing edge (blue), and OI (red) along the x axis. OIs were repositioned as the cells migrated, to maintain an approximately constant distance between the OI and the leading edge.
Direction of Immune Cell Migration Can Be Controlled Optically.
Because OI can be localized to any position on the surface of a cell, we examined whether the direction of migration can be controlled entirely by the location of the OI with reference to the cell. We found that switching the optical signal to the back of a migrating cell resulted in synchronized lamellipodia initiation at the back and retraction at the front (Fig. 3A and Movie S2). The cell then began to move in the reverse direction. Differential interference contrast (DIC) images of cells during a similar experiment are shown in Fig. 3 B and C. DIC images show more defined lamellipodia and the results show the rapid retraction of lamellipodia at the original front and the appearance of new lamellipodia at the newly created front once optical input is switched. When we tracked the migration of a cell that reversed its direction with reference to optical input, it was clear that the cell migrated precisely along the optical input trajectory and the response persisted as long as the signal persisted (Fig. 3C). A strong correlation between optical input and cell trajectories reveals precise directional sensing ability of immune cells (Fig. S1B). There is no apparent difference between forward and reverse average migration velocities (∼5 µm/min, Fig. S1C). The cells show a delay of ∼100 s to move in the reverse direction, suggesting that this may be the time taken to break down and reestablish polarity entirely.
Fig. 3.

Optically induced reversal of migration. (A) Confocal fluorescence images of a RAW 264.7 cell expressing bOpsin-mCh. Images were taken at 5-s intervals before and during optical activation. OIs were applied every 5 s. (B) DIC images of reversal of migration by optical activation of bOpsin (white box). Yellow arrow: lamellipodia appearance during initial activation. Orange arrows: retraction of the lamellipodia. Green arrow: initiation of lamellipodia appearance at new front. (C) Representative plot tracking the optical input (OI) and center of cell (x axis) (n = 10). Migration events are shown above, corresponding to different regions of the plot. OIs were repositioned as the cells migrated, to maintain an approximately constant distance between the OI and the leading edge.
Similarly, when a cell was activated sequentially in four opposing directions, the cell consistently responded with a migratory response toward the OI in each direction (Movie S3). By applying two independent OIs simultaneously to individual cells attached to each other, it was possible to direct their migration in opposing directions away from each other (Movie S4). These results demonstrate that optical control can sequentially induce the migration of a single cell in any chosen direction and it can control the migration of multiple cells in the field of vision simultaneously in any chosen direction (Fig. 2: 23:07). Potentially a single cell can be subjected to multiple inputs in different directions to examine the effect of competing gradients.
Molecular Responses Can Be Imaged During Optically Induced Migration.
We examined whether signaling molecules responded to light activation similarly to chemoattractants. Before migration initiation, an optical input evoked an increase in PIP3 at the front of a cell expressing bOpsin-mCh and PIP3 sensor Akt-PH-GFP (Fig. 4A and Movie S5) similar to that seen with chemoattractants (1). Migration of cells in response to chemoattractants is known to be mediated by Rac (13). When we introduced a dominant-negative Rac (T17N) (14) or wild-type Rac into bOpsin-expressing RAW cells, optical activation (OA)-induced migration was severely retarded by Rac (T17N) but not by Rac wild type (WT). This result showed that optically activated migration is mediated by a Rac-mediated pathway similar to chemoattractants. We then quantified the real-time actin dynamics during continued migration induced by confined optical activation of bOpsin-mCh and GFP-β-actin coexpressing cells. When migration was initiated, localized actin remodeling was detected at the front of the cell (Fig. 4B and Movie S6). This remodeling occurs on a similar timescale to that of PIP3 at the front of a cell (Fig. 4B, Right). Together these results suggest that a PIP3-Rac-actin pathway mediates optically induced migration similar to that in response to chemoattractants (Fig. 4C).
Fig. 4.

Monitoring secondary messenger activity during optically induced cell migration. (A) Confocal images of GFP fluorescence in RAW 264.7 cells expressing bOpsin-mCh and PH-Akt-GFP (PIP3 sensor). Images were taken at 5-s intervals before and during optical activation. OIs (green box) were applied every 5 s. (B) Confocal overlay images of a RAW cell transfected with bOpsin-mCh and GFP-β actin before and during optically induced migration. Plot shows actin accumulation dynamics proximal to the OI (n = 5). OIs (green solid box) were repositioned as the cells migrated, to maintain an approximately constant distance between the OI and the cell’s leading edge. Plot shows β-actin dynamics at the cell front. Responses are normalized from zero to one (n = 3, error bars: SD). (C) Schematic of signaling molecules involved in cell migration.
Optical Control Identifies an Enhanced Front-to-Back PIP3 Gradient Underlying Migration Initiation.
Modeling has suggested that the capability of migrating cells to sense very shallow gradients requires them to form steep internal gradients (15, 16). PIP3 is a candidate molecule for being this cue because PI3K activity is known to be stimulated at the leading edge of a migrating cell, resulting in localized PIP3 increase (17). However, in the absence of approaches that can introduce precise experimental control over directional cell migration, the relationship between the PIP3 response across a single cell and cell behavior has been difficult to examine. The ability to rapidly induce directional changes in cell migration using controlled optical functions allowed us to examine whether we could obtain direct evidence for a dynamic steep PIP3 gradient that mediates cell migration.
A RAW cell coexpressing bOpsin-mCh and a PIP3 sensor Akt-PH-GFP was continuously imaged before and during sequential optical activation of the front and the back (Fig. 5A). The cell was initially polarized randomly with PIP3 patches distributed along the cell periphery. On optical activation, PIP3 increased rapidly at the activated end, creating the leading edge (Fig. 5 A and B). Importantly, PIP3 levels decreased at the back of the cell (Fig. 5 B and C). Optical activation also resulted in lamellipodia formation and migration (Movie S7). Switching of the optical gradient resulted in rapid PIP3 gradient reversal (Fig. 5 A and B). We used repeatability of an identical optical input to the same cell to evoke a subsequent reversal cycle. This recapitulated the directionality of the PIP3 gradient change. These directional changes in the PIP3 gradient were associated with changes in migration direction.
Fig. 5.
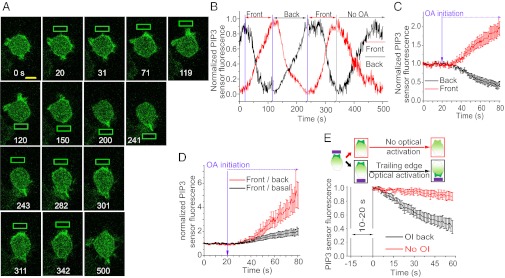
Monitoring PIP3 and cell migration dynamics during optically induced cell migration initiation and reversal. (A) Confocal images of GFP fluorescence in a RAW 264.7 cell expressing bOpsin-mCh and PH-Akt-GFP(PIP3 sensor). Images were taken at 1-s intervals before and during optical activation. OIs (green box) were applied every 1 s. The optical input after inducing PIP3 response at the front was switched to the back and the PIP3 gradient monitored. Green box: optical input. (B) Plot shows the normalized PIP3 sensor fluorescence intensity changes in the front (red) and the back (black). Vertical lines (purple) denote when the OI is switched in location. Vertical line (black) shows termination of optical activation. In each activation cycle individual front and back PIP3 responses are normalized from zero to one. (C) Baseline normalized front (red) and back (black) PIP3 sensor fluorescence during optical activation of front. (D) PIP3 sensor fluorescence front/back gradient (red) and front/basal gradient during optically induced migration initiation (n = 5). (E) Active and passive PIP3 breakdown at the optically activated cell front. Cartoons represent the experimental design, status of the cell, and location of the optical input. Plots show the corresponding PIP3 sensor mean fluorescence intensity at the front after terminating the OI (red, n = 6) or relocating the OI to the back of the cell (black, n = 4). Time = 0 is the point at which fluorescence starts to decrease. Error bars: Mean ± SEM.
The spatiotemporal dynamics of PIP3 in a single immune cell undergoing migration initiation, maintenance, and reversal have not been examined in real time. PIP3 increase at the front of an immune cell has previously been observed in response to a chemoattractant gradient (18). However, rapid concomitant decrease in PIP3 at the back has not been detected. Results show that decrease at the back increases the steepness of the front-to-back PIP3 gradient (Fig. 5D). There is an approximately fivefold higher rate of PIP3 degradation when optical activation is switched to the prior back of the cell compared with passive loss in the absence of the optical signal (Fig. 5E). This result suggests that activation of signaling in the front of the cell is communicated rapidly to the back through an unidentified mechanism that enhances PIP3 decrease. In contrast, in the absence of activation of the front, decrease in PIP3 at the back is likely due to passive basal PIP3 degradative mechanisms.
PIP3 Dynamics Across the Cell Are Switch-Like and Migration Initiation Occurs Midway Through the Sigmoidal PIP3 Response in Two Distinct Populations of Cells.
Systems-level analysis of a single migrating cell is difficult using present methods because it requires systematic control over the input over time across a cell while simultaneously imaging cellular and molecular responses. This makes it challenging to study signaling dynamics and quantify systems-level properties such as sensitivity in terms of the rapidity of the response and activation threshold for migration initiation. The ability to control migration with light pulses allowed us to quantify the PIP3 dynamics and migratory response in a single cell (Fig. 6A).
Fig. 6.

Single-cell PIP3 responses during optically induced migration initiation. (A) Schematic of the optical approach to classify a cell population on the basis of network properties of individual cells. Optical activation allows the molecular and migratory responses of a single cell to be followed while it is continually activated asymmetrically. The ability to induce migration with similar inputs using the optical method allows differences in responsiveness between individual cells to be identified. (B) Experimentally observed PIP3 responses at the cell front to increasing number of light pulses in individual cells (n = 18). Jagged lines, experimental data; solid lines, Hill fit [Hill coefficients (nH) varying from 2.6 to 7.7]. (C) Experimentally observed individual cell levels of enhancement of the PIP3 gradient (n = 23). Only cells with normalized PIP3 response A > 2 migrate. (D) Grouping of cells based on number of light pulses required for migration initiation shows diversity in activation threshold (K). A statistical distribution analysis was used to identify the groups (cut point = 46). Error bars: Mean ± SEM.
We applied identical continuous light pulses at every second to a confined region of a single cell to initiate cell migration and monitored the PIP3 response in 23 cells (Fig. 6B). We examined the dynamic response of PIP3 in the front of each of these cells during migration initiation. We fitted the PIP3 response to the Hill equation to quantify the sensitivity (Hill coefficients, nH ∼ 3 to ∼8) of individual cells (Fig. 6B). The values for the Hill coefficients suggest that the PIP3 response is highly ultrasensitive (19). Such switch-like dynamics help a cell make a dedicated all-or-none decision in response to changing environmental conditions as in the MAPK cascade mediation of oocyte maturation (20). The PIP3 response detected here is thus consistent with the decisive nature of migration initiation that shifts the cell from one state to another.
The ability of the optical method here to continuously provide identical inputs to multiple cells helped analyze the PIP3 response curves and the corresponding enhancement of the PIP3 gradient (A) in a heterogeneous cell population. We found that some cells showed a migratory response whereas others did not. Enhancement of the PIP3 gradient varied widely among responsive cells. None of the nonresponsive cells demonstrated significant enhancement of the PIP3 gradient (Fig. 6C). These results suggest that the PIP3 response has to cross a threshold (A = ∼2) to initiate the migratory response.
To characterize the cell-to-cell variation in a population we used the optical approach to examine the relationship between the activation threshold for PIP3 response (K, half-maximal constant from the Hill fit) and migration initiation (Nstart) in single cells. When 23 cells were examined, 18 cells showed a PIP3 and migratory response whereas 5 showed only the PIP3 response. The very low-intensity light of wavelengths in the visual spectrum used here does not have any damaging effects on cells. The nonresponsive cells are categorized as such only because they do not demonstrate obvious migration: extension of the leading edge and retraction of the trailing edge. PIP3 increase at the front of these cells demonstrates that the nonresponsive cells are viable.
Population distribution analysis showed that these cells could be grouped into two distinct pools of cells on the basis of migration initiation and the PIP3 response (Fig. S2A). Early migrants (Fig. 6D, blue, K = 37) required fewer stimuli for the initiation of migration and late migrants (Fig. 6D, red, K = 67) required more stimuli. However, there was no relationship between the expression level of the opsin receptor and the response, suggesting that the differences were due to intrinsic network properties of the cells and not receptor concentration.
Strikingly, migration initiation occurred in almost all of the cells close to the activation threshold for the PIP3 response, showing that migration is initiated at the switch-like region of the PIP3 increase at the front (Fig. 6D shows the population responses, Fig. S2B shows individual cell responses, and Table S1 shows the systems parameters for the PIP3 responses).
The differential tuning of migration initiation at the switch-like region of the PIP3 response in individual cells in a population further suggests that PIP3 is a critical second messenger mediator of macrophage cell migration. Overall these results show that the optical approach here allows single-cell analysis of signaling network properties that control cell migration.
Computational Modeling of PIP3 Dynamics During Migration Initiation.
The ability to confine activation of receptor and coupled G proteins to one edge of a cell allowed us to develop a two-compartment ordinary differential equation model for PIP3 accumulation in immune cells that is mediated by asymmetric GPCR activation. The model assumed the basic framework of the local excitation, global inhibition (LEGI) mechanism (21–23), which explains the ability of polarization of the second messenger across the cell during an asymmetric input stimulus. Here, we introduced an inhibition-activation mechanism by introducing antagonism between the inhibitor and the activator at the membrane as shown in Fig. 7A. The antagonistic mechanism proposed here is consistent with the balanced inactivation model proposed earlier (11, 24). The model thus comprises an incoherent feed-forward loop with faster activation kinetics of a membrane localized activator and slower recruitment kinetics of an inhibitor to the membrane from the cytosol (6, 12). Fig. 7A shows the reaction schematic used for the formulation of the model.
Fig. 7.
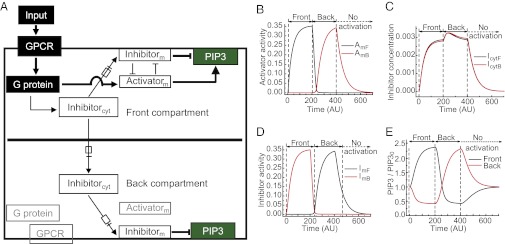
Two-compartment model for immune cell migration. (A) Simplified schematic representation of the biomolecular pathway at the cell front and back describing PIP3 regulation during immune cell migration. Arrows with a box indicate either recruitment to the membrane or translocation from front to back compartment. cyt, cytosolic; m, membrane bound. GPCR, G protein, and the activator in the back compartment are inactive and presented in gray. (B) Simulated activator dynamics in the front (AmF) and back (AmB) compartments of a single cell in response to switching the localized activation from front to back. (C) Simulated cytosolic inhibitor dynamics in the front (IytF) and back (IcytB) during corresponding activation switching. (D) Simulated membrane recruited inhibitor dynamics in the front (ImF) and back (ImB) during corresponding activation switching. (E) PIP3 dynamics at cell front and back controlled by the activity of activator (B) and inhibitor (D) activity.
When the signaling input to the cell is spatially confined to the front compartment, both the activator and the inhibitor are activated. The activator enhances PIP3 synthesis and the inhibitor decreases PIP3 levels. The activator and inhibitor act antagonistically on each other (Fig. 7A). The activator is confined to the plasma membrane whereas the diffusible inhibitor is present in both the membrane and the cytosol. Activation of the inhibitor leads to its membrane recruitment. The cytosolic inhibitor is capable of free exchange between the two compartments, whereas the activator and the membrane-bound inhibitor remain localized. The activator and recruited inhibitor act on PIP3 in two distinct compartments. We have made the assumption that all of the reacting species in the two compartments are well mixed individually. It should be noted that the inhibitor in the cytosol and that in the membrane are distinct variables.
The G-protein activation rate is directly proportional to the stimulus (S). The free G-protein concentration and deactivation of G protein follow first-order reaction kinetics. The total G-protein concentration (GT) is constant in each compartment.
The rate of accumulation of the activated G protein in the front compartment is
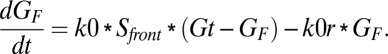 |
The rate of formation of the activator is assumed to follow Michaelis–Menten kinetics, the natural deactivation is assumed as first-order kinetics, and deactivation due to antagonism follows second-order kinetics involving both inhibitor and activator. The rate of accumulation of activator is
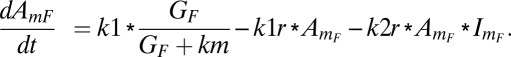 |
The rate of formation of inhibitor in the cytosol is assumed to follow Michaelis–Menten kinetics. Further, the inhibitor is recruited to the membrane with first-order kinetics. Moreover, the cytosolic inhibitor diffuses to the other compartment on the basis of its concentration gradient. The rate of accumulation of the inhibitor in the cytosol in the front compartment is
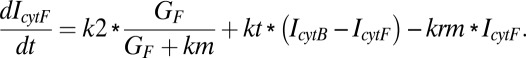 |
The accumulation rate of the membrane recruited inhibitor accounts for its rate of recruitment, the natural deactivation, and its antagonism with the activator:
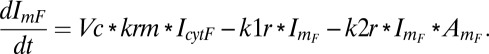 |
The formation of PIP3 is first order with respect to activator, whereas the disappearance of PIP3 depends on both the inhibitor and its own concentration in the front compartment. The accumulation rate of PIP3 is
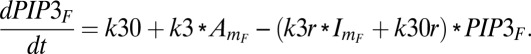 |
Similarly, equations for the back compartment are as follows (descriptions of model variables, model parameters, and initial conditions used in the study are shown in Tables S2–S4):
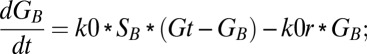 |
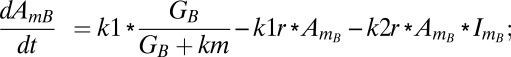 |
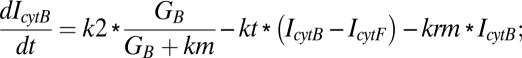 |
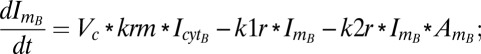 |
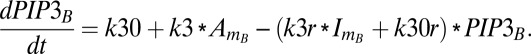 |
Fig. 7 B–D shows the simulated dynamic behavior of the activator and inhibitor. The front compartment of a cell is activated first (0 s), followed by the back compartment at 200 s, and then signal input is terminated at 400 s. In the front compartment, both the activator and the cytosolic inhibitors increase. The rapid diffusion increases the concentration of the cytosolic inhibitor in both compartments. The cytosolic inhibitor gets recruited at the membrane in both compartments at an equal rate. However, the lack of G-protein activation and consequent absence of activator at the back result in increasing inhibitor activity in the membrane at the back. At the front, the presence of activator at the membrane keeps the membrane-bound inhibitor activity low. This localization of activator and inhibitor activity in the front and back compartments is mainly due to the slower recruitment of the inhibitor to the membrane, deactivation of the inhibitor at the membrane front due to antagonism of the activator, and, finally, rapid diffusion of the cytosolic inhibitor between compartments. The polarization of the activator and inhibitor between the two compartments results in a PIP3 gradient.
When signal input is switched to the back, G-protein activation induces the activator at the back, leading to deactivation of inhibitor at the back (Fig. 7 B and D). The cytosolic inhibitor concentration at the back increases due to activation at this end (Fig. 7C). In the front compartment, the reduction in G-protein concentration reduces both activator and inhibitor. However, the rapid diffusion of cytosolic inhibitor from back to front ensures that recruitment of inhibitor at the front is maintained (Fig. 7C). Thus, the localized activity of activator and inhibitor gets reversed rapidly.
Termination of input at 400 s leads to the rapid decrease in the activator concentration at the “back” (Fig. 7B). However, in the absence of further switching of the optical signal to the opposite end of the cell, increased recruitment of the inhibitor to the back does not occur (Fig. 7 C and D). Thus, the dissipation of the PIP3 gradient occurs at a rate that is slower than that in the presence of the asymmetric input (Fig. 7E, black curve from 200 to 400 s vs. red curve from 400 to 650 s).
The model ensures localization of PIP3 decrease to the back compartment after initial activation and rapid PIP3 dynamics during reversal of input from front to back. It further accounts for the slow PIP3 decay during switch-off. Overall, the simulation thus captures the kinetics of PIP3 changes across the cell observed when executing the experimental paradigm (Fig. 6), thus validating the model.
Discussion
Considerable evidence from different approaches supports a role for PIP3 in normal migration of immune cells and Dictyostelium cells (1, 25–27). However, questions remain as to how PIP3 dynamically acts as an internal cue for migratory behaviors. Here we identify network-level properties governing PIP3 formation that underlie the cell’s ability to initiate migration and to alter direction, using an optical approach. This approach helped quantitate the relationship between signal input and the behavioral and spatially complex signaling output of a single cell in real time. The data generated using optical control were used to build a mechanistic model.
The PIP3 changes evoked by a continuous asymmetric signal are characterized by a sigmoidal response that, after an initial lag, allows a large output to a relatively small input (19, 28–30). The switch-like properties of an ultrasensitive response are well suited to mediate decisive changes in cellular states as in the case of the cell cycle (19). A typical Michaelis–Menten response (nH = 1) requires an 81-fold change in the input to bring about an output response from 10% to 90% activation. However, only a two- to fourfold change in input was required for this change in PIP3 production during migration initiation. Furthermore, the PIP3 gradient was enhanced in a switch-like manner by the decrease in PIP3 concentration at the back of the cell. Consistent with this switch-like behavior playing a role in the migratory decision, migration initiation occurs midway through the switch.
It is known that migratory cells can filter out background fluctuations in a signal (31) and demonstrate decisive state changes when encountering a chemoattractant gradient. The mechanistic bases for these properties have not been clear. The switch-like behavior of the PIP3 gradient provides a parsimonious mechanism that enhances the gradient, ensures that background noise is filtered, and allows initiation of migration to be decisive.
Optical control allows the extracellular input to be switched on or off and its location to be altered almost instantaneously. This facilitated migration to be redirected repeatedly and activation to be terminated rapidly. Because the optical signal emanates from the microscope objective, the approach also helps retain the cell within the field while continuously monitoring cellular and molecular response dynamics throughout the course of the experiment.
This approach is especially useful to address two-compartment models as demonstrated here. When the optical signal is reversed, local activation of a compartment overcomes the inhibition of PIP3 production and inhibition at the back ensures that PIP3 at the original front decays, creating a rapid gradient reversal. This is accompanied by reversal of cell migratory behavior. The dual action of an activator and inhibitor on the regulation of PIP3 provides flexibility in the tuning of dynamic switch-like responses (30). The consistency between the modeling and experimental results suggests that the presence of an activator and inhibitor with the properties suggested here can be pursued.
Cell populations have been traditionally studied to obtain valuable insights into the mechanistic basis of biological function. Single cells within a population are, however, likely to differ in their properties. Single-cell analysis so far has largely concentrated on transcripts, genes, secreted molecules, and metabolites (32–35). Differences between single cells in signaling network properties have been less studied. Information about such heterogeneity can help us achieve a better understanding of the mechanisms underlying cell function (36). Identifying network differences between cells may also help us evolve better strategies for personalized therapeutics.
It is difficult to quantify the network properties in a single living cell and classify a heterogeneous cell population on the basis of systems characteristics that govern cellular behavior such as migration (37). Ultrasensitivity in the cell cycle and in regulation by small GTPases has previously been identified in cell populations, using cell disruptive methods (30, 38). The approach here helps quantify network dynamics in a single living cell and identify systems-level properties that can otherwise be masked by ensemble measurements from a population of cells. This can suggest whether network control and the resultant cellular response vary probabilistically across cells or are deterministic, falling into specific classes. Predetermined variation among cells may form the basis of specific population response capabilities. Intrinsic differences in the signaling properties between cells can provide natural variants that can be used to identify the molecular basis of cell behavior. The results here distinguish immune cells on the basis of distinct differences in their PIP3 and migratory responsiveness to an extracellular signal. This result suggests again that the PIP3 gradient formation is central to the process of migration initiation. Variation in responsiveness among cells may ensure immune cell responses to a broader range of stimulus intensities.
We demonstrate here the ability to use a visual opsin to recruit endogenous signaling pathways and optically orchestrate complex cell behavior. The use of light as the stimulus affords high spatial and temporal resolution at subcellular levels over the signaling pathway that controls the behavior. The optical approach developed here can be applied to understand the basis of GPCR-stimulated signaling network control of other cell behaviors. This approach can also be developed to optically instruct migratory behaviors such as morphogenetic or tumor cell movements in whole animals because these are known to be controlled by asymmetric GPCR activity.
Methods
Cell Culture and Transfections.
Early passage RAW 264.7 cells (Tissue Culture Support Center at Washington University) were grown in DMEM with 10% (vol/vol) dialyzed FBS, antibiotics, and l-glutamine (2 mM). They were seeded at a density of 0.1 × 106 cells in 29-mm glass-bottom dishes and transfected the same day. Each 29-mm dish was transfected using 4 µL lipofectamine 2000 as per manufacturer’s protocol. Cells were incubated 4.5–5 h with transfection medium (Opti-MEM Reduced Serum Medium, lipofectamine, and ∼1 µg of each cDNA) and then replaced with regular medium. Cells were imaged after 16 h for optical activation. Before imaging, dishes were transferred to an incubator in a dark room and 11-cis retinal was added to the medium (3 ng/mL). After incubation with 11-cis retinal for 30 min, the medium was replaced with imaging buffer, Hanks’ balanced salt solution (HBSS) (with 1 g/L glucose prewarmed to 37 °C).
Confined Optical Activation of RAW Cells and Global Imaging of Response Dynamics.
Using a 63× oil objective, cells were focused to the appropriate confocal plane using 488-, 515-, or 595-nm wavelengths and the confocal plane was locked using Adaptive Focus Control (Leica) to prevent focal plane drift during cell migration. A detail of the imaging setup is described in SI Methods. Basal levels and changes in fluorescently tagged opsin distribution and fluorescence sensors (for second messengers) were imaged using laser beams with the following specifications: GFP, 1.2 µW/cm2, 10–50 ms; and mCh, 12.5 µW/cm2, 10–50 ms. These intensities and exposure conditions induce minimal or no blue opsin activation. A 445-nm laser with the intensity of 5 µW at the image plane was used to create versatile space and time variant optical inputs. During confined optical activation, a dual galvo scan head fluorescence recovery after photobleaching and photoactivation (FRAPPA) unit that bypasses the confocal scanner unit (CSU) was used to create the desired optical inputs.
Calculation of Cell and Optical Input Trajectories.
The trajectory of the optical input and coordinates of the migrating cells were tracked using the video analysis and modeling tool “Tracker” from the Open Source Physics Project.
Quantification of System Parameters in Single Cells.
Sensitivity and activation threshold.
Experimental PIP3 data obtained from optical activation of immune cells were fitted to a three-parameter Hill equation, 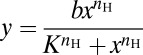 , where nH is the Hill coefficient or sensitivity and K is the half-maximal constant or activation threshold. Because different cells had different values of basal and peak PIP3 sensor fluorescence (on the membrane) and their peak response was reached after varying numbers of light pulses, we normalized the PIP3 response as the fractional change in PIP3 fluorescence (change in PIP3 sensor fluorescence from the basal level divided by peak PIP3 sensor fluorescence minus basal-level PIP3 sensor fluorescence). Alternatively, when combining data from different experiments and plotting a common curve for them, we used four-parameter Hill equations (18),
, where nH is the Hill coefficient or sensitivity and K is the half-maximal constant or activation threshold. Because different cells had different values of basal and peak PIP3 sensor fluorescence (on the membrane) and their peak response was reached after varying numbers of light pulses, we normalized the PIP3 response as the fractional change in PIP3 fluorescence (change in PIP3 sensor fluorescence from the basal level divided by peak PIP3 sensor fluorescence minus basal-level PIP3 sensor fluorescence). Alternatively, when combining data from different experiments and plotting a common curve for them, we used four-parameter Hill equations (18), 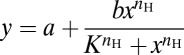 . This eliminated the possibility of underestimating sensitivity due to averaging curves.
. This eliminated the possibility of underestimating sensitivity due to averaging curves.
Factor for the enhancement of the PIP3 gradient.
We analyzed the enhancement of the PIP3 gradient, where the enhancement factor was calculated as 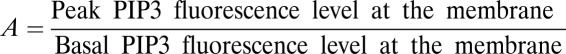 from the experimental PIP3 response curves. In these plots, the PIP3 response is normalized to the maximum PIP3 response obtained in 23 cells and the input level is normalized to the total number of light pulses required for maximal PIP3 response and termed fractional activation.
from the experimental PIP3 response curves. In these plots, the PIP3 response is normalized to the maximum PIP3 response obtained in 23 cells and the input level is normalized to the total number of light pulses required for maximal PIP3 response and termed fractional activation.
Supplementary Material
Acknowledgments
We thank P. O’Neill and V. Kalyanaraman for discussions. We thank P. Nanda and Andor Technology for customizing Andor IQ software; D. Oprian for cDNAs; and the National Eye Institute, the National Institutes of Health (NIH), and R. Crouch for 11-cis retinal. This work was supported by NIH Grants GM069027 and GM080558 (to N.G.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1220755110/-/DCSupplemental.
References
- 1.Cai H, Devreotes PN. Moving in the right direction: How eukaryotic cells migrate along chemical gradients. Semin Cell Dev Biol. 2011;22(8):834–841. doi: 10.1016/j.semcdb.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ambravaneswaran V, Wong IY, Aranyosi AJ, Toner M, Irimia D. 2010. Directional decisions during neutrophil chemotaxis inside bifurcating channels. Integr Biol 2(11–12):639–647. [DOI] [PMC free article] [PubMed]
- 3.Skoge M, et al. 2010. Gradient sensing in defined chemotactic fields. Integr Biol 2(11–12):659–668.
- 4.Li J, Lin F. Microfluidic devices for studying chemotaxis and electrotaxis. Trends Cell Biol. 2011;21(8):489–497. doi: 10.1016/j.tcb.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 5.Lin B, et al. Synthetic spatially graded Rac activation drives cell polarization and movement. Proc Natl Acad Sci USA. 2012;109(52):E3668–E3677. doi: 10.1073/pnas.1210295109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takeda K, et al. Incoherent feedforward control governs adaptation of activated ras in a eukaryotic chemotaxis pathway. Sci Signal. 2012;5(205):ra2. doi: 10.1126/scisignal.2002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu YI, et al. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature. 2009;461(7260):104–108. doi: 10.1038/nature08241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Karunarathne WKA, Giri L, Kalyanaraman V, Gautam N (2013) Optically triggering spatiotemporally confined GPCR activity in a cell and programming neurite initiation and extension. Proc Natl Acad Sci USA 110:E1565–E1574. [DOI] [PMC free article] [PubMed]
- 9.Janetopoulos C, Ma L, Devreotes PN, Iglesias PA. Chemoattractant-induced phosphatidylinositol 3,4,5-trisphosphate accumulation is spatially amplified and adapts, independent of the actin cytoskeleton. Proc Natl Acad Sci USA. 2004;101(24):8951–8956. doi: 10.1073/pnas.0402152101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jilkine A, Edelstein-Keshet L. A comparison of mathematical models for polarization of single eukaryotic cells in response to guided cues. PLOS Comput Biol. 2011;7(4):e1001121. doi: 10.1371/journal.pcbi.1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iglesias PA, Devreotes PN. Biased excitable networks: How cells direct motion in response to gradients. Curr Opin Cell Biol. 2012;24(2):245–253. doi: 10.1016/j.ceb.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levine H, Kessler DA, Rappel WJ. Directional sensing in eukaryotic chemotaxis: A balanced inactivation model. Proc Natl Acad Sci USA. 2006;103(26):9761–9766. doi: 10.1073/pnas.0601302103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang F. The signaling mechanisms underlying cell polarity and chemotaxis. Cold Spring Harb Perspect Biol. 2009;1(4):a002980. doi: 10.1101/cshperspect.a002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardiner EM, et al. Spatial and temporal analysis of Rac activation during live neutrophil chemotaxis. Curr Biol. 2002;12(23):2029–2034. doi: 10.1016/s0960-9822(02)01334-9. [DOI] [PubMed] [Google Scholar]
- 15.Chou CS, Bardwell L, Nie Q, Yi TM. Noise filtering tradeoffs in spatial gradient sensing and cell polarization response. BMC Syst Biol. 2011;5:196. doi: 10.1186/1752-0509-5-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Devreotes PN, Zigmond SH. Chemotaxis in eukaryotic cells: A focus on leukocytes and Dictyostelium. Annu Rev Cell Biol. 1988;4:649–686. doi: 10.1146/annurev.cb.04.110188.003245. [DOI] [PubMed] [Google Scholar]
- 17.Devreotes P, Janetopoulos C. Eukaryotic chemotaxis: Distinctions between directional sensing and polarization. J Biol Chem. 2003;278(23):20445–20448. doi: 10.1074/jbc.R300010200. [DOI] [PubMed] [Google Scholar]
- 18.Servant G, et al. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 2000;287(5455):1037–1040. doi: 10.1126/science.287.5455.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trunnell NB, Poon AC, Kim SY, Ferrell JE., Jr Ultrasensitivity in the regulation of Cdc25C by Cdk1. Mol Cell. 2011;41(3):263–274. doi: 10.1016/j.molcel.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrell JE, Jr, Machleder EM. The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science. 1998;280(5365):895–898. doi: 10.1126/science.280.5365.895. [DOI] [PubMed] [Google Scholar]
- 21.Ma L, Janetopoulos C, Yang L, Devreotes PN, Iglesias PA. Two complementary, local excitation, global inhibition mechanisms acting in parallel can explain the chemoattractant-induced regulation of PI(3,4,5)P3 response in dictyostelium cells. Biophys J. 2004;87(6):3764–3774. doi: 10.1529/biophysj.104.045484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiong Y, Huang CH, Iglesias PA, Devreotes PN. Cells navigate with a local-excitation, global-inhibition-biased excitable network. Proc Natl Acad Sci USA. 2010;107(40):17079–17086. doi: 10.1073/pnas.1011271107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levchenko A, Iglesias PA. Models of eukaryotic gradient sensing: Application to chemotaxis of amoebae and neutrophils. Biophys J. 2002;82(1 Pt 1):50–63. doi: 10.1016/S0006-3495(02)75373-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rappel WJ, Levine H. Receptor noise limitations on chemotactic sensing. Proc Natl Acad Sci USA. 2008;105(49):19270–19275. doi: 10.1073/pnas.0804702105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirsch E, et al. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 2000;287(5455):1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- 26.Sasaki T, et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287(5455):1040–1046. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- 27.Rickert P, Weiner OD, Wang F, Bourne HR, Servant G. Leukocytes navigate by compass: Roles of PI3Kgamma and its lipid products. Trends Cell Biol. 2000;10(11):466–473. doi: 10.1016/s0962-8924(00)01841-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giri L, Mutalik VK, Venkatesh KV. A steady state analysis indicates that negative feedback regulation of PTP1B by Akt elicits bistability in insulin-stimulated GLUT4 translocation. Theor Biol Med Model. 2004;1:2. doi: 10.1186/1742-4682-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldbeter A, Koshland DE., Jr An amplified sensitivity arising from covalent modification in biological systems. Proc Natl Acad Sci USA. 1981;78(11):6840–6844. doi: 10.1073/pnas.78.11.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lipshtat A, Jayaraman G, He JC, Iyengar R. Design of versatile biochemical switches that respond to amplitude, duration, and spatial cues. Proc Natl Acad Sci USA. 2010;107(3):1247–1252. doi: 10.1073/pnas.0908647107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tranquillo RT, Lauffenburger DA, Zigmond SH. A stochastic model for leukocyte random motility and chemotaxis based on receptor binding fluctuations. J Cell Biol. 1988;106(2):303–309. doi: 10.1083/jcb.106.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rubakhin SS, Romanova EV, Nemes P, Sweedler JV. Profiling metabolites and peptides in single cells. Nat Methods. 2011;8(4, Suppl):S20–S29. doi: 10.1038/nmeth.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang F, Lao K, Surani MA. Development and applications of single-cell transcriptome analysis. Nat Methods. 2011;8(4, Suppl):S6–S11. doi: 10.1038/nmeth.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fritzsch FS, Dusny C, Frick O, Schmid A. Single-cell analysis in biotechnology, systems biology, and biocatalysis. Annu Rev Chem Biomol Eng. 2012;3:129–155. doi: 10.1146/annurev-chembioeng-062011-081056. [DOI] [PubMed] [Google Scholar]
- 35.Itzkovitz S, van Oudenaarden A. Validating transcripts with probes and imaging technology. Nat Methods. 2011;8(4, Suppl):S12–S19. doi: 10.1038/nmeth.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Snijder B, Pelkmans L. Origins of regulated cell-to-cell variability. Nat Rev Mol Cell Biol. 2011;12(2):119–125. doi: 10.1038/nrm3044. [DOI] [PubMed] [Google Scholar]
- 37.Wang CJ, Bergmann A, Lin B, Kim K, Levchenko A. Diverse sensitivity thresholds in dynamic signaling responses by social amoebae. Sci Signal. 2012;5(213):ra17. doi: 10.1126/scisignal.2002449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim SY, Ferrell JE., Jr Substrate competition as a source of ultrasensitivity in the inactivation of Wee1. Cell. 2007;128(6):1133–1145. doi: 10.1016/j.cell.2007.01.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.