

Significance
Dengue virus infects more than 200 million people each year, and incidence of severe disease is increasing with no effective countermeasures. We demonstrate in this paper the engineering of an antibody that binds to all four serotypes of dengue virus with potent activity in vitro and in vivo. We also outline a distinct and widely applicable approach to antibody engineering that provides important information on the paratope/epitope interface in the absence of crystal structure data, enabling identification of antibody amino acids that could be mutated. We demonstrate experimentally the alteration of both specificity (enabling cross-serotype binding) and affinity of the engineered antibody.
Keywords: affinity enhancement, antibody engineering, computational docking, therapeutic, infectious diseases
Abstract
Affinity improvement of proteins, including antibodies, by computational chemistry broadly relies on physics-based energy functions coupled with refinement. However, achieving significant enhancement of binding affinity (>10-fold) remains a challenging exercise, particularly for cross-reactive antibodies. We describe here an empirical approach that captures key physicochemical features common to antigen–antibody interfaces to predict protein–protein interaction and mutations that confer increased affinity. We apply this approach to the design of affinity-enhancing mutations in 4E11, a potent cross-reactive neutralizing antibody to dengue virus (DV), without a crystal structure. Combination of predicted mutations led to a 450-fold improvement in affinity to serotype 4 of DV while preserving, or modestly increasing, affinity to serotypes 1–3 of DV. We show that increased affinity resulted in strong in vitro neutralizing activity to all four serotypes, and that the redesigned antibody has potent antiviral activity in a mouse model of DV challenge. Our findings demonstrate an empirical computational chemistry approach for improving protein–protein docking and engineering antibody affinity, which will help accelerate the development of clinically relevant antibodies.
Antibodies are of growing importance as therapeutic agents (1). Engineering improved affinity and specificity of these compounds can augment their potency and safety while decreasing required dosages. Production of antibodies with binding properties of interest typically relies on methods involving screening large numbers of clones generated by the immune system or by mutant libraries (2, 3). Alternatively, computer-based design offers the potential to rationally mutate available antibodies for improved properties, including enhanced affinity and specificity to target antigens. Recently, several successful examples of antibody affinity improvement by computational methods using physical modeling with energy minimization have been described (4–6). However, such approaches require a 3D structure of the antibody–antigen complex and rarely result in affinity gains greater than 10-fold. Further, these approaches are sensitive to precise atomic coordinates, rendering them inapplicable to computer-generated models. More significantly, enhancement of affinity in the context of an antibody that recognizes multiple antigens (i.e., cross-reactive) remains a particular challenge.
Dengue is the most medically relevant arboviral disease in humans, with an estimated 3.6 billion people at risk for infection. More than 200 million infections of dengue virus (DV) are estimated to occur globally each year (7). The incidence, geographical outreach, and number of severe disease cases of dengue are increasing (8, 9), making DV of increasing concern as a human pathogen. The complex of DVs is composed of four distinct serotypes (designated DV1–4) (10), which vary from one another at the amino acid level by 25–40%. The sequence and antigenic variability of DVs have challenged efforts to develop an effective vaccine or therapeutic against all serotypes (11). Currently, no licensed vaccine or specific therapy exists for dengue (12), and the leading vaccine candidate recently demonstrated protective efficacy of only 30% in a phase II study (13). The envelope (E) protein of DV is the major neutralizing target of the humoral immune response (14). Antibodies recognizing the highly conserved fusion loop on E protein demonstrate broad reactivity to all four serotypes; however, their neutralizing potency is limited due to this epitope being largely inaccessible in a mature dengue virion (15). In contrast, antibodies that recognize the “A” β-strand of E protein domain III (EDIII) have been shown to potently neutralize some—but rarely all four—serotypes (SI Appendix, Fig. S1) (16). We asked whether we could, through computational chemistry, redesign an A-strand-specific antibody, namely 4E11 (17, 18) (SI Appendix, Fig. S2), to potently neutralize all four serotypes by introducing rationally selected mutations to the antibody for increased affinity, thereby enhancing neutralizing activity. To computationally redesign 4E11 for potent neutralizing activity to all four serotypes, we faced multiple challenges: (i) to generate an accurate structural model of 4E11 with its multiple antigens and (ii) to design mutations that enhance affinity to one serotype while not detrimentally affecting affinity to the other serotypes. To overcome these challenges and design affinity-enhancing mutations, we explored the possibility of mining known antibody-antigen 3D structures to extract physicochemical information that may directly aide computational methods in discriminating native-like structures from decoys and predicting affinity-enhancing mutations.
Results
Physicochemical Features of Antigen–Antibody Interface Accurately Discriminate Native-Like Structures from Decoys.
In the absence of a cocrystal structure, computational protein–protein docking can be used to model an antibody–antigen interaction. Docking involves two components: a search algorithm that generates initial configurations of the protein–protein interaction and a scoring function that ranks the configurations based on an energy function. Docking can be especially effective when partial epitope and/or paratope residues are known. However, obtaining a native-like structure remains challenging due in part to limitations in energetic functions being able to reliably discriminate accurate from inaccurate structures (19–21). Docked structures having different antigen–antibody configurations often appear equally probable when evaluated in the context of an energy-scoring function. We hypothesized that key physicochemical features could be used to distinguish native-like structures from decoy structures and thus could help overcome limitations of using only energetic functions to rank poses.
In our analysis, an antigen–antibody interface is described using eleven features: six chemical and five physical (SI Appendix, Table S1 and Fig. S3). Notably, a knowledge-based pose-scoring metric, termed “ZEPII” (Standardized Epitope-Paratope Interface Index), which incorporates prior structural knowledge of protein–ligand interaction in the form of epitope–paratope pairwise amino acid interactions, was implemented and used as a molecular filter during the screening process to select native-like ligand-binding conformations (Materials and Methods and SI Appendix, Table S1). These atomic-level features are intended to capture the many geometrical and chemical properties that are the basis of molecular recognition. To test whether physicochemical features could distinguish native structures from decoys, a dataset comprising 84 nonredundant 3D structures of antigen–antibody complexes was assembled (Materials and Methods) and split into two parts: a training set consisting of 40 structures and a test set comprising the remaining 44 structures (SI Appendix, Tables S2 and S3). Corresponding to each crystal structure, we generated decoy structures using ZDOCK docking software (22), which uses an optimized scoring function (ZRANK) involving shape complementarity, desolvation, and electrostatic energy terms to rank poses (23), followed by a clustering procedure to select unique nonredundant conformations. In the clustering procedure, two decoy structures are considered similar (and therefore redundant) if the rmsd between the two sets of ligand interface atoms is less than 3 Å (Materials and Methods). This analysis yielded a total of 1,210 structures. Our expanded training dataset comprised 617 structures (40 native-like and 577 decoy structures) and testing dataset comprised 677 structures (44 native-like and 633 decoy structures). In the training phase, we used multivariate logistic regression analysis (MLR) to determine the relationship between each feature (explanatory variable) and the degree to which it can successfully discriminate native-like structures from decoy structures (outcome variable). The aim was to find a subset of features that, when combined, will yield the highest probability of success in discriminating native-like structures from decoys. To prevent nonuniform learning, which can lead to over- (or under-) estimation of the significance of the features, structures were grouped according to their Protein Data Bank (PDB) source, and input features for each structure were standardized (Z-score) with respect to the minimum values found within the respective groups. For the discrimination phase, we used the precomputed significant features to predict the probability that a structure in the test dataset is native-like.
Results from MLR analysis show that the relative dominance of individual features found to significantly affect the probability of accurately discriminating native versus decoy structures is in the order of ZEPII > density of H-bonds > buried surface area > density of cation-pi interactions (SI Appendix, Table S4 and SI Text1). Based on the logistic regression coefficients, H-bond density, and buried surface area are overestimated in the decoy structures. This trend is anticipated because increasing the values of these features tends to maximize the scoring function. On the contrary, ZEPII and cation–pi interactions appear to be underrepresented in the decoy structures. Cation–pi interactions do not contribute significantly to the energy scoring function. This feature was therefore not optimized in the decoy interfaces. Assessment of decoy structures further shows that the docking procedure does not authentically recapitulate the pairwise interactions common to dissociable antigen–antibody complexes; therefore, the decoy interfaces were found to have low ZEPII values.
We next used MLR to predict the native-like structures of 44 antigen–antibody complexes in the test dataset using the precomputed significant features and, in turn, compared the sensitivity and specificity of the MLR-based prediction to those of the ZRANK energy function (Materials and Methods). Overall, MLR is seen to be more sensitive at discriminating native-like structures from decoys than the ZRANK energy function (Fig. 1A), indicating that the MLR approach yields improvements over ZRANK in predicting native-like binding structures. Moreover, this trend is not affected by the rmsd threshold that is used for clustering ligand conformations (Fig. 1A). When the window size is one (i.e., when there is only one predicted positive per test case), more than half of the native-like structures (>65%) are correctly identified by the MLR method. On the other hand, the accuracy of ZRANK varies between 30% (rmsd = 5 Å) to 52% (rmsd = 3 Å) at window size one. The prediction accuracy of the MLR method is seen to be a logarithmically increasing function of window size with accuracy reaching ∼90% at window size three and 100% at window size 4. Conversely, ZRANK fails to predict 100% of the structures even when the window size is 5 (Fig. 1A).
Fig. 1.

Sensitivity and specificity of MLR and ZRANK methods evaluated on test dataset (44 native-like and 633 decoy structures). (A) Effect of window size on prediction accuracy. The window size represents the number of predicted positives. Prediction accuracy (or sensitivity) is determined by the number of test case structures (44 in total) correctly predicted. The rmsd threshold used for clustering ligand conformations was varied (3 Å, 5 Å, and 10 Å), and its effect on prediction accuracy was analyzed. (B) ROC curves at rmsd cutoffs 3 Å, 5 Å, and 10 Å for MLR and ZRANK predictions. The area under ROC curves at rmsd cutoffs 3 Å (MLR, 0.948; ZRANK, 0.862), 5 Å (MLR, 0.95; ZRANK, 0.718), and 10 Å (MLR, 0.943; ZRANK, 0.717) indicate that MLR is more efficient at recognizing native-like structures.
The prediction accuracy of the MLR method is also superior to ZRANK at various rmsd cutoffs, indicating that the likelihood of a false-positive prediction is lower when the poses are ranked according to MLR-based prediction probability (Fig. 1A). The superior classification capability of the MLR-based prediction probability approach is corroborated by the receiver operating characteristic (ROC) curve analysis (Fig. 1B). Closer examination of the decoy structures, their ZRANK scores, and MLR-based prediction probabilities reveals some interesting insights (SI Appendix, Fig. S4). Irrespective of the ranking of poses, ZDOCK identified native-like structures for 39 out of the 44 structures, indicating that the search algorithm can identify native-like structures but that the energy function cannot discriminate them from decoys. Specifically, (i) ZRANK scores vary significantly, even between similar structures (SI Appendix, Fig. S4); (ii) structures with substantially different modes of ligand engagement can receive highly similar scores, making it difficult to discriminate between solutions (SI Appendix, Fig. S4); and (iii) inaccurate solutions often receive better scores than native-like structures (SI Appendix, Fig. S4). In contrast, whereas MLR-based prediction probability also varies between similar structures, nonnative structures rarely receive high prediction probability—highlighting the ability to discriminate the native or near-native binding structure of the ligand from among decoy conformations. Accordingly, MLR-based prediction probability is seen to correlate better with rmsd compared with ZRANK score (SI Appendix, Fig. S4).
Failure to identify the native-like structure of the five test cases highlights both the limitations of the docking search algorithm and, more significantly, the challenges in predicting affinity-enhancing mutations in those cases.
Amino Acid Interface Fitness Captures Known Affinity-Enhancing Mutations.
Given the success of using physicochemical features to accurately discriminate native-like structures from decoys, we hypothesized that similar analyses could be productively implemented to predict affinity-enhancing mutations. To generate a scoring scheme for designing affinity-enhancing mutations, we developed a mathematical model to quantify the propensities of pairwise amino acid interactions (Materials and Methods). These statistical propensities are formulated as an interaction matrix that assigns a weight to each possible protein–protein amino acid pair (one paratope and one epitope). The fitness of a residue at a complementarity determining region (CDR) position, termed amino acid interface fitness (AIF), is the combined propensity of all interprotein pairwise contacts (defined as two amino acids within a certain distance of each other) involving that residue. Substitutions that lead to an improvement in AIF value without any structural consequences are considered candidates for affinity enhancement. The propensities are determined using known protein structures and statistics on amino acid contacts (Materials and Methods). Avoiding multiple distance cutoffs and energy minimization steps eliminates heavy dependencies on atomic coordinates.
Consistent with observations made in previous studies (24, 25), the propensity data show the dominance of tyrosine, tryptophan, serine, and phenylalanine over other residues in the paratope (SI Appendix, Table S5). We used the AIF metric to predict affinity-enhancing mutations of antibodies across three different systems for which published data can be used to validate the predictions. One of our test cases was the anti-epidermal growth factor receptor (EGFR) antibody drug cetuximab (Erbitux), where a 10-fold affinity improvement was engineered by three mutations on the light chain (6). Two of the mutations predicted by AIF, S26D and T31E, were shown to improve binding affinity as single mutations in cetuximab (6). AIF, however, did not identify the third mutation, N93A. Reportedly, the N93A mutation affects affinity through lower free energy from desolvation—an effect highly unlikely to be captured in pairwise interactions (6). Additionally, Ala is among a set of residues with weak contact propensities, and thus overall replacement potential (SI Appendix, Table S5). Another test case was the anti-lysozyme model antibody D44.1, where we predicted 18 mutations suitable for affinity enhancement. Four of the predicted mutations on the heavy chain, T28D, T58D, E35S, G99D, were part of a published high-affinity variant of D44.1 (6). Still another test case was the antibody E2, which targets cancer-associated serine protease MT-SP1. A previous in silico affinity enhancement study identified a single mutation, T98R, that improved the antibody affinity by 14-fold (26). AIF metric predicted eight mutations that included T98R. Our other AIF-based predictions in the above test systems remain to be tested.
Motivated by the success of our methods in accurately discriminating native-like structures from decoys and predicting affinity-enhancing mutations, we were interested to apply these approaches for ab initio modeling and affinity enhancement of the DV-neutralizing antibody 4E11.
MLR and AIF Methods Predict Affinity-Enhancing Mutations in the Cross-Reactive Antibody 4E11.
The cross-reactive antibody 4E11 exhibits high affinity and strong inhibitory potency to DV1–3 but low affinity and limited neutralizing activity to DV4 (SI Appendix, Fig. S2). This activity profile represents a particularly challenging case for engineering improved affinity: designed mutations must not only favorably contribute to DV4 binding but do so while not detrimentally affecting antibody interactions with three different antigens (i.e., DV1–3). Moreover, designs must be performed “blinded,” as there were no available crystal structures. The success of our design approach (SI Appendix, Fig. S6) therefore relied on three important factors: (i) to generate an accurate model of 4E11-EDIII interaction, (ii) to understand the serotype-specific structural elements and recapture the determinants of affinity and specificity, and (iii) to design substitutions that confer favorable interaction, and thus improved affinity, with DV4 while not negatively impacting binding to DV1–3.
In the absence of the antibody crystal structure, a structural model of the Fv region was generated (SI Appendix, SI Text2), and the modeled Fv was docked against EDIII of DV1 using ZDOCK software. Previously published functional data on the epitope and CDR H3 paratope (27, 28) were included as residues in the binding interface to ensure that docked structures did not deviate significantly from the native structure (SI Appendix, SI Text3). To thoroughly sample search space, ZDOCK was run five times with different combinations of input interface residues. The best ranking model from each run (SI Appendix, Fig. S7) was reranked using the MLR probabilities (Table 1). Interestingly, the top model predicted by the MLR approach did not match the best prediction of the ZRANK method.
Table 1.
Physicochemical properties, ZRANK scores, and MLR-based prediction probabilities of the top five docked structural models
| Pose | ZEPII | BSA | Cation-pi density | H-bond density | MLR probability | ZRANK score |
| 1 | 1.10 | 2,269 | 0.176 | 0.661 | 0.966 | −75.833 |
| 2 | 1.07 | 1,941 | 0.155 | 0.824 | 0.395 | −85.636 |
| 3 | 1.10 | 2,340 | 0.128 | 1.026 | 0.009 | −66.759 |
| 4 | 1.08 | 2,436 | 0.164 | 0.698 | 0.549 | −71.73 |
| 5 | 0.96 | 2,481 | 0.202 | 0.846 | 1.00e-5 | −72.775 |
| Regression coefficient | 9.05 | −2.796 | 2.066 | −6.384 |
To validate the top model predicted by the MLR approach, a comparison was performed between paratope hot spots computationally predicted by the web server KFC2 (29) and hot spots determined experimentally from Ala-scanning at each position in all CDR loops of 4E11 (Materials and Methods) with binding assessment by indirect EDIII-DV1 ELISA. Hot spot prediction of the selected model correctly identified 61% of experimentally determined hot spots whereas the remaining structures had hot spot prediction accuracies of <45% (range 28–44%), thus indicating that the selected structure was likely to reflect the true 4E11–EDIII binding configuration.
The top 4E11–EDIII (DV1) model was used to guide the modeling of the interaction between 4E11 and EDIII of a representative strain from each of the other three serotypes (SI Appendix, SI Text3). Using the four structural models, the mode of antibody binding to each of the serotypes was examined, and the molecular basis of poor affinity toward DV4 was explored using a combination of sequence and EDIII domain-level structural analysis. Multiple amino acid differences within and around the 4E11 binding interface between DV4 and other serotypes were identified. Notably, the orientation of the A-strand (residues 305–308) relative to neighboring β-strands is different in DV4 owing to a localized difference at position 307, a core epitope residue (Fig. 2). Critically, K307 of DV1 and DV2 form a salt bridge contact with E59 of VL; however, K307 is substituted by serine in DV4. Additionally, the 4E11–EDIII (DV4) interface possessed fewer H-bonds and salt bridge contacts, consistent with the low affinity and neutralizing potency to DV4.
Fig. 2.

Sequence and structural determinants of poor DV4 binding. (A) Sequence alignment of EDIII region of representative strains from each of the four serotypes. Putative antibody binding residues are highlighted in yellow. Residues at 307, 329, 361, 364, 385, 388, and 390 differentiate DV4 from the remainder of the sequences; these are marked in red and numbered. Residue contacts made by the five antibody mutations are boxed. (B) Structural model of 4E11–EDIII interaction. Sequence positions that discriminate DV4 from other strains are labeled, and the side chains of amino acids therein are represented as sticks.
Next we applied our AIF index to design mutations that would add new or improve favorable contacts to DV4, while not detrimentally affecting contacts with DV1–3. A conscious effort was taken to first designing affinity-enhancing mutations at CDR positions proximal to DV4 serotype-specific residues 307, 329, 361, 364, 385, 388, and 390 (Fig. 2). AIF metric was applied to 4E11–EDIII (DV1–4) models to select mutations that had potential to improve DV4 affinity while not being detrimental to other serotypes. Next, mutations that created new or improved favorable contacts to one or more serotypes including DV4 were also considered. To reduce any drastic changes to the binding, CDR mutations that affect interactions in the core of the antigen–antibody interface (with positions 308, 309, 312, 325, 387, 389, and 391 of the epitope) were selected only if their beneficial role was obvious from the structural model. These steps resulted in a set of 87 mutations spanning 23 CDR positions, with mutations representing amino acids of varied chemical properties. The choice of amino acid replacements was not always intuitive; for example, if the epitope region surrounding a paratope CDR position is negatively charged, Arg and Lys are not always statistically favored at that CDR position. In an effort to learn about the effects of point mutations on binding affinity, we did not restrict ourselves to substitutions with the highest probabilities of success.
Designed Mutations Result in a 450-Fold Affinity Gain with Enhanced Neutralizing Activity.
A total of 87 mutations were selected for experimental testing by indirect ELISA using purified recombinant EDIII of DV1–4 (Materials and Methods). Mutants were generated by site-directed mutagenesis, sequence-confirmed, and expressed from 293 cells by transient transfection (SI Appendix, SI Text6). Ten mutations were identified with enhanced EDIII–DV4 affinity with no or minimal reduction in binding to EDIII of DV1–3 (SI Appendix, Table S6). These 10 mutations spanned five CDR positions, with four in VL (R31, N57, E59, and S60) and one in VH (A55). Interestingly, 8 of the 10 mutations were in VL, with 7 being in CDR L2 alone. The successful mutations were mostly charged or polar in nature and found to reside at the periphery of the antibody–antigen interface area (SI Appendix, Fig. S8). Structural analysis revealed that the mutant side chains likely create new contacts with six different DV4 residue positions out of which four are conserved in DV-2 and three are conserved in DV1 and DV3 (Fig. 2 and SI Appendix, Table S7). Out of the six residues, residue at 329 is DV4–specific and exposed, whereas residues at 305, 310, 311, 323, and 327 are relatively buried in the interface. It is also interesting to note that these six residues do not overlap with the core epitope residues of the antigen–antibody interface.
To more accurately quantify the binding properties of these 10 single mutants, competition ELISA experiments were performed, which enabled determination of affinities at equilibrium and in solution (30) (Materials and Methods). Affinity results from five single mutant antibodies, representing those mutations that demonstrated the greatest EDIII–DV4 affinity enhancement while maintaining affinity to EDIII of DV1–3, are described in SI Appendix, Table S8. The extent of DV4 affinity enhancement ranged from 1.1-fold (VL-R31K) to 9.2-fold (VH-A55E). Somewhat unexpectedly, two mutations conferred increased affinity to other serotypes; VH-A55E resulted in a 16- and sevenfold affinity increase to EDIII–DV2 and EDIII–DV3, respectively, whereas VL-N57E demonstrated a threefold affinity increase to EDIII–DV2. Only 3 of the 15 affinities measured to serotypes 1–3 (with the five single mutant antibodies) showed a decrease greater than twofold, and only one antibody–EDIII affinity (VL-E59Q for EDIII–DV3) resulted in more than a threefold decrease in affinity.
The five affinity-enhancing positions structurally map to spatially distinct regions of the paratope (SI Appendix, Fig. S8). This observation led us to believe that additional enhancement could be achieved by combining successful single mutations. Multiple three-, four- and five-mutant combinations were tested, and a quintuple mutant antibody, termed 4E5A, showed the greatest increase in EDIII–DV4 affinity. Interestingly, 4E5A is composed of five substitutions representing the amino acid change at each position that conferred greatest affinity improvement to EDIII–DV4 as a single mutant. Compared with the parental mAb, 4E5A displayed 450-fold affinity improvement to EDIII–DV4 (KD = 91 nM) while maintaining affinity to EDIII of DV1 and DV3 and a 15-fold affinity increase to DV2 (Table 2 and SI Appendix, Table S9). This observation aligns with the degree of sequence conservation at the six new contact positions (Fig. 2). Significantly, we were able to increase affinity of the antibody from micromolar to near-nanomolar affinity. Surface plasmon resonance (SPR) was used to verify affinity measurements as well as to obtain kinetic binding parameters (Materials and Methods and SI Appendix, Table S10 and Fig. S9). Affinity values from SPR were in good quantitative agreement with those obtained by competition ELISA, with the exception that we could not detect specific binding of 4E11 wild type (WT) to EDIII–DV4, indicating a very low affinity, which is in general agreement with competition ELISA results (KD = 41 µM).
Table 2.
Binding affinities of 4E11 WT and 4E5A antibodies
| Method | mAb | EDIII–DV1 |
EDIII–DV2 |
EDIII–DV3 |
EDIII–DV4 |
||||
| KD, nM | Fold-change | KD, nM | Fold-change | KD, nM | Fold-change | KD, nM | Fold-change | ||
| Competition ELISA | 4E11 WT | 0.328 | — | 5.20 | — | 21.8 | — | 40,793 | — |
| 4E5A | 0.309 | 1.1 | 0.246 | 21.1 | 16.5 | 1.3 | 91.2 | 447.3 | |
| SPR | 4E11 WT | 0.50 | — | 6.20 | — | 7.58 | — | NB | — |
| 4E5A | 1.78 | 0.28 | 0.70 | 8.9 | 5.19 | 1.5 | 114 | — | |
To determine whether the increased affinity of 4E5A to EDIII–DV4 translated to enhanced activity, we used a focus reduction neutralization test (FRNT) assay (Materials and Methods). Compared with WT 4E11, 4E5A showed a >75-fold increase in neutralizing potency toward DV4, and it maintained potency to DV1–3 (Fig. 3). 4E5A exhibited strong inhibitory activity to all four serotypes, with FRNT50 values of 0.19, 0.028, 0.77, and 4.0 µg/mL for DV1–4, respectively. By contrast, the WT 4E11 had FRNT50 values of 0.062, 0.034, 0.52, and >300 µg/mL for DV1–4, respectively. To further extend our understanding of 4E5A activity, the antibody was assessed in an AG129 mouse model of DV2 challenge (31), which shows peak viremia at day 3 postinfection (Materials and Methods). AG129 mice have been widely used to evaluate therapeutic antibodies with viremia as a typical endpoint. Whereas there is active investigation of other mouse models of dengue that better captures elements of the pathology and immunology of dengue in humans, we used the AG129 mouse model to demonstrate that in vitro neutralization extended to in vivo protection. At both 1 mg/kg and 5 mg/kg, 4E5A caused a significant reduction in viremia, with 5 mg/kg treatment resulting in virus titer levels below the limit of detection (Fig. 4). Collectively, these results demonstrate that 4E5A exhibits strong inhibitory activity to all four serotypes of DV and has potent antiviral activity in vivo.
Fig. 3.

In vitro neutralizing activity of antibodies assessed by FRNT. Neutralization assays were performed with DV1–4 and antibodies 4E11 WT and 4E5A. Serial dilutions of antibody were mixed with equal amounts of virus and added to Vero cell monolayers followed by a viscous overlay. After 4–6 d, cells were fixed, and foci were immunostained and counted. Data points represent averages of duplicates with error bars representing SD. A standard four-parameter logistic model was fit to the data using least squares regression.
Fig. 4.

In vivo DV2 challenge model with prophylactic antibody administration. AG129 mice were administered 4E5A antibody (1 mg/kg or 5 mg/kg) or vehicle (PBS) 1 d before infection with DV2. Sera were collected 3 d postinfection, and virus was titered by quantitative PCR, with log10(CCID50/mL) titer extrapolated from a standard curve of a sample with known titer. The dashed line represents the approximate limit of detection, and error bars represent the SEM.
The engineered antibody 4E5A represents an interesting candidate that could be taken up for further rounds of optimization, including humanization. Cocrystal structures of 4E11–EDIII complex for each serotype were published (32) at the time we were testing our combination mutants with FRNT experiments, which allowed us to compare our structural models with the published complex structures. Consistent with our structural model leading to experimentally validated affinity-enhancing mutations, excellent correspondence exists between the crystal structures and predicted models, with Cα rmsd values of 1.3 Å (DV1), 1.2 Å (DV2), 1.5 Å (DV3), and 1.7 Å (DV4).
Discussion
Conventional approaches for discovering antibodies of therapeutic interest typically rely on experimental methods involving screening large numbers of immune system-generated clones or mutant libraries. These approaches can be expensive, technically challenging, and time consuming. For instance, the influenza FI6 antibody, which neutralizes clade 1 and 2 viruses, was identified by screening 104,000 B cells (33). An alternative strategy is to modify the properties of an existing antibody via rational engineering (34). In this study, computational methods for ab initio modeling and antibody redesign are presented. The MLR method incorporates prior structural knowledge of protein–ligand interaction “fingerprints” to identify native-like ligand conformations. In test runs, the sensitivity of the MLR-based approach in picking native-like structures (out of several decoy models) is shown to be superior to that of ZRANK. We further show that the AIF metric can capture known affinity-enhancing mutations across multiple antibody systems. Finally, we apply this framework to engineer greater affinity to a broad-spectrum anti-DV neutralizing antibody. Our approach and the results obtained in this study represent multiple findings: (i) this study used an empirical computational approach toward antibody redesign and affinity enhancement; (ii) affinity-enhancing mutations were predicted without a crystal structure of the antibody–antigen complex (i.e., blind prediction); and (iii) only one other study has attempted to improve the breadth of reactivity of an antibody by computational design, which was performed with limited success (26). Our application of a computational approach has led to a greater than ∼400-fold improvement in affinity (SI Appendix, Table S11). Given the simplicity of our computational methods, they can be broadly used for antibody engineering, and unlike physics-based energetic approaches, they are not affected by the precise location of the atom coordinates of the starting structure.
The top docking solution of 4E11–EDIII from ZRANK was structurally very different from the native-structure (ligand interface rmsd > 11 Å), indicating that any affinity enhancement efforts following the top ZRANK model would likely not have led to fruitful results. To complicate matters further, the ZRANK score of the native-like pose is in the same range as two additional decoy poses, pose 4 and pose 5 (Table 1). To determine whether an energetics-based approach would predict the affinity-enhancing mutations, we used the 4E11–EDIII (DV4) crystal structure (PDB: 3UYP) and an in-built binding energy scoring function in Discovery Studio to predict mutations. The scoring function performs amino-acid scanning mutagenesis on a set of selected CDR residues and evaluates the effect of single-point mutations on the binding affinity of molecular partners. Results highlight the challenges in free-energy discrimination of neutral and destabilizing mutations from stabilizing mutations (SI Appendix, Table S12). More significantly, the affinity-enhancing mutations N57E, N57S, and E59N were classified as destabilizing (SI Appendix, Table S12). The choice of docking software and energy scoring function influences the poses, their ranking, and, thus, the conclusion of our study. Detailed comparison against other docking algorithms, which goes beyond the scope of this study, should be performed for a systematic evaluation. Because ZRANK is widely used and has shown considerable success in critical assessment of predicted interactions (CAPRI) experiments (35, 36), we believe our method will perform comparatively well when evaluated against other top docking algorithms. The benchmarked test dataset contained a diverse set of 44 different targets that varied in size and in secondary and tertiary structures (SI Appendix, Table S3); these difficult test cases provided an opportunity to comprehensively validate the MLR-based structure discrimination. The fact that ZEPII appears as a highly significant feature in our MLR analysis indicates that amino acid composition and interresidue contacts could be used as an effective molecular filter during the screening process to select native-like ligand binding conformations. Interestingly, some geometrical features also have the predictive power to discriminate native interfaces from decoys. This finding correlates with the observation made in previous studies that antigen–antibody interfaces are more planar and significantly well-ordered or packed (37).
Methods that adopt CDR loop randomization strategies for affinity maturation (38) typically focus on heavy chain, especially CDR H3, because this loop accounts for most of the stabilizing contacts in many cases. Our results with 4E11 show that diversification strategies may benefit from a rational approach and that incorporating VL-loops for targeted diversification may further aid affinity maturation. The observation that the identified affinity-enhancing mutations are mainly polar in nature and lie at the periphery of the binding interface is consistent with other studies (5, 39, 40). The success rate at predicting mutations with targeted activities is 12% (10/87). These results are encouraging given the complexity of the design problem (i.e., involvement of four antigens) and considering that random mutations will have, on average, a detrimental effect on binding affinity.
No approved vaccine or specific therapy currently exists for dengue. The engineered antibody 4E5A exhibits strong inhibitory activity with a broad spectrum profile and is an interesting candidate for potential development for dengue treatment. A concern, however, in the use of antibodies as therapeutic or prophylactic agents for dengue rests in their potential to exacerbate disease by increasing the cellular uptake of viruses, resulting in higher viremia, a phenomenon termed antibody-dependent enhancement (ADE) (41). While ADE has been the leading theory to explain the observation of increased risk of severe disease upon a secondary infection from a heterologous DV serotype, recent studies in humans have called into question ADE as the principal mechanism of increased disease risk (13, 42, 43). Additionally, modifications to antibody Fc regions that disrupt antibody interaction with Fcγ receptors have been shown to be effective strategies in preventing ADE-mediated lethal disease in a mouse model (44). These lines of evidence support the possibility of using antibodies for dengue treatment with mitigated concern of enhanced disease. Additional in vitro and in vivo testing using multiple models needs to be considered to explore the potential of 4E5A as a broad-spectrum agent for dengue treatment.
Materials and Methods
Model for AIF and ZEPII Indices Derived from Pairwise Propensities of Epitope–Paratope Residues.
Briefly, in an antigen–antibody interface, a pair of residues will presumably interact if they have favorable energetics of interaction or by chance occurrence. The propensity of amino acid interaction is calculated by computing the number of interactions expected by chance, i.e., the expected frequency, and dividing the observed frequency by this number.
If two amino acids, one from each side of the antigen–antibody interface, are within 4.5 Å (i.e., shortest non-H atom distance is less than 4.5 Å) from each other, they are defined as pair residues. Suppose the total number of pairwise interactions between residues x (antigen) and y (antibody) at the interface is 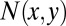 , then their concurrence frequency,
, then their concurrence frequency, 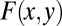 , can be defined as follows:
, can be defined as follows:
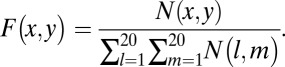 |
The denominator of the above equation indicates the summation of pairwise interactions of all residue pairs in the interface.
The frequency of occurrence of every amino acid at paratope and epitope must be calculated. The frequency of a particular amino acid x in the epitope, 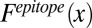 , can be defined as
, can be defined as
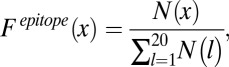 |
where 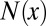 denotes the count of amino acid
denotes the count of amino acid 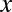 in the epitope. The denominator represents the total number of all amino acids in the epitope. Similarly, the frequency of occurrence of amino acid y in the paratope,
in the epitope. The denominator represents the total number of all amino acids in the epitope. Similarly, the frequency of occurrence of amino acid y in the paratope, 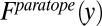 , can be defined as
, can be defined as
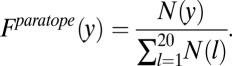 |
In the above equation, 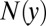 denotes the number of amino acid
denotes the number of amino acid 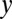 in the paratope. The denominator indicates the total number of all amino acids in the paratope. Parameters
in the paratope. The denominator indicates the total number of all amino acids in the paratope. Parameters 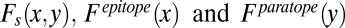 are determined using all of the 40 benchmarked antigen–antibody structures in the training dataset. Consistent with the observations made by previous studies (24, 45), tyrosine, serine, glycine, and asparagine are the most abundant paratope residues whereas lysine, arginine, leucine, and glycine are the most abundant epitope residues (SI Appendix, Fig. S5). If the occurrences of amino acids x and y are independent,
are determined using all of the 40 benchmarked antigen–antibody structures in the training dataset. Consistent with the observations made by previous studies (24, 45), tyrosine, serine, glycine, and asparagine are the most abundant paratope residues whereas lysine, arginine, leucine, and glycine are the most abundant epitope residues (SI Appendix, Fig. S5). If the occurrences of amino acids x and y are independent, 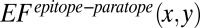 defined in the below equation is an expected frequency rate that amino acids x and y appear concurrently.
defined in the below equation is an expected frequency rate that amino acids x and y appear concurrently.
If the concurrence rate of the amino acids x and y at the interface for the antigen is more than the expected rate, the following ratio 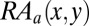 becomes greater than 1.
becomes greater than 1.
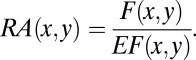 |
The pairwise propensities, 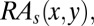 is a 20 × 20 matrix.
is a 20 × 20 matrix.
Applications of 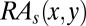 :
:
i) Using
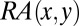 to determine the AIF of a CDR residue. The AIF of a CDR residue in the interface is defined as the sum of the
to determine the AIF of a CDR residue. The AIF of a CDR residue in the interface is defined as the sum of the 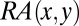 with its neighbors. Neighbors are defined by a distance criterion (4.5 Å).
with its neighbors. Neighbors are defined by a distance criterion (4.5 Å).ii) Determine the optimal choice of amino acid at an interface position (paratope reengineering): given an antigen–antibody complex, amino acid preferences at a CDR position can be computed using the AIF values. Specifically, at a given CDR position, the WT residue is systematically substituted by the remaining amino acids excluding glycine and proline (to avoid backbone conformation alterations), and the probability of replacement is evaluated at each instance using the AIF metric. Single mutations with replacement potential higher than WT residue are reevaluated computationally to find mutations that (i) do not bury polar groups and (ii) do not cause steric hindrance.
iii) Using
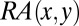 to quantify the strength of interaction of antigen–antibody interface [the “Epitope–Paratope Interface Index” (EPII)]. The interaction between an antigen and antibody results from the formation of numerous noncovalent bonds. Therefore, the interaction affinity is directly related to summation of the attractive and repulsive forces (van der Waals interactions, hydrogen bonds, salt bridges, and hydrophobic force). Herein, the strength of interaction of an antibody–antigen interface is investigated quantitatively by a linear combination of RAs for all combinations of amino acid pairs. An index expressing the strength of an antigen–antibody interface ‘i’ (called EPII) is defined by
to quantify the strength of interaction of antigen–antibody interface [the “Epitope–Paratope Interface Index” (EPII)]. The interaction between an antigen and antibody results from the formation of numerous noncovalent bonds. Therefore, the interaction affinity is directly related to summation of the attractive and repulsive forces (van der Waals interactions, hydrogen bonds, salt bridges, and hydrophobic force). Herein, the strength of interaction of an antibody–antigen interface is investigated quantitatively by a linear combination of RAs for all combinations of amino acid pairs. An index expressing the strength of an antigen–antibody interface ‘i’ (called EPII) is defined by
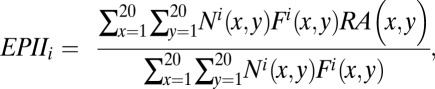 |
where
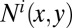 and
and 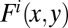 denote the number and concurrence frequency of amino acids x and y at interface i.
denote the number and concurrence frequency of amino acids x and y at interface i.iv) Using EPII to discriminate a true antigen-antibody interaction from docking decoys. To distinguish an interface with the most potential from other decoy interfaces generated by computational docking, the EPII values should be normalized by all of the interfaces in the protein. Z-scored EPII (ZEPII) are used for this purpose. If M interfaces are found in a protein, the ZEPII for interface i is calculated as follows:
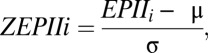 |
where
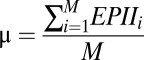 |
and
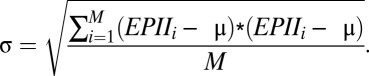 |
The ZEPIIscore is an indicator of the probability of antibody binding to a given interface. Interface with the highest ZEPII score in a protein is the most probable site for antibody binding.
Dataset of Nonredundant Antigen–Antibody Structural Complexes and Computational Docking to Generate Decoy Models.
We extracted a total of 568 antigen–antibody complexes from the Protein Data Bank. To ensure proper enumeration of geometric interface features (planarity, buried surface area, etc.), structures wherein the antigen length was less than 20 amino acids were excluded. Additionally, many structures contained the same or similar antigens, which could bias the studies, giving higher weight for factors derived from multiply represented protein antigen. To remove redundant structures from the dataset, structures that have homologous antigen (defined by BLAST (46); P value 10e27) and share 50% epitope residues were classified under the same group, and the structure with the highest resolution was selected as the representative. This analysis led to 84 nonredundant antigen–antibody complex structures.
We used ZDOCK (22) to generate decoy computational models of antigen–antibody interaction. The protocol for generating the decoy models was the same for all of the 84 structural complexes. Only the variable domain of the antibody was used for docking. The larger of the two molecules was considered the receptor whereas the smaller molecule was considered the ligand. The ligand orientation was rotated 6 degrees at each step to sample the various conformations. Because the initial docking procedure explores a relatively large area, we set up distance constraints between putative hotspot residues on epitope and paratope to ensure the generated models do not shift significantly from the native structure. We picked two hotspot residues on either side to ensure the challenges we faced with structure discrimination were equivalent to the 4E11 scenario. In all of the decoy models, the putative epitope and paratope hotspots were within 10 Å from each other. Hotspots were identified using the web server KFC2 (29). The initial docking procedure generates 100 structures that are then clustered based on an all-versus-all rmsd matrix, described by Comeau (47) and Lorenzen and Zhang (48). The rmsd between two docked structures is calculated based on the ligand atoms that are within 7 Å of the fixed receptor. Clustering procedure ensures that structures within a cluster have ligand interface rmsd < 3 Å whereas structures from different clusters have ligand interface rmsd ≥ 3 Å. Using a smaller value of ligand interface rmsd will typically increase the number of clusters and reduce the average number of structures per cluster. Overall, three different values of ligand interface rmsd were tested: (i) 3 Å, (ii) 5 Å, and (iii) 10 Å. Docked structures representing the cluster centers were taken up for further evalution. Among them, native-like structures are defined as those structures having ligand interface rmsd less than 3 Å from the ligand in the solved crystal structure. Native-like structures were replaced with their corresponding crystal structures for evaluating the sensitivity of the prediction methods. The remaining structures were considered as decoys. At rmsd = 3, a total of 617 (training) and 677 (testing) decoys were generated; at rmsd = 5, a total of 382 (training) and 454 (testing) decoys were generated; at rmsd = 10, a total of 195 (training) and 249 (testing) decoys were generated. ZDOCK uses shape complementarity along with desolvation and electrostatic energy terms (“ZRANK”) to rank the docked poses (23). Each of these decoys was further refined by Chemistry at Harvard Molecular Mechanics (CHARMM) using the CHARMm force field.
Indirect ELISA.
EDIII in PBS (0.1 µg per well) was adsorbed to Maxisorp 96-well plates (Nunc) at 4 °C overnight. Plates were blocked with PBS-T (PBS with 0.05% Tween) containing 1% BSA for 1 h. Serial dilutions of antibody were added to wells and incubated for 2 h, and, after washing, bound antibody was revealed by HRP-conjugated rabbit anti-human IgG (Jackson ImmunoResearch) followed by TMB substrate (KPL) addition.
Competition ELISA.
The affinities of antibodies to EDIII, in solution at equilibrium, were determined by competition ELISA (30). In 96-well plates, serial dilutions of EDIII were mixed with antibody at 0.2 nM in PBS-TB (PBS containing 0.01% Tween 20 and 0.01% BSA). The mixtures were incubated overnight to allow equilibrium to be reached. Subsequently, an optimized EDIII indirect ELISA, in which antibody concentration is linearly proportional to absorbance and equilibrium not significantly disturbed, was performed to measure the concentration of unbound or singly bound antibody. Briefly, maxisorp plates coated with EDIII-DV1 (2.5 ng per well, 4 °C overnight) were blocked with PBS-TB containing 1% BSA. After washing, equilibrium antibody–EDIII mixtures were added to the wells and incubated for 20 min. Bound antibody was detected by HRP-conjugated rabbit anti-human IgG (Jackson ImmunoResearch), followed by addition of TMB substrate (KPL), and 450 nm absorbance was recorded by a plate reader (Molecular Devices). The data were fit, by least squares regression in Excel (Microsoft), to the following model derived from mass action and as described (49), with adjustment to take into account antibody bivalence (50):
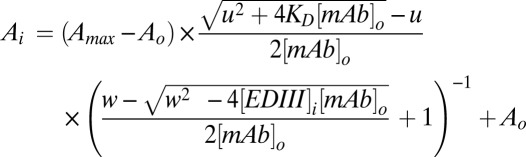 |
where
and
and [mAb]0 is the initial antibody concentration, [EDIII]i is the variable concentration of EDIII, Ai is the OD450 at [EDIII]i, Amax and A0 are the maximal and minimal OD450 (when [EDIII] = 0 and [EDIII] = ∞, respectively), and KD is the equilibrium dissociation constant. For data fitting, KD was the sole floating parameter.
Surface Plasmon Resonance.
SPR experiments were performed with a Biacore 3000 (GE Healthcare) instrument. Briefly, substoichiometrically biotinylated antibody (ligand) was applied to a CAPture Kit chip (GE Healthcare), and EDIII protein flowed as the analyte. Kinetic parameters (kon and koff) were determined by fitting resultant resonance units curves to a 1:1 binding model using BIAevaluation software (GE Healthcare). Because no binding of WT 4E11 was detected to EDIII–DV4, this interaction was also tested by steady-state experimental conditions, which increase sensitivity of detection to KD < 0.1 mM. KD was determined by the ratio koff/kon.
FRNT.
Serially diluted antibody was mixed with an equal volume of diluted virus (30 focus-forming units per well) and incubated for 2 h at 37 °C. The mixtures were then transferred to Vero cell monolayers in 24-well plates. Foci were detected as described for focus forming assay (SI Appendix, SI Text5). Each antibody concentration was run in duplicate. Data are expressed as the relative infectivity:
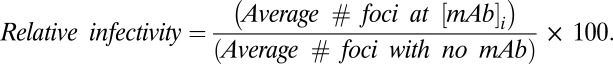 |
Four-parameter logistic model was fit to the data. The FRNT50 represents the concentration of antibody at 50% virus neutralization.
Ala-Scanning.
To identify paratope hot spots of binding interaction, Ala-scanning of 4E11 CDR loops was performed. Briefly, all residues in 4E11 CDR loops were individually mutated to Ala (or Ala→Gly), expressed, purified, and tested for binding to EDIII of DV1–3 by indirect ELISA, as described above. Energetic hot spots were defined as those in which KD increased by >100-fold for at least two serotypes.
In Vivo Experiments.
Mouse model studies were conducted at the Utah State University Laboratory Animal Research Facility (Institute for Antiviral Research) and were approved by the Institutional Animal Care and Use Committee (IACUC). AG129 mice (10 per group), which are deficient in IFN-α/β and IFN-γ receptors (51), were administered 4E5A antibody (1 mg/kg or 5 mg/kg) or vehicle (PBS) one day before challenge with 106.4 50% cell culture infectious dose (CCID50) New Guinea C virus per animal. Sera were collected from animals on day 3 postinfection, and viremia was quantified by quantitative RT-PCR (Stratagene). A virus stock of known titer was also extracted in parallel for use in quantification. The data are presented as averages, with error bars representing the SEM CCID50/mL from each group.
Supplementary Material
Acknowledgments
This work was funded by National Institutes of Health Grant R37 GM057073-13 and in part by the National Research Foundation Singapore through the Singapore-MIT Alliance for Research and Technology's Infectious Diseases Research Program.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303645110/-/DCSupplemental.
References
- 1.Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol. 2006;6(5):343–357. doi: 10.1038/nri1837. [DOI] [PubMed] [Google Scholar]
- 2.Griffiths AD, Duncan AR. Strategies for selection of antibodies by phage display. Curr Opin Biotechnol. 1998;9(1):102–108. doi: 10.1016/s0958-1669(98)80092-x. [DOI] [PubMed] [Google Scholar]
- 3.Hoogenboom HR. Designing and optimizing library selection strategies for generating high-affinity antibodies. Trends Biotechnol. 1997;15(2):62–70. doi: 10.1016/S0167-7799(97)84205-9. [DOI] [PubMed] [Google Scholar]
- 4.Clark LA, et al. Affinity enhancement of an in vivo matured therapeutic antibody using structure-based computational design. Protein Sci. 2006;15(5):949–960. doi: 10.1110/ps.052030506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marvin JS, Lowman HB. Redesigning an antibody fragment for faster association with its antigen. Biochemistry. 2003;42(23):7077–7083. doi: 10.1021/bi026947q. [DOI] [PubMed] [Google Scholar]
- 6.Lippow SM, Wittrup KD, Tidor B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol. 2007;25(10):1171–1176. doi: 10.1038/nbt1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guzman MG, et al. Dengue: A continuing global threat. Nat Rev Microbiol. 2010;8(12, Suppl):S7–S16. doi: 10.1038/nrmicro2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kyle JL, Harris E. Global spread and persistence of dengue. Annu Rev Microbiol. 2008;62:71–92. doi: 10.1146/annurev.micro.62.081307.163005. [DOI] [PubMed] [Google Scholar]
- 9.Gubler DJ. Dengue, urbanization and globalization: The unholy trinity of the 21st Century. Trop Med Health. 2011;39(4, Suppl):3–11. doi: 10.2149/tmh.2011-S05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McBride WJ, Bielefeldt-Ohmann H. Dengue viral infections: Pathogenesis and epidemiology. Microbes Infect. 2000;2(9):1041–1050. doi: 10.1016/s1286-4579(00)01258-2. [DOI] [PubMed] [Google Scholar]
- 11.Whitehead SS, Blaney JE, Durbin AP, Murphy BR. Prospects for a dengue virus vaccine. Nat Rev Microbiol. 2007;5(7):518–528. doi: 10.1038/nrmicro1690. [DOI] [PubMed] [Google Scholar]
- 12.Thomas SJ, Endy TP. Critical issues in dengue vaccine development. Curr Opin Infect Dis. 2011;24(5):442–450. doi: 10.1097/QCO.0b013e32834a1b0b. [DOI] [PubMed] [Google Scholar]
- 13.Sabchareon A, et al. Protective efficacy of the recombinant, live-attenuated, CYD tetravalent dengue vaccine in Thai schoolchildren: A randomised, controlled phase 2b trial. Lancet. 2012;380(9853):1559–1567. doi: 10.1016/S0140-6736(12)61428-7. [DOI] [PubMed] [Google Scholar]
- 14.Wahala WM, Silva AM. The human antibody response to dengue virus infection. Viruses. 2011;3(12):2374–2395. doi: 10.3390/v3122374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lai CY, et al. Antibodies to envelope glycoprotein of dengue virus during the natural course of infection are predominantly cross-reactive and recognize epitopes containing highly conserved residues at the fusion loop of domain II. J Virol. 2008;82(13):6631–6643. doi: 10.1128/JVI.00316-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lok SM, et al. Binding of a neutralizing antibody to dengue virus alters the arrangement of surface glycoproteins. Nat Struct Mol Biol. 2008;15(3):312–317. doi: 10.1038/nsmb.1382. [DOI] [PubMed] [Google Scholar]
- 17.Thullier P, et al. A recombinant Fab neutralizes dengue virus in vitro. J Biotechnol. 1999;69(2-3):183–190. doi: 10.1016/S0168-1656(99)00037-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thullier P, et al. Mapping of a dengue virus neutralizing epitope critical for the infectivity of all serotypes: insight into the neutralization mechanism. J Gen Virol. 2001;82(Pt 8):1885–1892. doi: 10.1099/0022-1317-82-8-1885. [DOI] [PubMed] [Google Scholar]
- 19.Pedotti M, Simonelli L, Livoti E, Varani L. Computational docking of antibody-antigen complexes, opportunities and pitfalls illustrated by influenza hemagglutinin. Int J Mol Sci. 2011;12(1):226–251. doi: 10.3390/ijms12010226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohan V, Gibbs AC, Cummings MD, Jaeger EP, DesJarlais RL. Docking: Successes and challenges. Curr Pharm Des. 2005;11(3):323–333. doi: 10.2174/1381612053382106. [DOI] [PubMed] [Google Scholar]
- 21.Simonelli L, et al. Rapid structural characterization of human antibody-antigen complexes through experimentally validated computational docking. J Mol Biol. 2010;396(5):1491–1507. doi: 10.1016/j.jmb.2009.12.053. [DOI] [PubMed] [Google Scholar]
- 22.Chen R, Li L, Weng Z. ZDOCK: An initial-stage protein-docking algorithm. Proteins. 2003;52(1):80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 23.Pierce B, Weng Z. ZRANK: Reranking protein docking predictions with an optimized energy function. Proteins. 2007;67(4):1078–1086. doi: 10.1002/prot.21373. [DOI] [PubMed] [Google Scholar]
- 24.Fellouse FA, Wiesmann C, Sidhu SS. Synthetic antibodies from a four-amino-acid code: A dominant role for tyrosine in antigen recognition. Proc Natl Acad Sci USA. 2004;101(34):12467–12472. doi: 10.1073/pnas.0401786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fellouse FA, et al. Molecular recognition by a binary code. J Mol Biol. 2005;348(5):1153–1162. doi: 10.1016/j.jmb.2005.03.041. [DOI] [PubMed] [Google Scholar]
- 26.Farady CJ, Sellers BD, Jacobson MP, Craik CS. Improving the species cross-reactivity of an antibody using computational design. Bioorg Med Chem Lett. 2009;19(14):3744–3747. doi: 10.1016/j.bmcl.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lisova O, Hardy F, Petit V, Bedouelle H. Mapping to completeness and transplantation of a group-specific, discontinuous, neutralizing epitope in the envelope protein of dengue virus. J Gen Virol. 2007;88(Pt 9):2387–2397. doi: 10.1099/vir.0.83028-0. [DOI] [PubMed] [Google Scholar]
- 28.Bedouelle H, et al. Diversity and junction residues as hotspots of binding energy in an antibody neutralizing the dengue virus. FEBS J. 2006;273(1):34–46. doi: 10.1111/j.1742-4658.2005.05045.x. [DOI] [PubMed] [Google Scholar]
- 29.Darnell SJ, Page D, Mitchell JC. An automated decision-tree approach to predicting protein interaction hot spots. Proteins. 2007;68(4):813–823. doi: 10.1002/prot.21474. [DOI] [PubMed] [Google Scholar]
- 30.Friguet B, Chaffotte AF, Djavadi-Ohaniance L, Goldberg ME. Measurements of the true affinity constant in solution of antigen-antibody complexes by enzyme-linked immunosorbent assay. J Immunol Methods. 1985;77(2):305–319. doi: 10.1016/0022-1759(85)90044-4. [DOI] [PubMed] [Google Scholar]
- 31.Johnson AJ, Roehrig JT. New mouse model for dengue virus vaccine testing. J Virol. 1999;73(1):783–786. doi: 10.1128/jvi.73.1.783-786.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cockburn JJ, et al. Mechanism of dengue virus broad cross-neutralization by a monoclonal antibody. Structure. 2012;20(2):303–314. doi: 10.1016/j.str.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 33.Corti D, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333(6044):850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 34.Kuroda D, Shirai H, Jacobson MP, Nakamura H. Computer-aided antibody design. Protein Eng Des Sel. 2012;25(10):507–521. doi: 10.1093/protein/gzs024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fleishman SJ, et al. Community-wide assessment of protein-interface modeling suggests improvements to design methodology. J Mol Biol. 2011;414(2):289–302. doi: 10.1016/j.jmb.2011.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hwang H, Vreven T, Pierce BG, Hung JH, Weng Z. Performance of ZDOCK and ZRANK in CAPRI rounds 13-19. Proteins. 2010;78(15):3104–3110. doi: 10.1002/prot.22764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones S, Thornton JM. Principles of protein-protein interactions. Proc Natl Acad Sci USA. 1996;93(1):13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barderas R, Desmet J, Timmerman P, Meloen R, Casal JI. Affinity maturation of antibodies assisted by in silico modeling. Proc Natl Acad Sci USA. 2008;105(26):9029–9034. doi: 10.1073/pnas.0801221105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Selzer T, Albeck S, Schreiber G. Rational design of faster associating and tighter binding protein complexes. Nat Struct Biol. 2000;7(7):537–541. doi: 10.1038/76744. [DOI] [PubMed] [Google Scholar]
- 40.Schreiber G, Fersht AR. Rapid, electrostatically assisted association of proteins. Nat Struct Biol. 1996;3(5):427–431. doi: 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- 41.Murphy BR, Whitehead SS. Immune response to dengue virus and prospects for a vaccine. Annu Rev Immunol. 2011;29:587–619. doi: 10.1146/annurev-immunol-031210-101315. [DOI] [PubMed] [Google Scholar]
- 42.Laoprasopwattana K, et al. Dengue Virus (DV) enhancing antibody activity in preillness plasma does not predict subsequent disease severity or viremia in secondary DV infection. J Infect Dis. 2005;192(3):510–519. doi: 10.1086/431520. [DOI] [PubMed] [Google Scholar]
- 43.Endy TP, et al. Relationship of preexisting dengue virus (DV) neutralizing antibody levels to viremia and severity of disease in a prospective cohort study of DV infection in Thailand. J Infect Dis. 2004;189(6):990–1000. doi: 10.1086/382280. [DOI] [PubMed] [Google Scholar]
- 44.Beltramello M, et al. The human immune response to Dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe. 2010;8(3):271–283. doi: 10.1016/j.chom.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramaraj T, Angel T, Dratz EA, Jesaitis AJ, Mumey B. Antigen-antibody interface properties: Composition, residue interactions, and features of 53 non-redundant structures. Biochim Biophys Acta. 2012;1824(3):520–532. doi: 10.1016/j.bbapap.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 47.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: An automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20(1):45–50. doi: 10.1093/bioinformatics/btg371. [DOI] [PubMed] [Google Scholar]
- 48.Lorenzen S, Zhang Y. Identification of near-native structures by clustering protein docking conformations. Proteins. 2007;68(1):187–194. doi: 10.1002/prot.21442. [DOI] [PubMed] [Google Scholar]
- 49.Martineau P. Affinity measurements by competition ELISA. In: Kontermann R, Dubel S, editors. Antibody Engineering. Vol 1. Berlin: Springer; 2010. pp. 657–665. [Google Scholar]
- 50.Bobrovnik SA. Determination of antibody affinity by ELISA. Theory. J Biochem Biophys Methods. 2003;57(3):213–236. doi: 10.1016/s0165-022x(03)00145-3. [DOI] [PubMed] [Google Scholar]
- 51.van den Broek MF, Müller U, Huang S, Aguet M, Zinkernagel RM. Antiviral defense in mice lacking both alpha/beta and gamma interferon receptors. J Virol. 1995;69(8):4792–4796. doi: 10.1128/jvi.69.8.4792-4796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.