

Abstract
Single-molecule super-resolution microscopy is an emerging technique for nanometer-scale fluorescence imaging, but in vitro single-molecule imaging protocols typically require a constant supply of reagents, and such transport is restricted in constrained geometries. In this paper, we develop Single-Molecule Micelle-Assisted Blink (MAB) microcopy to enable sub-diffraction-limit imaging of nanochannels with better than 40-nm accuracy. The method, based on micelles and thiol-related photoswitching, is used to measure nanochannels formed in polydimethyl-siloxane (PDMS) through tensile cracking. These conduits are reversibly size-adjustable from a few nanometers up to a micron, and enable filtering of small particles and linearization of DNA. Unfortunately, conventional techniques cannot be used to measure widths, characterize heterogeneities or discover porosity in situ. We overcome the access barriers by using sodium dodecyl sulfate (SDS), an ionic surfactant, to facilitate delivery of Cy5 dye and beta-mercaptoethanol reducing agent in the confined geometry. These SDS micelles and admicelles have the further benefit of slowing diffusion of Cy5 to improve localization accuracy. We use MAB microscopy to measure nanochannel widths, to reveal heterogeneity along channel lengths and between different channels in the same device, and to probe biologically relevant information about the nano-environment, such as solvent accessibility.
Keywords: Fluorescence, diffraction limit, microfluidics, photoswitching, atomic force microscopy
INTRODUCTION
Fluidic devices made from soft materials are useful for manipulating fluids and biological cells, and these devices allow sorting and small-volume confinement. Once limited by traditional optical lithography to micron-scale dimensions, novel non-lithographic techniques have more recently enabled polymer-based devices with nanometer-scale features to be produced with the same versatility, ease of fabrication and low cost as their microfluidic counterparts.1 In order to fully explore the capabilities of elastomeric nanodevices, it is important to characterize feature sizes, heterogeneity and responses to force. Confocal fluorescence microscopy can permit in situ observation of microfluidic channels, including accurate measurements of particulate flow velocities,2, 3 but confocal imaging has a resolution limited by the diffraction limit of light to ~250 nm, whereas the details of interest in characterizing nanofluidic devices are an order of magnitude smaller. Electron and atomic-force microscopies, on the other hand, are well-matched to probing small features, but the former technique typically requires an electrically conductive substrate and is therefore of limited use for soft materials, while the latter method relies on physical contact between the probe and the features of interest, and therefore cannot be applied directly to confined spaces.
By localizing individual, isolated fluorescent molecules and fitting the point-spread function (PSF), single-molecule fluorescence (SMF) enables non-invasive, non-destructive imaging with nanometer resolution in a conventional optical microscope.4 To achieve sub-diffraction limited images, the low emitter concentration required by SMF can be attained via photoactivation (F/PALM),5, 6 photoswitching (STORM),7, 8 dynamics (PAINT),9, 10 and chemical blinking (ROXS).11 In all these methods, the position of each molecule is recorded and localized sequentially, and a super-resolution image is reconstructed from the sum of all fits. Unfortunately, none of these single-molecule super-resolution methods are appropriate for the particular challenges of imaging inside confined spaces. Here, we develop Single-Molecule Micelle-Assisted Blink (MAB) Microscopy to address the unique challenges of imaging size-constrained, hard-to-access environments: a difficulty introducing sufficient reagents for super-resolution imaging into the small spaces under investigation.
In single-molecule blink microscopy,11, 12 a thiol such as beta-mercaptoethanol (BME) restores emission from reversibly non-fluorescent dye molecules, giving rise to a stochastic distribution of isolated emitters visualized over time. Under constant CW excitation in a blinking buffer (BME in Tris), Cy5 has been shown to photoswitch efficiently and repeatedly between a bright (fluorescent) state and a dark (non-fluorescent) state.13 This blink microscopy method requires a constant supply of fresh BME to be delivered to the Cy5 dye molecules, and though isolated single dye molecules were detected in larger flow cells as early as 1990,14 the constant delivery condition is not easily attained in nanofluidic devices. The ionic surfactant molecule sodium dodecyl sulfate (SDS) is regularly used in capillary electrophoresis and microfluidic experiments to modify surface and interfacial tension, and in the present study, we incorporate SDS into the imaging buffer to improve delivery of Cy5 and BME into nanochannels to enhance the SMF imaging resolution. As well, in the presence of SDS, Cy5 diffuses more slowly upon incorporation in micelles, thereby yielding improved localization accuracy. Indeed, though individual Cy5 molecules cannot be resolved in free solution with typical single-molecule fluorescence detection rates of 10–100 frames/s, here in the presence of micelles, Cy5 diffusion is slow enough that the PSF is not discernibly blurred by motion during the 27-ms imaging frame (i.e., the motion is slower than ~1 μm/s). We apply single-molecule MAB microscopy to nanochannel devices fabricated within a polydimethylsiloxane (PDMS) substrate through tensile nanocracking.15, 16 These nanoscale conduits are reversibly size-adjustable from a few nanometers to about a micron, and enable filtering of small particles15 and linearization of DNA molecules.17 In this system, MAB microscopy can directly probe the nano-environment and reveal biologically relevant information about the sizes of the nanochannels, the solution-accessible volume therein, and the heterogeneity along channel lengths and between individual channels in the same device.
EXPERIMENTAL SECTION
Nanochannel device preparation
Arrays of polydimethylsiloxane (PDMS) nanochannels were generated by tensile cracking with a 10% applied strain (Figure 1) as described in reference 16. Briefly, a PDMS slab containing two 1-cm microchannels spaced 100- or 300-μm apart (for “short” and “long” devices, respectively) and a 160-μm-thick PDMS membrane were cleaned under vacuum and plasma oxidized (Harrick Plasma, 30 W). These two surfaces spontaneously bonded upon contact. The bonded system was loaded into a stretcher, and a uniaxial applied force generated cracks that tunnel through the brittle layer at the bonding interface (Figure 1). Bulk electrical impedance and fluorescence measurements of these devices16, 17 have shown that cracks begin to appear at strains above 10 % (as indicated by a large decrease in resistance), that these cracks collapse monotonically as the strain is released and eventually heal when there is no more applied force (resistance returns to the high level measured before cracking), and that cracks re-appear at the same location as the original cracks when strain is applied again. In the current system, the nanochannels were spaced on average 14 μm apart (approximately 700 nanochannels per device).17 After cracking, the PDMS device was loaded with de-aerated Tris buffer and then de-aerated by exposure to six cycles of vacuum (5 inHg) and nitrogen purging at 1 atm and room temperature. After de-aeration, the sample was stored in N2 and imaged under a constant flow of N2.
Figure 1.

Micelle-Assisted Blink (MAB) Microscopy. Top: Uniaxial tensile cracking under applied strain causes nanochannels to form between a pair of parallel lithographically-defined microchannels. Bottom: Amphiphilic SDS molecules (grey) assemble into micelles within the nanochannels and admicelles along the nanochannel walls. Cy5 (red) and BME (blue) molecules can be free in solution, associated with the micelles, or associated with the admicelle. A blinking event (pink star) occurs when non-fluorescent Cy5 is reactivated by nearby BME.
Micelle-Assisted Blink (MAB) Microscopy
50 mM pH 8.5 Tris buffer was de-aerated by 2 min sonication in vacuum. Cy5-NHS ester (Lumiprobe) was stored for one week in water and thus hydrolyzed (as verified by mass spectrometry; Supporting Figure S10) to yield unreactive Cy5 dye. A blinking buffer was prepared by adding SDS (Sigma-Aldrich) and BME (American Bioanalytical) to the de-aerated Tris buffer to final concentrations of 36 mM and 50 mM, respectively. Blinking buffer containing 10 nM Cy5 was loaded into the de-aerated PDMS device, and flow of reagents into the nanochannels was regulated by applying 1–5 V between the two microchannel reservoirs (Tenma 72–6610 DC power supply) via tinned copper leads cleaned in Tris buffer. Resistance in the nanochannels limited the current to < 0.1 A. The channels were imaged in a standard epifluorescence configuration (Olympus IX71) with a 60x 1.20-NA water-immersion objective (Olympus) and a 3.3× beam expander. Cy5 fluorescence was excited with 3–10 kW/cm2 639-nm CW laser excitation (Coherent Cube), and the emission was recorded on an EMCCD detector (Photometrics Evolve) at a 37-Hz frame rate and 84 nm/pixel image size.
AFM Characterization
UV-curable epoxy (EPO-TEK UVO-114, Epoxy Technology) was introduced into nanochannels under each strain condition. After exposure to UV light to cure the epoxy replica, the PDMS nanochannel device was then soaked in pre-saturated 1 M tetrabutylammonium fluoride (Sigma-Aldrich) in tetrahydrofuran (Sigma-Aldrich) for six hours. The device was briefly washed with distilled water, and then the partially melted PDMS membrane was carefully peeled off. The surface layer of the epoxy replicate was probed by AFM (Veeco Instrument Inc.) in tapping mode.
Nanochannel Wall Functionalization
PDMS devices were rinsed with 70% ethanol, and then filled with 1% 3-aminopropyltriethoxysilane (APTES) (Fisher Scientific). The APTES-filled nanochannels were maintained open at 10% strain for 30 min and then closed (0% strain) for 2 h at room temperature. After rinsing with deionized water, microchannels were loaded with 0.6 μM Cy5-NHS ester (Lumiprobe) in 20 mM pH 8 phosphate buffer. The reaction was again maintained at room temperature for 30 min at 10% strain and then closed for 2 h. Labeled devices were imaged in the SDS/BME blinking buffer as described above, but without the added free Cy5. Reagent flow was regulated as described above, but with 6–10 V applied to the device to overcome the increased resistance introduced by surface functionalization.
Image Processing
Individual Cy5 dye fluorescence point spread functions (PSFs) were fit to two-dimensional Gaussian functions with the Matlab routine nlinfit. The localization error was estimated by the 95% confidence error on the fit. Super-resolution reconstructions of the molecules were generated by representing each fit molecule by a Gaussian with width equal to the localization error and amplitude proportional to the number of detected photons. The channel widths were obtained from the super-resolution images, each of which is composed of 7000–11000 single-molecule localizations. For each observation, a 10-μm length of the nanochannel was imaged, and the variance of the width was obtained by comparing the average widths for every 1-μm subsection.
RESULTS AND DISCUSSION
Above the critical micelle concentration of 8 mM (Supporting Figure S7), the SDS surfactant establishes an adsorbed monolayer, or admicelle, along the channel wall, and also self-associates into micelles to reduce the free energy.18, 19 Micelles are thermodynamically stable and dynamically equilibrate with free surfactant, and the development of micelles is a spontaneous and reversible cooperative entropy-driven process.20 Micellar liquid chromatography, in which micelles form a mobile phase21, 22 and admicelles constitute the stationary phase23, 24 has been widely applied. The solutes in such systems have been found to interact electrostatically with the charged outer layer of the ionic micelle and hydrophobically with their core.25 Similarly, in MAB microscopy, the redox reagent (BME) and the dye (Cy5) can be partitioned into micelles and transported by an applied electric field. Cy5 and BME are both distributed thermodynamically between the surface admicelle, the micelles, and the free solution, and can react with one another to activate the blinking process (Figure 1).
Single-Molecule MAB Microscopy of Nanochannels with Free Cy5
Parallel arrays of fluidic nanochannels with diamond-shaped cross-sections were generated between two lithographically-defined microfluidic channels by fracturing of oxidized PDMS (Supporting Figure S6).15, 16 A de-aerated imaging mixture (20 nM Cy5, 36 mM SDS and 50 mM BME in pH 8.5 Tris buffer) was added to the microchannel of a nitrogen-purged device. The imaging mixture was introduced into the nanochannels with a flow regulated by an applied electric field (1–5 V) to ensure a constant, controllable supply of Cy5 and BME. At the low concentration of 20 nM Cy5, each micelle contains at most one dye molecule at any time (Supporting Figure S7), making the setup suitable for SMF imaging. Though typical blink microscopy experiments are done with an enzymatic oxygen scavenger,11 we found that this enzyme blocked the nanochannel openings, and instead performed experiments in a nitrogen-purged environment. Individual Cy5 molecules in solution, in micelles, or adsorbed to the surface in the admicelle, were imaged under 639-nm excitation (3–10 kW/cm2). Over time, imaged Cy5 molecules turned dark and the fluorescence of other previously dark Cy5 molecules was restored by reaction with BME. In addition, fresh Cy5 dye molecules from the microchannel were also delivered by the applied field. The excitation intensity and the concentrations of Cy5 and BME were chosen such that only one fluorescent molecule was visible in a diffraction-limited spot at any time. Fluorescence images were recorded at a frame rate of 37 Hz and, in a post-processing step, molecules were localized by PSF fitting.
As depicted in Supporting Figure S1, the positions of all Cy5 molecules localized with precision better than 40 nm (as given by the statistical 95% confidence interval of a fit of the PSF to a 2D Gaussian function)8 were used to form a reconstructed image. For each strain condition, 7000 – 11000 molecular positions surpassed this precision threshold and were used to create the super-resolution image. Considered together, the precision of all the < 40-nm fits formed a left-skewed distribution with median 34.1 nm and mean 32.6 nm. To generate an image of each nanochannel, each molecule is plotted with a Gaussian profile with fixed standard deviation of 40 nm, and with amplitude proportional to the inverse of the localization precision; that is, molecules that are localized better are given more weight in the reconstruction. Two sorts of nanochannel arrays were investigated: “short” devices having a length of 100 μm, and “long” devices having a length of 300 μm, and in each device, multiple nanochannels were imaged at their middle and at their end (close to the microchannel) at strains between 1 % and 10 %. The images in Figures 2a, 2c, 2e, and 2g are super-resolution reconstructions of short channels at their middle, short channels at their end, long channels at their middle, and long channels at their end, respectively. The width of each channel at its middle and its end was found from the reconstructed images; these widths are plotted as a function of strain in Figures 2b, 2d, 2f, and 2h, which correspond to Figures 2a, 2c, 2e, and 2g, respectively. The error bars in Figures 2b, 2d, 2f, and 2h show the variance of the width along the ten 1-μm subsections of the 10-μm section of the nanochannel region that is imaged.
Figure 2.

Single-molecule MAB microscopy of Cy5 dye in nanochannels. (a,c,e,g) Super-resolution reconstruction images under 1.67 – 10% applied strain (left to right) of a) a 100-μm nanochannel at the center, c) a 100-μm nanochannel at the edge, e) a 300-μm nanochannel at the center, and g) a 300-μm nanochannel at the edge. The corresponding panels (b,d,f, and h, respectively) show the channel widths measured from the super-resolution image at each strain, and the error bars give the variance along the imaged length. Dotted lines are linear fits to the data. Insets indicate the region of interest investigated.
In Figures 2b, 2d, 2f, and 2h, the width vs. strain relationship follows a sigmoidal relation. The width is generally linearly proportional to the strain for intermediate applied strains (3%–7%), but the majority of devices demonstrate a sub-linear behavior at high strains (8%–10%). Interestingly, when considering nanochannels at zero applied strain by extrapolating a linear fit, we observe some loss of elasticity: the nanochannels do not fully close in the absence of strain. These apparent gaps, perhaps attributable to mechanical defects, are 179±28 nm and 151±15 nm for the end and middle of the short channels, respectively, and 180±36 nm and 73±58 nm for the end and middle of the long channels, respectively (Supporting Table S1), and may limit the size of molecule that can be excluded from the nanochannels in filtering applications. Knowledge of the gap size also provides a measure of the forces that would be exerted on a biomolecule trapped inside a strain-free nanochannel. Comparing the nanochannel end to its middle, the zero-strain gap is observed to be greater at the edge than at the center for both lengths of devices. This heterogeneity along the length of the nanochannel is very important since the ability to use a nanochannel as a molecular filter will depend on the smallest gap size. At non-zero applied strains, however, comparing the nanochannel centers to their edges shows that in general, the width of the nanochannels is greater at the center than at the edge. This is always true for the shorter devices (Figure 2a,b vs. Figure 2c,d), indicating that these 100-μm nanochannels pucker rather than opening in parallel. For the longer devices (Figure 2e,f vs. Figure 2g,h), the difference is more subtle, indicating that such long devices open in parallel at low strains and then begin to pucker at higher applied strains.
To verify that the Cy5 molecules were virtually immobile during the 27-ms imaging frames such that their images (point-spread functions, PSF) were not blurred by dye molecule motion, the data was reprocessed with fits to an asymmetric Gaussian function where widths parallel and perpendicular to the flow were allowed to differ. Though the full width at half maximum along the nanochannel axis (average = 479 nm, standard deviation = 62 nm) was found to be subtly greater than that perpendicular to the nanochannel axis (average = 438 nm, standard deviation = 41 nm), 87 % of the point-spread functions have a less than 1.25 ratio of parallel width to perpendicular width.
Comparison to Atomic Force Microscopy
Recent studies have shown that the resolution of single-molecule blink microscopy can approach that of atomic force microscopy (AFM).26, 27 Therefore, in confined environments such as the nanochannels studied in the present paper, where AFM cannot be applied directly, single-molecule MAB microscopy emerges as an excellent alternative. Though the present super-resolution fluorescence measurements cannot be directly compared to in situ AFM characterization, we validate our results by indirectly characterizing by AFM a set of 300-μm nanochannels made by an identical process. Here, UV-curable epoxy was used to form replicates of the nanochannels under different strains, and the cross-section of the replicates was measured by tapping mode AFM. Figure 3 compares the width vs. strain relationship measured by MAB microscopy at the center of eight different 300-μm nanochannels at each of six different strain conditions (48 measurements) in a single device (closed circles) to the width vs. strain relationship determined from AFM analysis of 273 such nanochannels, each fixed at a single applied strain (open circles). At 0 % strain, AFM measures an average channel width of 239±91 nm, agreeing with our finding from MAB microscopy that the channels remain open even after strain is released. In both methods, the error bars, corresponding to the standard error of the mean, indicate that in addition to the heterogeneity along the length of each individual nanochannel, there is also significant variability from one nanochannel to the next. Bulk studies based on electrical resistance can accurately estimate the cross-section of such PDMS nanochannels,16, 17 but these measurements fail to address the heterogeneity among the channels, a spread which increases with applied strain. Despite this heterogeneity, the comparison in Figure 3 finds that the width measurements obtained by AFM are generally larger than the MAB microscopy measurements.
Figure 3.

Comparison of width vs. strain characterization for 300-μm nanochannels by single-molecule MAB microscopy (closed circles) and by atomic force microscopy (open circles). Dotted lines are linear fits to the data.
Single-Molecule MAB Microscopy of Nanochannels with Surface-Immobilized Cy5
The difference between AFM and MAB microscopy may be explained in several ways. In AFM, the viscosity of the injected epoxy, as well as the thermal expansion of the epoxy upon curing, may lead to an overestimation of channel width. The indirect method of epoxy injection and curing followed by nanochannel removal and then AFM may also introduce artifacts in the measurement. On the other hand, if Cy5 or BME are excluded from the channel edges by the hydrophobicity of the nanochannels, the MAB microscopy technique could under-sample the channel width. To eliminate the possibility that the difference between AFM and optical microscopy is due to exclusion of the dye from the nanochannel edges, nanochannels covalently functionalized with Cy5 dye were prepared. Due to the oxidation process used to generate the nanochannels, a large number of free hydroxyl groups exist on the nanochannel corners, and so 300-μm nanochannels were labeled with Cy5 via an N-Hydroxysuccinimide (NHS) ester-primary amine coupling reaction based on functionalizing the hydroxyl groups as depicted in Figure 4.28, 29
Figure 4.
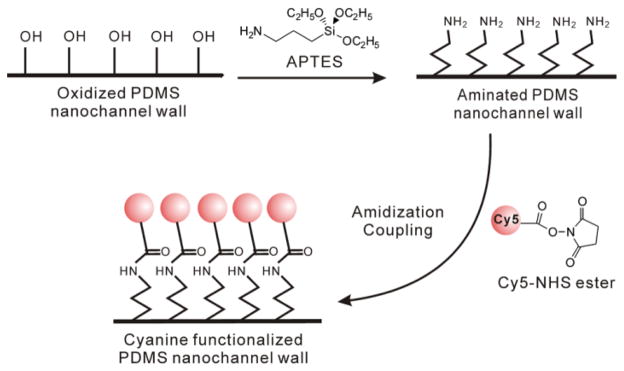
Cy5 labeling of PDMS nanochannel walls via an N-Hydroxysuccinimide (NHS) ester-primary amine coupling reaction based on functionalizing the hydroxyl groups.
The Cy5-functionalized nanochannels were imaged similarly to the unlabeled nanochannels described in Figures 1–3, but with free Cy5 omitted from the imaging buffer. These labeled devices had extremely hydrophobic walls, and so a higher electric field (6–10 V) was applied to induce flow of the blinking buffer and thus deliver a steady supply of BME to the immobilized Cy5 dye molecules. In fact, no flow could be achieved at all in the absence of SDS for the labeled devices, and even with SDS, only nanochannels with strains greater than 3% could be imaged. In these labeled devices, BME was dynamically incorporated into and released from the free micelles and the admicelle monolayer on the channel surface, enabling delivery of this reducing agent to the Cy5 labels for the blinking reaction (Supporting Figure S2).
A 15-μm length of the labeled nanochannels was photobleached by a 15-minute exposure to 639-nm CW laser illumination. Initially dark, fluorescence from subsets of dark Cy5 molecules was restored as the molecules reacted with BME, and the fluorescence images were collected and analyzed as for the unlabeled nanochannels. Figure 5b compares the strain vs. width relationship measured at the center of a long nanochannel labeled with Cy5 by NHS ester coupling (open circles, from Figure 5a) with the measurements of a long unlabeled nanochannel with introduced free Cy5 (closed circles, based on Figure 5c, which corresponds to Figure 2c). Interestingly, there is no significant difference between the two measurements in 5b, showing that the true accessible area imaged in situ with MAB microscopy with free Cy5 dye does not underestimate the width of PDMS nanochannels. This investigation of solution-accessible area in the nanochannel highlights the ability of MAB microscopy for providing biologically relevant information, such as local hydrophobicity, in these constrained geometries.
Figure 5.

Super-resolution images of the centers of 300-μm nanochannels imaged by single-molecule MAB microscopy with a) Cy5-functionalized walls and c) free Cy5 dye. b) Comparison of width vs. strain characterization for the two cases (open circles: labeled channels, closed circles: free dye). The error bars give the variance along the imaged length.
CONCLUSIONS
In this paper, we have introduced Single-Molecule Micelle-Assisted Blink (MAB) microscopy as a novel technique to probe the shape and size of structures with dimensions below the optical diffraction limit and geometries inaccessible to common alternatives like atomic-force microscopy (AFM). Here, we have characterized the size-strain relationship and heterogeneity of PDMS nanochannels, with ramifications for applications in sorting and macromolecule immobilization.15, 17 Importantly for these applications, we find that the nanochannels are smaller at their ends than in their middle if the strain is over 8% for long nanochannels and over 3% for short nanochannels. Additionally, after strain is relieved, all the nanochannels investigated retain a non-zero opening. As well, we determine that characterization of these channels by indirect AFM via epoxy replicates overestimates channel sizes. Single-molecule MAB microscopy was performed both with Cy5 dye in solution and with Cy5 dye covalently attached to the channel walls, and in both cases, these measurements give an accurate measurement of the channel widths. The free-dye method, in particular, can be easily extended in the future to other novel nanomaterials that have similarly constrained geometries which limit conventional single-molecule blink microscopy. The MAB technique, inspired by traditional applications of micelles like micellar liquid chromatography, should be widely applicable in imaging engineered nanomaterials, such as microporous coordination polymers,30 as well as biological nanostructures like enzyme clusters and viruses.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge support from a Burroughs Wellcome Career Award at the Scientific Interface and NIH grants HG004653 and 1-R01-EB006789-03.
Footnotes
Supporting Information Available: Raw data movie of single Cy5 molecules in a nanochannel, description of parallel experiments with covalently labeled nanochannels, in vitro investigations of Cy5 fluorescence in SDS, and mass spectroscopy data for the hydrolyzed Cy5 dye. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Quake SR, Scherer A. Science. 2000;290:1536–1539. doi: 10.1126/science.290.5496.1536. [DOI] [PubMed] [Google Scholar]
- 2.Edel JB, Hill EK, de Mello AJ. Analyst. 2001;126:1953–1957. doi: 10.1039/b106559a. [DOI] [PubMed] [Google Scholar]
- 3.Knight JB, Vishwanath A, Brody JP, Austin RH. Phys Rev Lett. 1998;80:3863–3866. [Google Scholar]
- 4.Thompson RE, Larson DR, Webb WW. Biophys J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 6.Hess ST, Girirajan TPK, Mason MD. Biophys J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rust MJ, Bates M, Zhuang X. Nat Meth. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biteen JS, Thompson MA, Tselentis NK, Bowman GR, Shapiro L, Moerner WE. Nat Meth. 2008;5:947–949. doi: 10.1038/NMETH.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharonov A, Hochstrasser RM. Proc Natl Acad Sci U S A. 2006;103:18911–18916. doi: 10.1073/pnas.0609643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biteen JS, Goley ED, Shapiro L, Moerner WE. ChemPhysChem. 2012;13:1007–1012. doi: 10.1002/cphc.201100686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vogelsang J, Cordes T, Forthmann C, Steinhauer C, Tinnefeld P. Proc Natl Acad Sci U S A. 2009;106:8107–8112. doi: 10.1073/pnas.0811875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinhauer C, Forthmann C, Vogelsang J, Tinnefeld P. J Am Chem Soc. 2008;130:16840–16841. doi: 10.1021/ja806590m. [DOI] [PubMed] [Google Scholar]
- 13.Stein IH, Capone S, Smit JH, Baumann F, Cordes T, Tinnefeld P. ChemPhysChem. 2012;13:931–937. doi: 10.1002/cphc.201100820. [DOI] [PubMed] [Google Scholar]
- 14.Shera EB, Seitzinger NK, Davis LM, Keller RA, Soper SA. Chem Phys Lett. 1990;174:553–557. [Google Scholar]
- 15.Huh D, Mills KL, Zhu X, Burns MA, Thouless MD, Takayama S. Nat Mater. 2007;6:424–428. doi: 10.1038/nmat1907. [DOI] [PubMed] [Google Scholar]
- 16.Mills KL, Huh D, Takayama S, Thouless MD. Lab Chip. 2010;10:1627–1630. doi: 10.1039/c000863j. [DOI] [PubMed] [Google Scholar]
- 17.Matsuoka T, Kim BC, Huang J, Douville NJ, Thouless MD, Takayama S. Nano Lett. doi: 10.1021/nl304063f. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukerjee P. Advan Colloid Interface Sci. 1967;1:241–275. [Google Scholar]
- 19.Yeskie MA, Harwell JH. J Phys Chem. 1988;92:2346–2352. [Google Scholar]
- 20.Puvvada S, Blankschtein D. J Chem Phys. 1990;92:3710–3724. [Google Scholar]
- 21.Terabe S, Otsuka K, Ando T. Anal Chem. 1985;57:834–841. [Google Scholar]
- 22.Rocklin RD, Ramsey RS, Ramsey JM. Anal Chem. 2000;72:5244–5249. doi: 10.1021/ac000578r. [DOI] [PubMed] [Google Scholar]
- 23.Dorsey JG, DeEchegaray MT, Landy JS. Anal Chem. 1983;55:924–928. [Google Scholar]
- 24.Hu W, Takeuchi T, Haraguchi H. Anal Chem. 1993;65:2204–2208. [Google Scholar]
- 25.Armstrong DW, Nome F. Anal Chem. 1981;53:1662–1666. [Google Scholar]
- 26.Cordes T, Strackharn M, Stahl SW, Summerer W, Steinhauer C, Forthmann C, Puchner EM, Vogelsang J, Gaub HE, Tinnefeld P. Nano Lett. 2010;10:645–651. doi: 10.1021/nl903730r. [DOI] [PubMed] [Google Scholar]
- 27.Duim WC, Chen B, Frydman J, Moerner WE. ChemPhysChem. 2011;12:2387–2389. doi: 10.1002/cphc.201100392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee KS, Ram RJ. Lab Chip. 2009;9:1618–1624. doi: 10.1039/b820924c. [DOI] [PubMed] [Google Scholar]
- 29.Xia Y, Ramgopal Y, Li H, Shang L, Srinivas P, Kickhoefer VA, Rome LH, Preiser PR, Boey F, Zhang H, Venkatraman SS. ACS Nano. 2010;4:1417–1424. doi: 10.1021/nn901167s. [DOI] [PubMed] [Google Scholar]
- 30.Liao Y, Yang SK, Koh K, Matzger AJ, Biteen JS. Nano Lett. 2012;12:3080–3085. doi: 10.1021/nl300971t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.