

Abstract
Aromatase inhibitors (AIs) are important drugs for treating postmenopausal patients with hormone receptor (HR)-positive breast cancer. However, acquired resistance to AI therapies is a significant problem. Our study has revealed that the histone deacetylase inhibitor LBH589 treatment abrogated growth of AI-resistant cells in vitro and in vivo, causing cell cycle G2/M arrest, and induced apoptosis. LBH589 treatment also reduce the levels of NF-κB1 which overexpresses when AI-resistance develops. Analyzing paired tumor specimens from 12 patients, we found that NF-κB1 expression was increased in recurrent AI-resistant tumors as compared to the paired primary tumors before AI treatment. This finding was consistent with up-regulated NF-κB1 expression seen in a collection of well-established AI-resistant cell lines. Furthermore, knockdown of NF-κB1 expression significantly suppressed the proliferation of AI-resistant cells. Treatment of AI-resistant cell lines with LBH589 repressed NF-κB1 mRNA and protein expression. In addition, LBH589 treatment abrogated growth of AI-resistant tumors in mice, and was associated with significantly decreased levels of NF-κB1 in tumors. In all, our findings strongly support further investigation of LBH589 as a novel therapeutic strategy for patients with AI-resistant breast cancer, in part by suppressing the NF-κB1 pathway.
Keywords: Acquired aromatase inhibitor resistance, LBH589, NF-κB1
Introduction
Since the third-generation aromatase inhibitors (AIs) (i.e., anastrozole, letrozole, and exemestane) have been shown to be more effective in prolonging disease free survival than tamoxifen [1], these AIs are now the first choice endocrine therapy for initial adjuvant therapy or for metastatic disease instead of tamoxifen, especially for postmenopausal patients with hormone receptor (HR)-positive breast cancer. Unfortunately, approximately 20–25% of patients will eventually develop resistance to AIs within a decade after adjuvant treatment [2–4]. Furthermore, most of patients with metastatic disease develop resistance to AIs around 9–10 months. Thus, acquired AI-resistance is a major problem in the management of HR-positive breast cancer, and it is critical to find new strategy to treat acquired AI-resistance.
The mechanisms of acquired AI resistance are poorly understood. There are two main reasons for insufficient progress in this area. The research has been delayed due to the lack of suitable preclinical models. A significant number of experiments to study AI resistance have been performed using models without the presence of aromatase and estrogen receptor (ER), i.e., non-physiologically relevant models. Our laboratories have prepared ER- and aromatase-positive breast cancer cell lines (i.e., MCF-7aro and T47Daro) and generated a series of acquired AI as well as tamoxifen-resistant cell lines [5]. These cell lines allow us to evaluate the molecular differences between AI- and tamoxifen resistance in a simultaneous manner [6], and to identify essential players involved in acquired AI resistance. Another difficulty to study acquired AI resistance is the shortage of paired tumor tissues from the same patients before AI treatment and after cancer recurrence, for confirming the findings from preclinical studies. The lack of these paired samples is due to unpredictability for patients to acquire AI resistance, and surgery is not typical option when cancer recurs.
Using our models, we have found that LBH589 (panobinostat), a histone deacetylase (HDAC) inhibitor, is a potentially effective drug to treat acquired AI resistance. HDACs are enzymes involved in the remodeling of chromatin, and have a key role in the epigenetic regulation of gene expression [7]. Small-molecule HDAC inhibitors can function as anticancer drugs to suppress a variety of HDAC-regulated activities, including apoptosis, cell-cycle arrest in G2/M phase, and cell differentiation [8]. LBH589 is a novel cinnamic hydroxamaic acid analog HDAC inhibitor. It has been evaluated through phase I/II clinical trials on multiple myeloma, and hematological and solid tumors [9].
In this paper, we will also present results to support nuclear factor-κB1 (NF-κB1) as an important target of LBH589 and to play an important role in acquired AI resistance through studies using our AI-resistant cell models and analyses of a collection of paired clinical specimens from the same patients before AI treatment and after cancer recurrence following AI treatment. Several studies indicate that NF-κB is activated in breast cancer cells, where it enhances cell proliferation and suppresses apoptosis [10]. The Rel or NF-κB family can form hetero- or homo-dimeric combination of five members, NF-κB1 (p50 and its precursor p105), NF-κB2 (p52 and its precursor p100), RelA (p65), RelB and c-Rel. Activation of the classic NF-κB complex, composed of NF-κB1/RelA heterodimers, is detected in a portion of breast cancers [11]. Furthermore, several studies have demonstrated that some HDAC inhibitors suppress NF-κB pathway signaling [12, 13]. Moreover, DNA microarray profiling of colon cancer cells treated with the HDAC inhibitors varinostat and LBH589 revealed that this treatment caused significant down-regulation of NF-κB1 expression [14]. Therefore, down-regulation of NF-κB1 is thought to be one mechanism by which HDAC inhibitors induce apoptotic effects in cancer cells.
Our data in this paper support the further evaluation of LBH589 as a new therapeutic option for AI-resistant breast cancer. In addition, our results indicate that NF-κB1 is potentially an important gene involved in AI resistance and a marker of LBH589-mediated inhibition.
Materials and Methods
Cell culture and reagents
The ER-positive aromatase-overexpressing MCF-7 and T47D cell lines, MCF-7aro and T47Daro, respectively, were generated in our laboratory as previously described [15]. We also used AI-resistant cell lines derived from MCF-7aro, which are resistant to anastrozole (Ana-R), letrozole (Let-R), or exemestane (Exe-R), as well as a long-term estrogen deprived MCF-7aro line (LTEDaro) [6,16]. A long-term estrogen deprived T47Daro (T47DaroLTED) line was recently generated and also used. T47DaroLTED cells were cultured in RPMI-1640, without phenol red, and supplemented with 10% charcol dextran treated FBS. LBH589 was provided by Novartis Pharmaceutical Inc. TNFα was purchased from R&D Systems.
Cell proliferation assay, western blotting and cell cycle analysis
Please see supplementary materials for procedures.
Animal studies
Female, 6- to 7-week-old ovariectomized, BALB/c Nu-Nu athymic mice were purchased from the National Cancer Institute, and housed/maintained on a 12 h light/dark cycle in the City of Hope Animal Facility. All animal research procedures were approved by the City of Hope Institutional Animal Care and Use Committee. The design of the animal study was shown in Fig. 6A. At 8 weeks-of-age, mice were s.c. injected in the hind flank with MCF7aro cells (2 × 107 cells/injection site) mixed with an equal volume of Matrigel (BD Bioscience). Mice were also s.c. implanted with 7.5 mg/60 days time-release androstenedione pellets (Innovative Research of America). Tumor size and body weights were monitored three times per week as an indicator of overall health. All mice received food and water ad libitum. To establish the exemestane resistant xenograft, mice started to be s.c injected with exemestane (250 μg/mouse) daily when tumors became 400 mm3. Tumors responded to exemestane treatment initially; however, they eventually became resistant in several weeks. When resistant tumors reached 700 mm3, mice were randomly divided into two groups of 7 mice each, then treated with exemestane only (daily) or exemestane (daily) and LBH589 (20 mg/kg, three times per week, intraperitoneally), as a design to evaluate the effect of a new drug (i.e., LBH589) together with an established drug (i.e., exemestane). After three weeks of LBH589 treatment, mice were euthanized via CO2 asphyxiation. Tumors were removed, weighted, and sent for H&E histologic staining through the City of Hope Pathology Core Facility (Please see supplementary materials for procedures for detail methods).
Fig. 6. Overexpression of NF-κB1 induced AI-resistance and plays an important role for cell proliferation.
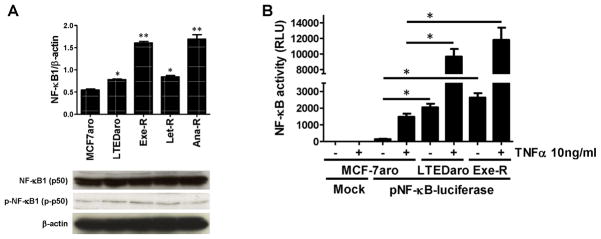

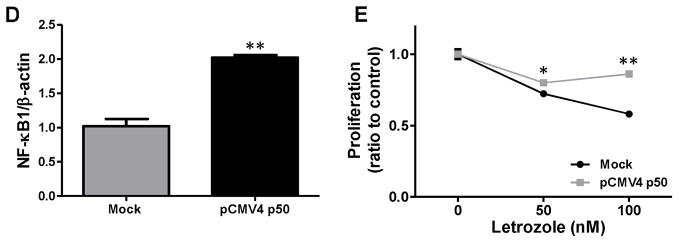
(A) Basal NF-κB1 mRNA and protein expressions were shown in AI-responsive MCF-7aro cells and AI-resistant cells. mRNA levels were determined by real-time PCR. *, p < 0.01. Protein levels were evaluated by western blotting using indicated antibodies against NF-κB1 and p-NF-κB1. (B) The basal transcriptional activity of NF-κB is higher in AI-resistant cells (LTEDaro and Exe-R) than MCF-7aro, and remarkably higher after TNFα (10 ng/ml) stimulation for an hour. NF-κB activity was evaluated via the pNF-κB-luciferase reporter assay. Luciferase activity was assayed after 24 hours and normalized to total protein concentration. Data are expressed as relative luciferase units (RLU). Columns, mean; bars, SE. *, p < 0.01. (C) siRNA-mediated knockdown of NF-κB1 knockdown significantly suppresses the proliferation of AI-resistant cells. MCF-7aro, LTEDaro, Exe-R, Let-R and Ana-R cells were transfected with control siRNA or NF-κB1 siRNA. Cell viability was assessed by MTT assay for 7 days after transfection. Five replicates were performed for each measurement. *, p < 0.01. (D) Real-time PCR analysis of NF-κB1 mRNA expression after pCMV4 p50 transfection. Gene expression was normalized to β-actin. **, p < 0.01. (E) Over-expression of NF-κB induces AI resistance in AI-responsive MCF-7aro cells. The cells were transfected with mock or pCMV4 p50 (NF-κB1 over-expression) plasmid and treated with letrozole at the indicated concentrations for four days. Cell viability was assessed by MTT assay in triplicate, and the mean and standard error were calculated. Data are shown as a ratio of treated samples to untreated control, mean ± SE.
Real-time PCR analysis
Please see supplementary materials for procedures.
Microarray analysis, statistical processing and Ingenuity Pathway Analysis (IPA)
Please see supplementary materials for procedures
Clinical samples
Three hospitals (Kumamoto City Hospital, Hamanomachi Hospital, and Hokkaido Cancer Center) and one clinic (Shimada Breast & Surgery Clinic) were involved in enrolling the subset of 12 patients with primary breast tumors who developed recurrent tumors following AI therapy. All patients gave informed consent, and the study was approved by the appropriate institutional review boards. Patient characteristics are shown in Table 1. For immunohistochemical analysis, the tumor specimen slides (3–4μm) were stained using NF-κB1 antibody (H-119, Santa Cruz Biotechnology) (Please see supplementary materials for procedures for detail methods).
Table 1.
Clinical characteristics of primary and recurrent tumors from 12 patients receiving AI-adjuvant therapy.
| Patient | Age | Primary Tumor (before AJ therapy)
|
Adjuvant HTE | Time to recurrence (Month) | Recurrent Tumor (After AI therapy)
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| pTNMA | ERB | PRC | HER2D | Site | ER | PR | HER2 | ||||
| #1 | 50 | pT4cN1M0 | + | − | 2 | LetF | 30 | Supraclavicular LNI | + | + | 0 |
| #2 | 67 | pT2N1M0 | + | + | 3 | AnaG | 18 | Intra-pectoral LN | + | + | 1 |
| #3 | 72 | pT1N0M0 | + | − | 2 | ExeH | 43 | Subcutaneous nodule | + | − | 0 |
| #4 | 69 | pT1N0M0 | + | − | 3 | Ana | 58 | Subcutaneous nodule | + | + | 3 |
| #5 | 60 | pT2N1M0 | + | − | 1 | Ana | 27 | Subcutaneous nodule | + | − | 2 |
| #6 | 52 | pT2N1M0 | + | + | 0 | Ana | 33 | Subclavicular LN | + | + | 0 |
| #7 | 59 | pT4bN1M0 | + | + | 1 | Ana | 53 | Supraclavicular LN | − | − | 1 |
| #8 | 78 | pT1N1M0 | + | + | 1 | Ana | 72 | Supraclavicular LN | + | + | 2 |
| #9 | 53 | pT2N0M0 | + | + | 0 | Exe | 72 | Subclavicular LN | + | + | 0 |
| #10 | 53 | pT2N1M0 | + | + | 1 | Exe | 68 | Subcutaneous nodule | + | + | 1 |
| #11 | 78 | pT3N1M0 | + | − | 0 | Exe | 24 | Subcutaneous nodule | + | − | 0 |
| #12 | 53 | pT2N2M0 | + | + | 0 | Ana | 41 | Ipslateral breast | + | +/− | 1 |
pTNM: pathological tumor-lymph nodes-metastasis classification according to the Union Internationale Contra le Cancer (UICC).
ER: Estrogen receptor status.
PR: Progesterone receptor status.
HER2/neu status: HER2.
HT: Hormone therapy.
Let: Letrozole.
Ana: Anastrozole.
Exe: Exemestane.
LN: Lymph nodes.
Luciferase assay, NF-κB1 siRNA treatment and NF-κB1 transfection
Please see supplementary materials for procedures.
Statistics
To assess statistical significance, values were compared with controls by either Student’s t test or one-way ANOVA, followed by Dunnett’s multiple range test (α = 0.05) using Prism GraphPad 5 software (GraphPad Software, Inc.).
RESULTS
LBH589 inhibits the proliferation of AI resistance breast cancer cells
To study cellular response to AIs and the mechanisms of acquired AI-resistance, we used the previously generated AI-responsive cell line MCF-7aro [15] and AI-resistant variants of MCF-7aro created following in vitro selection against each AI (i.e., Exe-R, Let-R and Ana-R) or long-term culture in the absence of estrogen (i.e., LTEDaro) [16]. Our molecular characterization has implicated LTEDaro as a model of late-stage AI resistance as it fails to respond to any of the three AIs [16].
MCF-7aro, LTEDaro, and three AI-resistant cell lines were exposed to increasing concentrations of LBH589. This drug inhibited proliferation of all cell lines in a dose-dependent manner (Fig. 1A). To confirm the effect of LBH589, we used another ER-positive aromatase-overexpressing cell line, T47Daro [15], which was derived from T47D, and T47DaroLTED, which is long-term estrogen deprived T47Daro. T47Daro proliferates in response to testosterone and estrogen treatment and T47DaroLTED proliferates without the need of testosterone or estrogen (Fig. 1C). The proliferation of T47Daro was inhibited by letrozole treatment, while T47DaroLTED cells were resistant to letrozole treatment (Fig. 1D). Similar to its effect on AI-resistant MCF-7aro cell lines, LBH589 effectively suppressed growth of both AI-responsive T47Daro and AI-resistant T47DaroLTED cells in a dose-dependent manner (Fig. 1B).
Fig. 1. Inhibitory effect of LBH589 on the proliferation of AI-resistant cell lines.

(A) LBH589 decreased cell proliferation of all cell lines in a dose-dependent manner. Cells were treated with the indicated concentrations of LBH589 (LBH) or DMSO (vehicle control) for 6 days and medium was replaced every 72 h. Cell viability was assessed by MTT assay. Five replicates were performed for each measurement, and the mean and standard error were calculated. Data are shown as a ratio of treated samples to untreated controls. (B) LBH589 treatment significantly reduced proliferation of T47Daro and T47DaroLTED cells. Cells were treated same as in (A). (C) Proliferation of the AI-resistant cell line T47DaroLTED was not stimulated by hormone treatment (1 nM estradiol [E2] or 1 nM testosterone [T]). (D) Proliferation of T47Daro cells, but not AI-resistant T47DaroLTED cells, was inhibited by letrozole. Cell proliferation after six days of treatment with the indicated concentrations of letrozole (Let) was measured by MTT assay. *, p < 0.05; **, p < 0.01.
LBH589 induces apoptosis and cell cycle arrest in AI resistance cell lines
Western blotting analyses further demonstrated that LBH589 treatment induced expression of apoptosis-associated proteins, as indicated by elevated levels of cleaved-poly (ADP-ribose) polymerase (cleaved-PARP, a hallmark of apoptosis) and the pro-apoptotic protein Bax, as well as by reduced levels of the pro-survival protein Bcl-xL, and this induction was dose-dependent (Fig. 2A). In addition, levels of p21WAF1/CIP1, were enhanced by LBH589 treatment, confirming that LBH589 inhibits cell cycle progression.
Fig. 2. LBH589 induced apoptosis and cell cycle arrest in AI-resistance cells.

(A) LBH589 suppresses the expression of these apoptosis and cell cycle related molecules in a dose-dependent manner. Western blot analyses were performed on MCF-7aro and Exe-R cells that were treated with DMSO (control) or the indicated concentrations of LBH589 for 48 hours. (B) LBH589 induces cell cycle arrest. Flow cytometric analysis of DNA content of MCF-7aro and Exe-R cells treated with LBH589 (20 nM) or DMSO for 48 hours. Both non-adherent and adherent particles and cells were stained with propidium iodide.
To confirm LBH589-induced apoptosis, flow cytometry was applied to examine the effects of LBH589 treatment on the cell cycle distribution of MCF-7aro and Exe-R cells. Both cell lines displayed a significant G2/M arrest accompanied by a sharp reduction in cells in the G1 and S phases after treatment with LBH589 (Fig. 2B); the percentage of cells in S phase decreased from 18.0% in untreated controls to 2.6% for MCF-7aro and from 9.5% to 1.6% for Exe-R.
LBH589 inhibits the Exe-R tumor growth in vivo
To evaluate the inhibitory effects of LBH589 on AI resistance in vivo, we used the exemestane-resistant MCF7aro xenograft model (described in Materials and Methods) (Fig. 3A). Initially, to confirm the biochemical effect of LBH589 in vivo, 4 mice from both exemestane alone and exemestane+LBH589 groups were sacrificed 48 hours after the first injection of LBH589. The remaining mice (7 mice per group) were treated for 22 days. LBH589 treatment significantly inhibited the growth of exemestane-resistant tumors; tumor weight at the end of experiment was significantly less in mice treated with LBH589 than in control mice (Fig. 3B). No mice in the LBH589 treatment groups showed significant body weight loss (Fig. 3C), indicating that the LBH589 treatment was well tolerated. Consistent with the effect of LBH589 on gross characteristics of the tumors, proliferation (assessed by Ki-67 staining) of tumor cells was significantly decreased in LBH589-treated mice and apoptosis (assessed by staining for cleaved PARP) of tumor cells was significantly increased (Fig. 3D).
Fig. 3. Evaluation of the in vivo activity of LBH589.

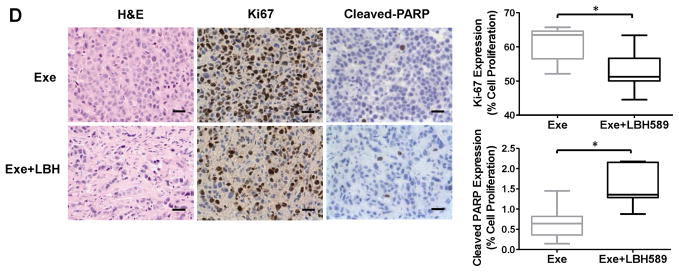
(A) Experimental design to evaluate the LBH589 effects using Exe-resistant mouse xenograft. Exe, exemestane; LBH, LBH589. (B) Tumor volumes (left) and weights (right) of control (exemestane only) and treated (exemestane- and LBH589-treated) mice. (C) Body weights of control and LBH589-treated mice. (D) Immunohistochemical analysis of cell proliferation (Ki-67) and apoptosis (cleaved PARP) in tumors of LBH589-treated mice. Bar, 10 μm in reprehensive picture. Graph showed that percentage of positive stain cells. n=7/group *, p < 0.05
Identification of differentially expressed genes in LBH589-treated cells
To identify the effective targets of LBH589 treatment, as a nonbiased approach, we compared gene expression profiles of three types of human cancer cell lines (H295R, HeLa and MCF-7her2), with and without LBH589 treatment (50 nM, 24 hours). We compared the LBH589-modulated genes identified from these cell lines and analyzed their functional grouping by IPA. These microarray datasets contain probes for 20,140 unique genes, of which 335 were down-regulated in all three cell lines after treatment with LBH589 (Fig. 4A). The IPA networks of those genes most consistently down-regulated by LBH589 were enriched for genes associated with cell death, cellular function and maintenance, and the cell cycle. In particular, the most down-regulated network (Fig. 4B) contained NF-κB1 and CFLAR (encoding cFLIP). NF-κB1 expression was significantly down-regulated (FDR < 0.05) in all three cell lines after treatment with LBH589 (−2.2-fold change in H295R, −3.6-fold change in HeLa, and −4.6-fold change in MCF-7her2). Moreover, CFLAR, a downstream gene regulated by NF-κB, is an apoptosis regulator that was significantly down-regulated (FDR < 0.05) in all three cell lines (−3.4-fold change in H295R, −2.5-fold change in HeLa, and −3.3-fold change in MCF-7her2). Therefore, these results indicate that LBH589 suppresses the levels and function of NF-κB1.
Fig. 4. Comparison of global gene expression profiles of three LBH589-treated cancer cell lines.

(A) Three cell lines (H295R, HeLa and MCF7her2) were treated with 50 nM LBH589 for 4 hours and gene expression analyzed by Affymetrix Human Gene 1.0 ST Array. Expression of 335 genes changed among all three cell lines after treatment. Genes with an FDR-adjusted p-value < 0.05 were considered to be differentially expressed and subjected to Venn analysis. Venn analysis was first performed by analyzing cell-line-specific alterations in differentially expressed genes in each cell line, and then by analyzing overlap between gene lists from different lines. (B) Gene network of down-regulated genes by LBH589. The most down-regulated network identified by Ingenuity Pathway Analysis contains 27 genes.
Levels of NF-κB1 are elevated in AI-resistant breast cancers compared to corresponding primary tumors
To define NF-κB1 expression changes that occur during the development of resistance, major efforts were made to identify paired (primary and acquired AI-resistant) specimens from the same patients. Potential participants were identified from three hospitals and one clinic in Japan, between 2003 and 2011, 2,205 breast cancer patients were postmenopausal and had ER-positive cancer, while 1,454 were treated with upfront AI. Ninety-four patients had experienced cancer recurrence by the end of 2011. With extensive search, we were able to obtain tissue specimens from 12 patients prior to AI treatment and at the time of cancer recurrence, during which cancer recurrence was diagnosed at the same hospitals. The time to recurrence ranged from 18 to 72 months. Three patients had recurrence at 12 months after adjuvant AI therapy for five years. The clinical characteristics for these patients, whose recurrent tumors were removed by a core needle or excisional biopsy, are listed in Table 1. These specimens are valuable because they are truly paired tissues from the same patients before and after they acquired AI resistance, allowing for a direct comparison of molecular features associated with changes of the response to AIs. Tumor specimens were stained using NF-κB1 antibody to detect nuclear NF-κB1 protein levels as an activation marker (Fig. 5A). The nuclear staining scores of 9/12 samples were significantly higher in recurrent tumor cells in comparison with those of paired primary tumor cells (Wilcoxon matched-pairs signed rank test; p=0.0248, n=12), 2 were not changed and 1 decreased (Fig. 5B).
Fig. 5. NF-κB1 expression in paired primary and recurrent AI-resistant tumors from the same patients.

(A) Photomicrographs of tissue samples immunostained by NF-κB1 antibody showing representative intensity scores. Positive cells show a dark brown or black nuclear signal. These representative tumors obtained a total IHC score of 0.95 (left, proportion score=1.0, intensity score=0.95), 1.9 (middle, proportion score=0.95, intensity score=2), and 2.7 (right, proportion score=0.9, intensity score=3). Scores were calculated from proportion and intensity scores obtained from immunohistochemical evaluation of nuclear NF-κB1. Bar, 10 μm. (B) Graph shows immunohistochemical (IHC) scores of NF-κB1 expression in recurrent tumors and paired primary tumors (*, p < 0.05).
NF-κB1expression is constitutively up-regulated and NF-κB activity is increased in AI-resistant cells
We found that AI-resistant cells exhibited higher levels of NF-κB1 mRNA compared with AI-responsive MCF-7aro cells (Fig. 6A; p<0.01). Expression of the RelA and NF-κB2 subunits did not differ between AI-responsive and -resistant cell lines (data not shown). The levels of NF-κB1 and p-NF-κB1 were also examined by western blot analysis. While the results indicate that the levels of p-NF-κB1 were higher in AI-resistant cells, to demonstrate higher transcriptional activity of NF-κB in resistant cells, we performed a luciferase reporter assay in which MCF-7aro and AI-resistant cells were co-transfected with a pNF-κB-luciferase reporter plasmid or mock control. The NF-κB promoter activity was 10-fold greater (p<0.01) in AI-resistant cells compared with the AI-sensitive MCF-7aro cells (Fig. 6B). Moreover, the transcriptional activity of NF-κB was remarkably elevated after a one-hour treatment with TNF (10 ng/ml), especially in AI-resistant cells such as LTEDaro and Exe-R.
NF-κB1 knockdown suppresses the proliferation of AI-resistant cells
To confirm the functional significance of NF-κB1, we transfected MCF-7aro, LTEDaro, and three AI-resistant cell lines with siRNA against NF-κB1. As the expression of NF-κB1 was effectively suppressed by siRNA in all cell lines (Supplementary Fig. 1), siRNA treatment significantly suppressed the proliferation of LTEDaro and three AI-resistant cell lines, as assessed by MTT assay, but proliferation of MCF-7aro was less affected (Fig. 6C). These data suggest that NF-κB1 plays a more indispensable role in AI-resistant cells than in AI-responsive cells, such as MCF-7aro.
NF-κB1 over-expression induces AI resistance
To confirm that over-expression of NF-κB1 induces AI resistance, we assessed the response of NF-κB1-over-expressing MCF-7aro cells to letrozole. Real time PCR analysis confirmed that NF-κB1 expression was higher in cells transfected with the NF-κB1 plasmid versus mock transfected cells (Fig. 6D). MCF-7aro cells transfected with an NF-κB1 over-expression plasmid were significantly less responsive to letrozole (50 nM and 100 nM) treatment as shown by an increase in cell proliferation relative to mock transfected cells (Fig. 6E). Taken together, these results strongly confirm an essential role of NF-κB1 in acquisition of AI resistance.
LBH589 reduces NF-κB1 expression in breast cancer
NF-κB1 expression was significantly reduced in all five cell lines after treatment with LBH589 (Fig. 7A). Also, NF-κB1 expression was significantly reduced in T47Daro and T47DaroLTED cells after LBH589 treatment (Fig. 7B). Base-line NF-κB1 expression was found to be significantly higher in T47DaroLTED cells than in AI-responsive T47Daro cells (Fig. 7B).
Fig. 7. Gene expression changes in the NF-κB signaling pathway in AI-resistant cell lines.
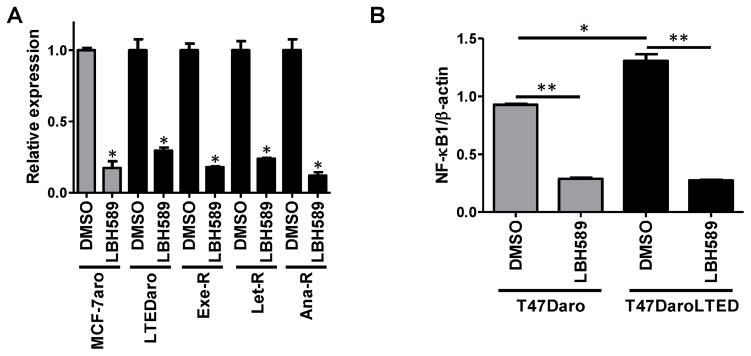
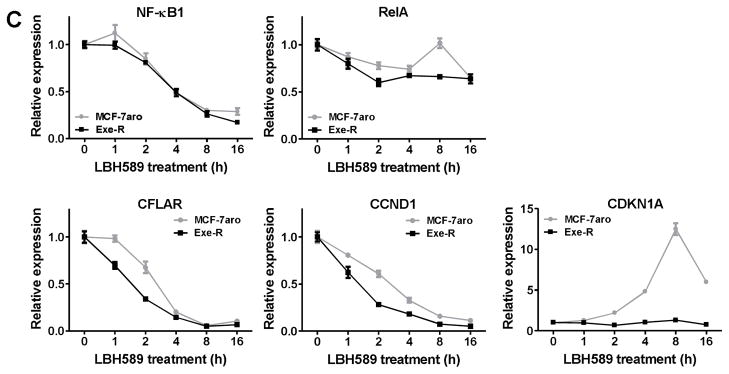
(A) LBH589 treatment significantly reduced NF-κB1 mRNA expression. MCF-7aro, LTEDaro, Exe-R, Let-R and Ana-R cells were treated with 20 nM LBH589 for 24 hours. Real-time PCR was performed to evaluate changes in gene expression. Gene expression was normalized to β-actin. *, p < 0.01. (B) Base-line NF-κB1 mRNA expression was higher in T47DaroLTED cells than in T47Daro cells, and decreased in both cell lines after LBH589 treatment. Cells were treated with 20 nM LBH589 for 24 hours, and NF-κB1 mRNA expression was analyzed by real time PCR. Gene expression was normalized to β-actin. *, p < 0.05; **, p < 0.01. (C) MCF-7aro and Exe-R were treated with 20 nM LBH589 for 16 hours, and total RNA was extracted at the indicated time points. Real-time PCR was used to assess expression of NF-κB1, RelA, NF-κB target genes (CFLAR and CCND1), and CDKN1A. Gene expression was normalized to β-actin mRNA. Data are expressed as a ratio of treated samples to untreated controls and shown as mean ± SE.
LBH589 changed the expression of NF-κB1-targeted genes
Real-time PCR throughout a 16-hour time course after LBH589 treatment revealed that expression of NF-κB1-targeted genes (CFLAR, and CCND1 [encoding Cyclin D1]) decreased in both cell lines (Fig. 7C). However, neither cell line showed a remarkable change in RelA expression. Expression of CDKN1A (encoding p21WAF1/CIP1, cyclin-dependent kinase inhibitor 1) increased dramatically in response to LBH589 treatment in AI-responsive cells and peaked after eight-hour treatment, but not in AI-resistant cells (Fig. 7C).
LBH589 inhibits the Exe-R tumor growth through suppression of NF-κB1 pathway in vivo
NF-κB1 mRNA expression was significantly decreased in tumors from mice treated with LBH589 for 48 hours (Fig. 8A) and at the end of experiment (Fig. 8B). To further demonstrate the inhibitory effect of LBH589 on AI resistance, we established an exemestane-resistant cell line, LTET (long-term exemestane-treated), from the above resistant tumors (Please see supplementary materials for procedures for detail methods). We confirmed the exemestane resistance of the LTET cell line, compared to the parental cell line MCF-7aro. Again, LBH589 was effective at suppressing the proliferation of the parental cell line MCF-7aro and LTET cells in a dose-dependent manner (Fig. 8C). Also, NF-κB1 expression was significantly higher in LTET cells than MCF-7aro cells (Fig. 8D). These results serve as additional evidence that AI-resistant cells, derived from resistant tumors, express higher levels of NF-κB1 and are responsive to LBH589 treatment.
Fig. 8. NF-κB1 expression analysis in xenograft tumors treated with LBH589.

(A) Tumors from LBH589-treated or control mice (four mice/group) with exemestane-resistant MCF-7aro tumors were harvested 48 hours after mice received a single injection of LBH589. Real time PCR was performed to quantify the levels of NFκB mRNA in tumors. (B) Real time PCR analysis of levels of NF-κB mRNA present in tumors at the end of experiment. n = 7 mice/group. (C) LBH589 decreased cell proliferation in the parental cell line, MCF7aro, in a dose-dependent manner but not in long-term exemestane-treated LTET cells established from MCF-7aro tumors from mice treated with exemestane. The LTET and MCF-7aro cells were treated with the indicated concentrations of exemestane or LBH589 for 6 days and proliferation analyzed by MTT assay. (D) Baseline NF-κB1 mRNA expression was significantly increased in LTET cells as compared to the parental MCF7aro cells, as determined by real-time PCR analysis. *, p < 0.05; **, p < 0.01.
LBH589 decreased the NF-κB1 (p50) protein and its phosphorylated form in nuclear fraction
LBH589 suppressed expression of NF-κB1(p50) and reduced levels of phosphorylated NF-κB1 (p-p50), which is the form that enhances transcriptional activity of the NF-κB complex in MCF7aro and Exe-R cells treated with LBH589 in a dose dependent manner (Fig. 9A). Protein levels of the HSP90 client proteins (ER, and AKT) and the NF-κB downstream molecule (cFLIP) were reduced in a dose-dependent manner (Fig. 9A).
Fig. 9. Protein expression changes of NF-κB1 and related molecules by LBH589 treatment.
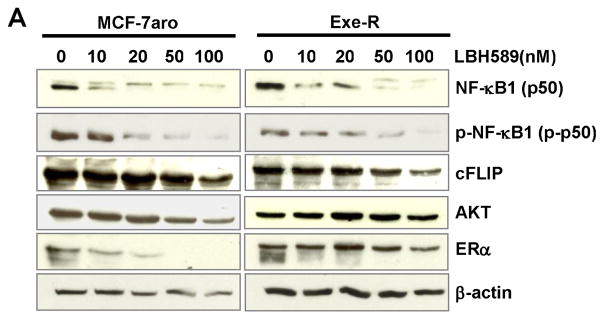

(A) LBH589 suppressed NF-κB1 (p50) and it phosphorylated form protein expression in a dose-dependent manner. Also, blots were probed with the indicated antibodies against NF-κB downstream molecules (cFLIP) and LBH589 targets (AKT and ER ). LBH589 suppresses the expression of these molecules in a dose-dependent manner. Western blot analyses were performed on MCF-7aro and Exe-R cells that were treated with DMSO (control) or the indicated concentrations of LBH589 for 48 hours. (B) Levels of NF-κB1 and p-NF-κB1 were decreased in both nuclear and cytoplasm in a dose-dependent manner. Cells were fractionated into cytoplasm and nuclear fractions after treatment with DMSO or LBH589 for 48 hours, and protein levels of interesting proteins in the cytoplasm and nucleus were examined by western blotting. Lamin A/C (70kD) was used as a nuclear marker.
To gain further insight on molecular action of LBH589, we fractionated and extracted cytoplasmic and nuclear proteins. Levels of NF-κB1 (p50) and p-NF-κB1(p-p50) were decreased in both the cytoplasm and nuclear fractions after LBH589 treatment, but those of RelA and p-RelA remained relatively constant (Fig. 9B), which was consistent with the time course study of mRNA expression (Fig. 7C). Lamin A/C, a nuclear membrane structural protein, is cleaved by caspase-6 and serves as a marker for caspase-6 activation. In addition, cleavage of Lamin A/C into a large (40–45 kDa) and a small (28 kDa) fragment, results in nuclear disregulation and apoptosis. Therefore, cleaved Lamin A/C is a marker of nuclear fraction and of apoptosis.
Discussion
AIs are effective in treating postmenopausal patients with ER-positive breast cancer. However, resistance to these drugs remains a major problem in the management of this cancer. In this study, we showed that LBH589 inhibited growth of AI-resistant cancer cells, both in vitro and in vivo, by inhibiting NF-κB1 expression, inducing apoptosis and cell cycle arrest. Moreover we demonstrated enhanced NF-κB1 expression in acquired AI-resistance tumor specimens and in our acquired AI-resistance models, determined that NF-κB1 promotes the growth of AI resistant cells.
Vorinostat was the first HDAC inhibitor approved by the FDA for the treatment of cancer. The combination of vorinostat and tamoxifen is well tolerated and exhibited promising activity in hormone resistance breast cancer [17]. Recently, it was reported that the histone deacetylase inhibitor Entinostat together with exemestane significantly improves the progression free survival of postmenopausal women with ER-positive advanced breast cancer who had ≤ 1 prior chemotherapy and had progressed on a non-steroidal AI [18]. LBH589 is a highly potent HDACi with demonstrated antitumor activities at low nanomolar concentration in several preclinical studies and its clinical efficacy is currently under investigation in several clinical trials such as recurrent high-grade glioma and refractory Hodgkin Lymphoma [19–21]. Not all HDAC inhibitors function identically; thus, it is not unexpected that some HDAC inhibitors activate [22–24] and some inhibit [12,13,25–28] NF-κB transcription. It has been found that more genes are repressed than activated in tumor samples treated with LBH589 [29]. Our current study shows that NF-κB1 is significantly suppressed by LBH589 treatment, similar to that reported previously in colon cancer [14].
It is essential to recognize the difficulty of collecting paired tumor tissues from the same patients before AI treatment and after the acquisition of AI resistance. We were able to collect the primary tumor specimens and those after cancer recurred from 12 patients, even after up to six years of treatment with AIs. We also found that the rate of acquired AI resistance (6.5%) to the adjuvant therapy in Japanese patients is lower than that in patients from Western countries. All recurrent tumors, except one (#7), remained ER-positive, supporting that ER-mediated pathways remain important in AI-resistant tumors, as suggested by our preclinical studies [6]. Although the primary tumors from two of twelve patients were HER2-positive, these patients responded to AI treatment for 18 and 58 months, respectively. Therefore, there were no obvious correlations between HER2 levels and time to recurrence. Using those samples, we have found that tumors from patients who acquired AI resistance had higher expression of NF-κB1 compared to primary tumors from the same patients before AI treatment. This is the first report to show that NF-κB1 is expressed at higher levels in acquired AI-resistant patient samples. In support of these findings, NF-κB1 is over-expressed in AI-resistant cell lines. It is worthwhile to note that expression of NF-κB1 is not elevated in tumors after a two week-treatment or three-month treatment with AIs [30], indicating that a short term treatment of AI does not change the expression of NF-κB1.
NF-κB forms an inducible transcription factor complex that plays a crucial role in regulating the inflammatory, immune, and anti-apoptotic reactions. Notably, most signaling pathways that lead to tamoxifen resistance share a common mechanistic link with activation of the gene-regulating NF-κB complex [31–33]. Higher NF-κB1 DNA-binding values were associated with significantly reduced disease free survival among a collection of 81 ER-positive primary breast cancers [32]. Patients whose primary breast cancers over-expressed NF-κB1 were significantly more likely to develop distal metastases and experienced shorter metastasis-free survival than those whose cancers did not [32]. As Zhou et al. have suggested the role of NF-κB1 in de novo tamoxifen resistance, we now report the functional significance of NF-κB1 in acquired AI resistance. It should be pointed out that RelA has been also reported to play an important role in tamoxifen resistance [31]. Activate NF-κB1 (p50) homodimer can be transcriptional repressors or activators both [34]. Our nuclear expression results showed that LBH589 could reduce expression of NF-κB1 (p50) and its phosphorated form which was promoting NF-κB1 in nuclear fraction, potentially to interact with RelA in the AI-resistance cells. Our results suggested that LBH589 inhibited tumor growth through reduction of NF-κB1 (p50) /RelA dimers.
Our preclinical studies have revealed that NF-κB1 is more important in promoting the growth of AI-resistant cells than of AI-responsive cells. NF-κB1 knockdown further showed that NF-κB1 is indispensable in AI-resistant cells but less important in responsive cells. Since LBH589 suppresses the proliferation of both AI-resistant and -responsive cells, our data indicate that LBH589 can suppress the proliferation of ER- and aromatase-positive breast cancer through more than one mechanism. As a comparison, LBH589 is less effective in the suppression of MCF-10A, a noncancerous breast epithelial cells (supplementary Fig. 2). Previous studies have also indicated CDKN1A is readily up-regulated in response to HDAC inhibitor treatment in vitro [35]. CDKN1A was found to be most up-regulated at a relatively early phase (eight hours) after LBH589 treatment in AI-sensitive cells, but not in AI-resistant cells. Thus, cell cycle signaling via p21WAF1/CIP1 is likely to be more sensitive to LBH589 in AI-responsive cells than AI-resistant cells. Our in vitro results suggest that LBH589 inhibits ER-positive AI-responsive cells, which is consistent with our previous finding that combination therapy of an AI and LBH589 can synergistically inhibit the proliferation of AI-responsive cells [36]. In the current study, we focused on elucidating the mechanism underlying LBH589’s effects on AI-resistant cell lines, and showed that LBH589 inhibits the NF-κB1 pathway. After patients become AI resistance, there are no effective way to treat them, thus LBH589 may be potential choice for the patients who acquire AI resistance.
LBH589 inhibits HDAC6, which has also been identified as a major deacetylase of heat shock protein (HSP) 90 [37]. HSP90 is a cytoplasmic chaperone whose client proteins include nuclear receptors and kinases important for oncogenesis. HSP90 acts to prevent their ubiquitinylation and proteasomal degradation. LBH589 induces tumor cell apoptosis that HSP90 and its co-chaperones modulate through down-reglation on AKT, TNF-α and NF-κB function [38]. Also, HDAC inhibitors potentiate anti-estrogen therapy and/or lead to a reversal of hormone therapy resistance through the interaction of HDAC6 with the ER and HSP90 chaperone complex [39]. Our results further show that AKT and ER protein levels are reduced in cells treated with LBH589. It has been reported that NF-κB activity can be up-regulated by acetylation of RelA/p65 in the nucleus, which is mediated by histone acetyl transferases (e.g., P300, CBP, and PCAF) [24]. Interleukin-1 receptor-associated kinase 1 (IRAK1) is the upstream gene that stimulates the NF-κB pathway [40]. Our microarray analyses indicated that expression of CREBBP (encoding CBP/p300) and IRAK1 was decreased, consistent with previous reports [14]. These factors may contribute the mechanisms by which LBH589 inhibits the NF-κB pathway.
The induced NF-κB has been reported to increases aromatase expression by enhancing transcription of several proinflammatory mediators [41,42] and synergistically activates ER [43]. Interestingly, our previous studies have demonstrated that LBH589 can suppress aromatase expression through the down regulation of promoters I.3 and II [36]. Thus, LBH589 can overcome AI resistance through multiple pathways. For triple negative breast cancer, it was reported that LBH589 suppressed the proliferation [44], and moreover HDAC inhibitor increased ER expression [45,46], suggesting that the inhibitory mechanisms of this drug are different in ER+ and ER− breast cancers.
Recent exciting clinical observations from other groups, together with our preclinical data for LBH589, strongly suggests that LBH589 offers a novel therapeutic strategy to improve the response of hormone-responsive cancers to AIs and thereby overcome AI resistance.
Supplementary Material
Acknowledgments
The research was supported by Susan G. Komen for the Cure (KG080161) and by Grant Number P30 CA033572 from the National Cancer Institute. The authors would like to thank Ms. Sophia Loera, Ms. Lucy Brown, and Dr. Keely Walker for pathologic technical assistance, flow cytometry, and editing this manuscript, and Molly Storer, Meng Wu, and Peter Nguyen for helping with the cell proliferation, animal, and NF-κB1 plasmid experiment, respectively.
Footnotes
Conflict of interest: There are no potential conflicts of interest.
References
- 1.Josefsson ML, Leinster SJ. Aromatase inhibitors versus tamoxifen as adjuvant hormonal therapy for oestrogen sensitive early breast cancer in post-menopausal women: meta-analyses of monotherapy, sequenced therapy and extended therapy. Breast (Edinburgh, Scotland) 2010;19 (2):76–83. doi: 10.1016/j.breast.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 2.Cuzick J, Sestak I, Baum M, Buzdar A, Howell A, Dowsett M, Forbes JF. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial. Lancet Oncol. 2010;11 (12):1135–1141. doi: 10.1016/S1470-2045(10)70257-6. [DOI] [PubMed] [Google Scholar]
- 3.van de Velde CJ, Rea D, Seynaeve C, Putter H, Hasenburg A, Vannetzel JM, Paridaens R, Markopoulos C, Hozumi Y, Hille ET, Kieback DG, Asmar L, Smeets J, Nortier JW, Hadji P, Bartlett JM, Jones SE. Adjuvant tamoxifen and exemestane in early breast cancer (TEAM): a randomised phase 3 trial. Lancet. 2011;377 (9762):321–331. doi: 10.1016/S0140-6736(10)62312-4. [DOI] [PubMed] [Google Scholar]
- 4.Regan MM, Neven P, Giobbie-Hurder A, Goldhirsch A, Ejlertsen B, Mauriac L, Forbes JF, Smith I, Lang I, Wardley A, Rabaglio M, Price KN, Gelber RD, Coates AS, Thurlimann B. Assessment of letrozole and tamoxifen alone and in sequence for postmenopausal women with steroid hormone receptor-positive breast cancer: the BIG 1–98 randomised clinical trial at 8.1 years median follow-up. Lancet Oncol. 2011;12 (12):1101–1108. doi: 10.1016/S1470-2045(11)70270-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong C, Chen S. The development, application and limitations of breast cancer cell lines to study tamoxifen and aromatase inhibitor resistance. The Journal of steroid biochemistry and molecular biology. 2012 doi: 10.1016/j.jsbmb.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masri S, Phung S, Wang X, Wu X, Yuan YC, Wagman L, Chen S. Genome-wide analysis of aromatase inhibitor-resistant, tamoxifen-resistant, and long-term estrogen-deprived cells reveals a role for estrogen receptor. Cancer research. 2008;68 (12):4910–4918. doi: 10.1158/0008-5472.CAN-08-0303. [DOI] [PubMed] [Google Scholar]
- 7.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5 (9):769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 8.Scuto A, Kirschbaum M, Kowolik C, Kretzner L, Juhasz A, Atadja P, Pullarkat V, Bhatia R, Forman S, Yen Y, Jove R. The novel histone deacetylase inhibitor, LBH589, induces expression of DNA damage response genes and apoptosis in Ph- acute lymphoblastic leukemia cells. Blood. 2008;111 (10):5093–5100. doi: 10.1182/blood-2007-10-117762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prince HM, Bishton MJ, Harrison SJ. Clinical studies of histone deacetylase inhibitors. Clin Cancer Res. 2009;15 (12):3958–3969. doi: 10.1158/1078-0432.CCR-08-2785. [DOI] [PubMed] [Google Scholar]
- 10.Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25 (51):6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 11.Cogswell PC, Guttridge DC, Funkhouser WK, Baldwin AS., Jr Selective activation of NF-kappa B subunits in human breast cancer: potential roles for NF-kappa B2/p52 and for Bcl-3. Oncogene. 2000;19 (9):1123–1131. doi: 10.1038/sj.onc.1203412. [DOI] [PubMed] [Google Scholar]
- 12.Kramer OH, Baus D, Knauer SK, Stein S, Jager E, Stauber RH, Grez M, Pfitzner E, Heinzel T. Acetylation of Stat1 modulates NF-kappaB activity. Genes Dev. 2006;20 (4):473–485. doi: 10.1101/gad.364306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu J, Colburn NH. Histone deacetylase inhibition down-regulates cyclin D1 transcription by inhibiting nuclear factor-kappaB/p65 DNA binding. Mol Cancer Res. 2005;3 (2):100–109. doi: 10.1158/1541-7786.MCR-04-0070. [DOI] [PubMed] [Google Scholar]
- 14.LaBonte MJ, Wilson PM, Fazzone W, Groshen S, Lenz HJ, Ladner RD. DNA microarray profiling of genes differentially regulated by the histone deacetylase inhibitors vorinostat and LBH589 in colon cancer cell lines. BMC Med Genomics. 2009;2:67. doi: 10.1186/1755-8794-2-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun XZ, Zhou D, Chen S. Autocrine and paracrine actions of breast tumor aromatase. A three-dimensional cell culture study involving aromatase transfected MCF-7 and T-47D cells. The Journal of steroid biochemistry and molecular biology. 1997;63 (1–3):29–36. doi: 10.1016/s0960-0760(97)00068-x. [DOI] [PubMed] [Google Scholar]
- 16.Chen S, Masri S, Wang X, Phung S, Yuan YC, Wu X. What do we know about the mechanisms of aromatase inhibitor resistance? The Journal of steroid biochemistry and molecular biology. 2006;102 (1–5):232–240. doi: 10.1016/j.jsbmb.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, Melisko M, Ismail-Khan R, Rugo H, Moasser M, Minton SE. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. British journal of cancer. 2011;104 (12):1828–1835. doi: 10.1038/bjc.2011.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yardley D. Entinostat, a novel histone deacetylase inhibitor, added to exemestane improves PFS in advanced breast cancer in a randomized, phase II, double-blind study. Cancer Res. 2011;71 (24 Suppl):118s. [Google Scholar]
- 19.Drappatz J, Lee EQ, Hammond S, Grimm SA, Norden AD, Beroukhim R, Gerard M, Schiff D, Chi AS, Batchelor TT, Doherty LM, Ciampa AS, Lafrankie DC, Ruland S, Snodgrass SM, Raizer JJ, Wen PY. Phase I study of panobinostat in combination with bevacizumab for recurrent high-grade glioma. Journal of neuro-oncology. 2012;107 (1):133–138. doi: 10.1007/s11060-011-0717-z. [DOI] [PubMed] [Google Scholar]
- 20.Jones SF, Bendell JC, Infante JR, Spigel DR, Thompson DS, Yardley DA, Greco FA, Murphy PB, Burris HA., 3rd A phase I study of panobinostat in combination with gemcitabine in the treatment of solid tumors. Clin Adv Hematol Oncol. 2011;9 (3):225–230. [PubMed] [Google Scholar]
- 21.Dickinson M, Ritchie D, DeAngelo DJ, Spencer A, Ottmann OG, Fischer T, Bhalla KN, Liu A, Parker K, Scott JW, Bishton M, Prince HM. Preliminary evidence of disease response to the pan deacetylase inhibitor panobinostat (LBH589) in refractory Hodgkin Lymphoma. British journal of haematology. 2009;147 (1):97–101. doi: 10.1111/j.1365-2141.2009.07837.x. [DOI] [PubMed] [Google Scholar]
- 22.Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293 (5535):1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 23.Mayo MW, Denlinger CE, Broad RM, Yeung F, Reilly ET, Shi Y, Jones DR. Ineffectiveness of histone deacetylase inhibitors to induce apoptosis involves the transcriptional activation of NF-kappa B through the Akt pathway. J Biol Chem. 2003;278 (21):18980–18989. doi: 10.1074/jbc.M211695200. [DOI] [PubMed] [Google Scholar]
- 24.Dai Y, Rahmani M, Dent P, Grant S. Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol Cell Biol. 2005;25 (13):5429–5444. doi: 10.1128/MCB.25.13.5429-5444.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lerebours F, Vacher S, Andrieu C, Espie M, Marty M, Lidereau R, Bieche I. NF-kappa B genes have a major role in inflammatory breast cancer. BMC Cancer. 2008;8:41. doi: 10.1186/1471-2407-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imre G, Gekeler V, Leja A, Beckers T, Boehm M. Histone deacetylase inhibitors suppress the inducibility of nuclear factor-kappaB by tumor necrosis factor-alpha receptor-1 down-regulation. Cancer research. 2006;66 (10):5409–5418. doi: 10.1158/0008-5472.CAN-05-4225. [DOI] [PubMed] [Google Scholar]
- 27.Furumai R, Ito A, Ogawa K, Maeda S, Saito A, Nishino N, Horinouchi S, Yoshida M. Histone deacetylase inhibitors block nuclear factor-kappaB-dependent transcription by interfering with RNA polymerase II recruitment. Cancer Sci. 2011;102 (5):1081–1087. doi: 10.1111/j.1349-7006.2011.01904.x. [DOI] [PubMed] [Google Scholar]
- 28.Song W, Tai YT, Tian Z, Hideshima T, Chauhan D, Nanjappa P, Exley MA, Anderson KC, Munshi NC. HDAC inhibition by LBH589 affects the phenotype and function of human myeloid dendritic cells. Leukemia. 2011;25 (1):161–168. doi: 10.1038/leu.2010.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prystowsky MB, Adomako A, Smith RV, Kawachi N, McKimpson W, Atadja P, Chen Q, Schlecht NF, Parish JL, Childs G, Belbin TJ. The histone deacetylase inhibitor LBH589 inhibits expression of mitotic genes causing G2/M arrest and cell death in head and neck squamous cell carcinoma cell lines. J Pathol. 2009;218 (4):467–477. doi: 10.1002/path.2554. [DOI] [PubMed] [Google Scholar]
- 30.Miller WR, Larionov A. Molecular effects of oestrogen deprivation in breast cancer. Mol Cell Endocrinol. 2011;340 (2):127–136. doi: 10.1016/j.mce.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 31.Nehra R, Riggins RB, Shajahan AN, Zwart A, Crawford AC, Clarke R. BCL2 and CASP8 regulation by NF-kappaB differentially affect mitochondrial function and cell fate in antiestrogen-sensitive and -resistant breast cancer cells. Faseb J. 2010;24 (6):2040–2055. doi: 10.1096/fj.09-138305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Y, Eppenberger-Castori S, Marx C, Yau C, Scott GK, Eppenberger U, Benz CC. Activation of nuclear factor-kappaB (NFkappaB) identifies a high-risk subset of hormone-dependent breast cancers. The international journal of biochemistry & cell biology. 2005;37 (5):1130–1144. doi: 10.1016/j.biocel.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 33.Zhou Y, Eppenberger-Castori S, Eppenberger U, Benz CC. The NFkappaB pathway and endocrine-resistant breast cancer. Endocrine-related cancer. 2005;12(Suppl 1):S37–46. doi: 10.1677/erc.1.00977. [DOI] [PubMed] [Google Scholar]
- 34.Pereira SG, Oakley F. Nuclear factor-kappaB1: regulation and function. The international journal of biochemistry & cell biology. 2008;40 (8):1425–1430. doi: 10.1016/j.biocel.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 35.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S A. 2000;97 (18):10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen S, Ye J, Kijima I, Evans D. The HDAC inhibitor LBH589 (panobinostat) is an inhibitory modulator of aromatase gene expression. Proc Natl Acad Sci U S A. 2010;107 (24):11032–11037. doi: 10.1073/pnas.1000917107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18 (5):601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 38.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5 (10):761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 39.Thomas S, Munster PN. Histone deacetylase inhibitor induced modulation of anti-estrogen therapy. Cancer Lett. 2009;280 (2):184–191. doi: 10.1016/j.canlet.2008.12.026. [DOI] [PubMed] [Google Scholar]
- 40.Vig E, Green M, Liu Y, Donner DB, Mukaida N, Goebl MG, Harrington MA. Modulation of tumor necrosis factor and interleukin-1-dependent NF-kappaB activity by mPLK/IRAK. J Biol Chem. 1999;274 (19):13077–13084. doi: 10.1074/jbc.274.19.13077. [DOI] [PubMed] [Google Scholar]
- 41.Morris PG, Hudis CA, Giri D, Morrow M, Falcone DJ, Zhou XK, Du B, Brogi E, Crawford CB, Kopelovich L, Subbaramaiah K, Dannenberg AJ. Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer prevention research (Philadelphia, Pa. 2011;4 (7):1021–1029. doi: 10.1158/1940-6207.CAPR-11-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Subbaramaiah K, Howe LR, Bhardwaj P, Du B, Gravaghi C, Yantiss RK, Zhou XK, Blaho VA, Hla T, Yang P, Kopelovich L, Hudis CA, Dannenberg AJ. Obesity is associated with inflammation and elevated aromatase expression in the mouse mammary gland. Cancer prevention research (Philadelphia, Pa. 2011;4 (3):329–346. doi: 10.1158/1940-6207.CAPR-10-0381. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Baumgarten SC, Frasor J. Minireview: Inflammation: An Instigator of More Aggressive Estrogen Receptor (ER) Positive Breast Cancers. Mol Endocrinol. 2012;26 (3):360–371. doi: 10.1210/me.2011-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tate CR, Rhodes LV, Segar HC, Driver JL, Pounder FN, Burow ME, Collins-Burow BM. Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat. Breast Cancer Res. 2012;14 (3):R79. doi: 10.1186/bcr3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sabnis GJ, Goloubeva O, Chumsri S, Nguyen N, Sukumar S, Brodie AM. Functional activation of the estrogen receptor-alpha and aromatase by the HDAC inhibitor entinostat sensitizes ER-negative tumors to letrozole. Cancer research. 2011;71 (5):1893–1903. doi: 10.1158/0008-5472.CAN-10-2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou Q, Atadja P, Davidson NE. Histone deacetylase inhibitor LBH589 reactivates silenced estrogen receptor alpha (ER) gene expression without loss of DNA hypermethylation. Cancer biology & therapy. 2007;6 (1):64–69. doi: 10.4161/cbt.6.1.3549. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.