

Abstract
Neuroinflammation is an important component of Alzheimer's disease (AD) pathogenesis and has been implicated in neurodegeneration. Interleukin-1 (IL-1), a potent inflammatory cytokine in the CNS, is chronically upregulated in human AD and believed to serve as part of a vicious inflammatory cycle that drives AD pathology. To further understand the role of IL-1β in AD pathogenesis, we used an inducible model of sustained IL-1β overexpression (IL-1βXAT) developed in our laboratory. The triple transgenic mouse model of AD, which develops plaques and tangles later in its life cycle, was bred with IL-1βXAT mice, and effects of IL-1β overexpression on AD pathology were assessed in F1 progeny. After 1 and 3 months of transgene expression, we found robust increases in tau phosphorylation despite an ∼70–80% reduction in amyloid load and fourfold to sixfold increase in plaque-associated microglia, as well as evidence of greater microglial activation at the site of inflammation. We also found evidence of increased p38 mitogen-activated protein kinase and glycogen synthase kinase-3β activity, which are believed to contribute to tau phosphorylation. Thus, neuroinflammation regulates amyloid and tau pathology in opposing ways, suggesting that it provides a link between amyloid accumulation and changes in tau and raising concerns about the use of immunomodulatory therapies in AD.
Introduction
Alzheimer's disease (AD) is the most common form of dementia in the elderly. In addition to deposition of amyloid plaques and neurofibrillary tangles, neuroinflammation has been recognized as an important component of AD pathology (McGeer et al., 1987; Griffin et al., 1989; Rogers et al., 1999; Akiyama et al., 2000; Heneka and O'Banion, 2007), which parallels disease severity (Sheng et al., 1997a,b). One neuroinflammatory mediator upregulated in AD is interleukin-1 (IL-1), a major proinflammatory cytokine in the brain. IL-1-positive activated microglia and S100β-positive reactive astrocytes are closely associated with amyloid plaques in the human AD brain (Griffin et al., 1989), an association recapitulated in murine AD models (Benzing et al., 1999; Kitazawa et al., 2005). Based on the ability of IL-1 and amyloid precursor protein (APP) to mutually regulate the expression and processing of each other in vitro, numerous studies have suggested that IL-1 and β-amyloid (Aβ) participate in a vicious cytokine cycle that, once induced, drives AD pathology (Goldgaber et al., 1989; Gray and Patel, 1993; Sheng et al., 1996; Barger and Harmon, 1997; Griffin et al., 1998; Meda et al., 1999; Liao et al., 2004).
To experimentally investigate the effects of IL-1 on AD pathology, our laboratory developed the first in vivo model of conditional IL-1β overexpression (Shaftel et al., 2007b). This model displays a robust neuroinflammatory phenotype with prominent gliosis and leukocyte recruitment in the brain alongside elevations in other proinflammatory cytokines, which is mediated by IL-1 receptor (IL-1R) type 1 (Shaftel et al., 2007a; Shaftel et al., 2007b). We previously reported an abrogating effect of IL-1β on amyloid burden without overt neurodegeneration in APP/presenilin-1 (PS-1) mice, suggesting an adaptive role for IL-1β in AD (Shaftel et al., 2007b, 2008; Matousek et al., 2012).
In the current study, our goal was to investigate the role of sustained overexpression of IL-1β on both amyloid and tau pathology in vivo. To this end, we used the triple transgenic (3xTgAD) murine model of AD, which harbors human mutant APP, PS1, and TAU transgenes and develops both senile plaques and neurofibrillary tangles later in life (Oddo et al., 2003). 3xTgAD mice were crossed to IL-1βXAT mice, and human IL-1β (hIL-1β) expression was induced in the subiculum of 15-month-old F1 progeny. After 1 or 3 months of transgene expression, we found a 70–80% reduction in amyloid load in 3xTgAD/IL-1βXAT mice in the subiculum, consistent with our previous results. Interestingly, we also found twofold to fourfold elevated phospho-tau at different epitopes in the hippocampus and evidence for increased activity of glycogen synthase kinase-3β (GSK3β) and p38 mitogen-activated protein kinase (MAPK), both of which have been implicated in tau phosphorylation (Sheng et al., 2000, 2001; Cho and Johnson, 2003; Li et al., 2003). These data indicate that neuroinflammation affects amyloid and tau pathology differentially in our model and suggests that the interrelationship between amyloid and tau is complex. It also lends credence to the idea that neuroinflammation can initiate tau phosphorylation as a bystander effect in an attempt to clear amyloid.
Materials and Methods
Transgenic mice.
All animal procedures were reviewed and approved by the University Committee on Animal Resources of the University of Rochester Medical Center for compliance with federal regulations before the initiation of the study. Two lines of transgenic mice were used in the present study. The construction and characterization of the IL-1βXAT mice on a C57BL/6 background has been described previously (Shaftel et al., 2007a,b). The 3xTgAD mice (Oddo et al., 2003) express mutated human APP, PS1, and TAU genes under the control of the Thy1.2 regulatory element and develop plaques and tangles later in life. The 3xTgAD mice were bred to IL-1βXAT mice, and 15-month-old progeny were used for this study with littermate controls.
FIV–Cre.
The construction and packaging of the feline immunodeficiency virus (FIV) has been described previously (Lai et al., 2006). Briefly, the FIV–Cre virus encodes a modified version of Cre recombinase with a nuclear localization signal and a V5 epitope tag under the control of a cytomegalovirus promoter. FIV–Cre was packaged to a final titer of ∼1 × 107 viral particles/ml.
Stereotactic activation of the IL-1β transgene.
Mice were anesthetized with 1.75% isoflurane in 30% oxygen and 70% nitrogen and secured in a David Kopf Instruments stereotactic apparatus using ear bars. The scalp was thoroughly rubbed with betadine. An incision was made on the top of the skull, a 0.5 mm burr hole was drilled in the skull at 2.0 mm horizontal from lambda, and a 33 gauge needle connected to a 10 μl syringe preloaded with virus was lowered 1.25 mm from the brain surface over 2 min. A Micro-1 microsyringe pump controller (World Precision Instruments) was used to inject 1.5 μl of virus at a constant rate over 10 min. After a 5 min viral diffusion (∼1.5 × 104 infectious viral particles), the needle was raised slowly over 2 min, the burr hole was sealed with Ethicon bone wax, and soft tissues were sutured using 6-0 Dermalon. For this study, all mice received a unilateral injection of FIV–Cre in the subiculum to control for viral transduction. For the 16 month time point, a total of 34 (17 3xTgAD and 17 3xTgAD/IL-1βXAT) animals were injected (eight males and nine females in each group). For the 18 month time point, a total of 16 animals (eight 3xTgAD and eight 3xTgAD/IL-1βXAT) were injected (four males and four females in each group).
Immunohistochemistry.
Mice were deeply anesthetized with a mixture of ketamine (100 mg/kg, i.p.) and xylazine (10 mg/kg, i.p) and perfused intracardially with 0.15 m PBS containing 0.5% sodium nitrite (w/v) and 2 IU heparin/ml, followed by 4% ice-cold paraformaldehyde (PFA), pH 7.2, in 0.15 m PBS. Brains were collected, fixed for 2 additional hours in 4% PFA, pH 7.2, at 4°C, equilibrated in 30% sucrose in PBS overnight, frozen in cold isopentane, and stored at −80°C. They were then cryosectioned into 30 μm sections on a −25°C freezing stage microtome, and free-floating sections were stored in a cryoprotectant solution until assayed. For most immunohistochemical protocols, sections were washed to remove cryoprotectant, incubated in 3% H2O2, washed, blocked with 10% normal goat serum, and incubated in primary antibody for 48 h. They were then washed and incubated with secondary antibody for 2 h at room temperature and developed with Elite ABC kit and 3,3-diaminobenzidine (Vector Laboratories). Sections were mounted, cleared, and coverslipped in DPX (VWR). For fluorescent immunostaining, sections were incubated with secondary antibodies bound to Alexa/Dylight fluorophores (Invitrogen) and coverslipped in Prolong gold (Vector Laboratories). For biotinylated primary antibodies, the secondary antibody incubation was omitted and an Alexa fluorophore conjugated to streptavidin was used. Congo red staining was performed per kit instructions (Sigma). Primary and secondary antibody dilutions included the following: biotinylated 6E10 (Covance) at 1:2000; GFAP (Dako) at 1:2000; Iba-1 (Wako) at 1:5000; V5 (Novus) at 1:100; CD68 (Serotec) at 1:1000; AT180 and HT7 (biotinylated) (Thermo Fisher Scientific) at 1:10; anti-pT205 and pS396 (Invitrogen) at 1:5000; biotinylated goat anti-rabbit (Vector Laboratories) at 1:2000; goat anti-rat IgG Alexa Fluor 488 (Invitrogen) at 1:400; goat anti-rabbit IgG Alexa Fluor 647 (Invitrogen) at 1:400; donkey anti-rat IgG Alexa Fluor 594 (Invitrogen) at 1:400; donkey anti-rabbit IgG Alexa Fluor 488 (Invitrogen) at 1:400; donkey anti-goat IgG Alexa Fluor 488 (Invitrogen) at 1:400; and streptavidin conjugated to Alexa Fluor 647 (Invitrogen) at 1:400. Sections were blocked in 3% normal donkey serum whenever secondary antibodies prepared in donkeys were used.
Image acquisition and analyses.
Photomicrographic images were captured by a Carl Zeiss Axioplan IIi microscope equipped with a SPOT camera (Advanced Diagnostics) linked to a SPOT advanced software running on a 64-bit Microsoft Windows XP computer. For 6E10 histological analysis, 8-bit grayscale images were captured from sections closest (<360 μm) to the injection site using a 5× objective. Subicular boundaries were defined in NIH ImageJ (http://rsb.info.nih.gov/ij/), and images were converted into binaries, after which area fractions were determined using a threshold to minimize artifact. Phospho-tau and total tau stained sections were imaged using a 10× objective and analyzed similarly. Representative fluorescent images were captured by an Olympus FV1000 confocal microscope using a 40×/100× oil-immersion objective and analyzed in Fluoview 3.0 software.
The number of Iba1-positive cells overlapping with plaques were quantified as follows. Sections <120 μm distant from the FIV–Cre injection site were chosen for each animal. A low-resolution photomicrograph showing Congo-Red-positive plaques was used as a map of the subicular area for each section analyzed. Numbers of Iba1/DAPI double-positive cells in direct contact with each Congophilic plaque ≥10 μm were counted using a Carl Zeiss Axiolplan IIi microscope under a 40× dry objective lens by adjusting focus through the z-axis. The number of Iba1 and DAPI double-positive cells associated with each plaque was recorded in the map, and a mean number of Iba1-positive cells was calculated for each hemisphere in every section; this was ultimately used to calculate an ipsilateral/contralateral (I/C) ratio for every section. We analyzed three sections bearing the subiculum in each animal and derived a mean I/C ratio for every animal that was used in the statistical comparison of different genotypes.
For quantitative CD68 analysis, subicular sections were coimmunostained with 6E10, Iba1, and DAPI. Plaques ≥10 μm were imaged with a 40× dry objective on the Carl Zeiss Axioplan IIi attached to a Sensicam (Cooke Corporation), using Slidebook 5.0 software (Intelligent Imaging Innovations). Z-stacks were deconvolved, and projection images were analyzed for area fraction determination in NIH ImageJ. Briefly, a 500 pixel circular ROI was drawn around the plaque, discarding all pixel information outside the circle. The merged image within this 500 pixel ROI was split into individual fluorescent channels and converted into individual 8-bit images. Images were then thresholded to minimize artifact and area fractions calculated. Six to seven plaques were analyzed per hemisphere. In some animals, ipsilateral hemispheres had less than six plaques, in which case all plaques present were analyzed. A minimum of two subicular sections were analyzed per animal. Mean area fraction values for CD68/6E10 and CD68/Iba1 per hemisphere were used to calculate a I/C ratio/section.
Final images and layout were created using Adobe Photoshop CS5 and Adobe Illustrator CS5 (Adobe Systems).
ELISA and Western blots.
Mice were deeply anesthetized with a mixture of ketamine (100 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.), perfused intracardially with 0.15 m PBS containing 0.5% sodium nitrite (w/v) and 2 IU heparin/ml. Hippocampi were quickly dissected, frozen in isopentane, and stored at −80°C until additional processing. Hippocampi were homogenized in Tper (50 mg/ml; Thermo Fisher Scientific) with protease and phosphatase inhibitor cocktails (Calbiochem), vortexed, and sonicated. Aβ peptide concentrations were determined per kit instructions (Invitrogen). Briefly, lysates were centrifuged at 100,000 × g for 1 h to separate monomeric and oligomeric forms of Aβ from the larger, fibrillar deposits, which remain in the pellet. The supernatant was carefully collected and stored at −80°C. This was analyzed as the soluble fraction, bearing both monomeric and oligomeric forms of Aβ. The pellet, bearing insoluble, fibrillar Aβ, was extracted in guanidinium-HCl, pH 8.0 (150 mg/ml), and centrifuged at 100,000 × g for 1 h. The supernatant was stored in −80°C to be analyzed as the insoluble fraction. Soluble samples were diluted 1:5 in kit buffer for ELISA. Insoluble samples were diluted 1:200 in kit buffer. For analyzing Aβ oligomers, the soluble fraction was diluted 1:1 in kit buffer. All dilutions were established empirically. Mouse IL-1β (mIL-1β) levels were determined per kit instructions (R&D Systems) using hippocampal lysates in Tper.
For Western blot, hippocampal lysates were diluted in 2× sample buffer (125 mm Tris-HCl, 4% SDS, and 20% glycerol), and protein concentration was determined by a BCA assay (Thermo Fisher Scientific). Protein (12 μg/lane for most blots) was electrophoresed on a Tris-HCl polyacrylamide gel and transferred to a nitrocellulose membrane (Bio-Rad) for 90 min at 4°C. After 1 h in Western blocking reagent (Roche Diagnostics), membranes were incubated overnight with primary antibodies. After rinsing, blots were incubated with peroxidase-linked secondary antibodies (provided in Supersignal West Dura Kit; Thermo Fisher Scientific) and treated with the ECL substrate included in the kit, and bands were visualized using either Biomax XAR film or imaged using the Image Station 440 CF (Eastman Kodak). List of primary antibodies used included the following: APP (clone 6E10; Millipore Bioscience Research Reagents) at 1:10,000; Tubulin (Calbiochem) at 1:5000; β-secretase activity of the β-site APP-cleaving enzyme (BACE) (Cell Signaling Technology) at 1:1000; β-C-terminal fragment (β-CTF) (Sigma-Aldrich) at 1:1000; Anti-Tau (Dako) at 1:10,000; AT180 (Thermo Fisher Scientific) 1:100; pT205 (Invitrogen) at 1:1000; PHF1 (gift from Dr. Peter Davies) at 1:100; and phospho-Ser9 GSK3β, GSK3β, phospho-p38MAPK, and p38MAPK (Cell Signaling Technology) at 1:1000.
Statistics.
All Statistical comparisons were performed using Prism 5.0 (GraphPad Software). p ≤ 0.05 was considered significant. Student's t test was used when two group means were compared. Results in which more than two group means were compared were analyzed with one-way or two-way ANOVA depending on the number of parameters. A Bonferroni's/Tukey's post hoc test was used to establish significance between individual groups in such instances.
Results
3xTgAD/IL-1βXAT mice display a robust neuroinflammatory phenotype after hIL-1β transgene activation
The IL-1βXAT mouse is a somatic mosaic model of inducible chronic IL-1β overexpression in the CNS, which makes use of an excisionally activated transgene (XAT) cassette to achieve gene expression (Shaftel et al., 2007a,b). The hIL-1β construct engineered into IL-1βXAT mice consists of the sequence for mature hIL-1β with the 5′ signal sequence of IL-1RA for direct secretion without caspase-1-dependent processing (Shaftel et al., 2007b). A stop signal flanked by LoxP sites lying upstream of the hIL-1β gene holds transgene expression in check in these mice. After delivery of Cre recombinase, the stop signal is excised out and transgene expression starts locally under the control of a murine GFAP promoter (Fig. 1A). To deliver Cre, we use an infection-competent, replication-incompetent FIV vector. This model enables us to exercise both spatial and temporal control over the expression of the IL-1β transgene. IL-1βXAT mice were bred to the triple transgenic AD mice (3xTgAD) that harbor three mutations in APP, PS1, and TAU genes and consequently develop plaques and tangles in their brain as they age (Oddo et al., 2003). Extracellular plaque pathology is established in the 3xTgAD mice by 15 months of age (Mastrangelo and Bowers, 2008). Therefore, 15-month-old 3xTgAD/IL-1βXAT mice and littermate 3xTgAD controls were used for this study. All animals received a unilateral stereotactic injection of FIV–Cre in the right subiculum regardless of genotype, which controls for inflammation arising from the surgery and viral transduction. Furthermore, having an intact contralateral hemisphere in every animal provides an internal control. We chose to overexpress IL-1β in the subiculum because it is one of the first areas affected in AD and, in the 3xTgAD mice, plaque deposition starts in the subiculum before the hippocampus (Mastrangelo and Bowers, 2008). Brains were analyzed after 1 and 3 months of IL-1β overexpression (at 16 and 18 months of age) for amyloid and tau pathology, as well as markers of glial activation and neuroinflammation (Fig. 1B). Previously, we reported a persistent increase of murine IL-1β mRNA in the IL-1βXAT mice up to 12 months from the date of transgene activation (Shaftel et al., 2007b). At 16 months of age (1 month of transgene expression), we detected robust expression of mIL-1β in the ipsilateral hippocampus (919.2 ± 84.0 pg/mg protein) compared with the contralateral hippocampus (114.4 ± 13.8 pg/mg protein) in the 3xTgAD/IL-1βXAT mice by ELISA measurements (Fig. 1C). The levels of mIL-1β observed in the ipsilateral hippocampus of 3xTgAD/IL-1βXAT mice was also significantly more compared with the ipsilateral hippocampus of 3xTgAD mice (116.7 ± 17.1 pg/mg protein) (one-way ANOVA test with Tukey's post hoc test, F(3,20) = 46.42, p < 0.0001, n = 3–9 per group). We thus clearly show a ∼8.3-fold increase in mIL-1β at the site of transgene activation, without any effect of stereotactic surgery on mIL-1β expression. As additional evidence of sustained neuroinflammation, we found approximately twofold to threefold greater activation of microglia and astrocytes in via Iba1 and GFAP immunostaining in the ipsilateral subiculum of 3xTgAD/IL-1βXAT mice compared with 3xTgAD mice (Fig. 1D–G). Neuroinflammation and gliosis persisted at 18 months of age (3 months of transgene expression) in the ipsilateral 3xTgAD/IL-1βXAT subiculum (data not shown). We further investigated the spread of the FIV–Cre construct near the injection site by staining for V5, a viral epitope tag present in the construct. We could only detect V5-positive cells in the ipsilateral hemisphere near the injection site in the 3xTgAD/IL-1βXAT subiculum at 16 months (Fig. 1H). Coimmunostaining with GFAP and DAPI showed the presence of the V5 tag in the nuclei of astrocytes only in the ipsilateral 3xTgAD/IL-1βXAT subiculum in high-power photomicrographs (Fig. 1I) as expected. To quantify the number of FIV–Cre-positive cells in the ipsilateral hemisphere, we used 8-month-old IL-1βXAT mice who received an unilateral injection of FIV–Cre in the dentate gyrus of the hippocampus. After 1 month of transgene activation, we could find an average of six to seven V5-positive cells in the ipsilateral dentate. The contralateral dentate gyrus was consistently devoid of any cellular V5 staining (n = 3). Thus, we demonstrated that viral transduction did not spillover to the contralateral hemisphere in our model of unilateral activation of the IL-1β transgene.
Figure 1.
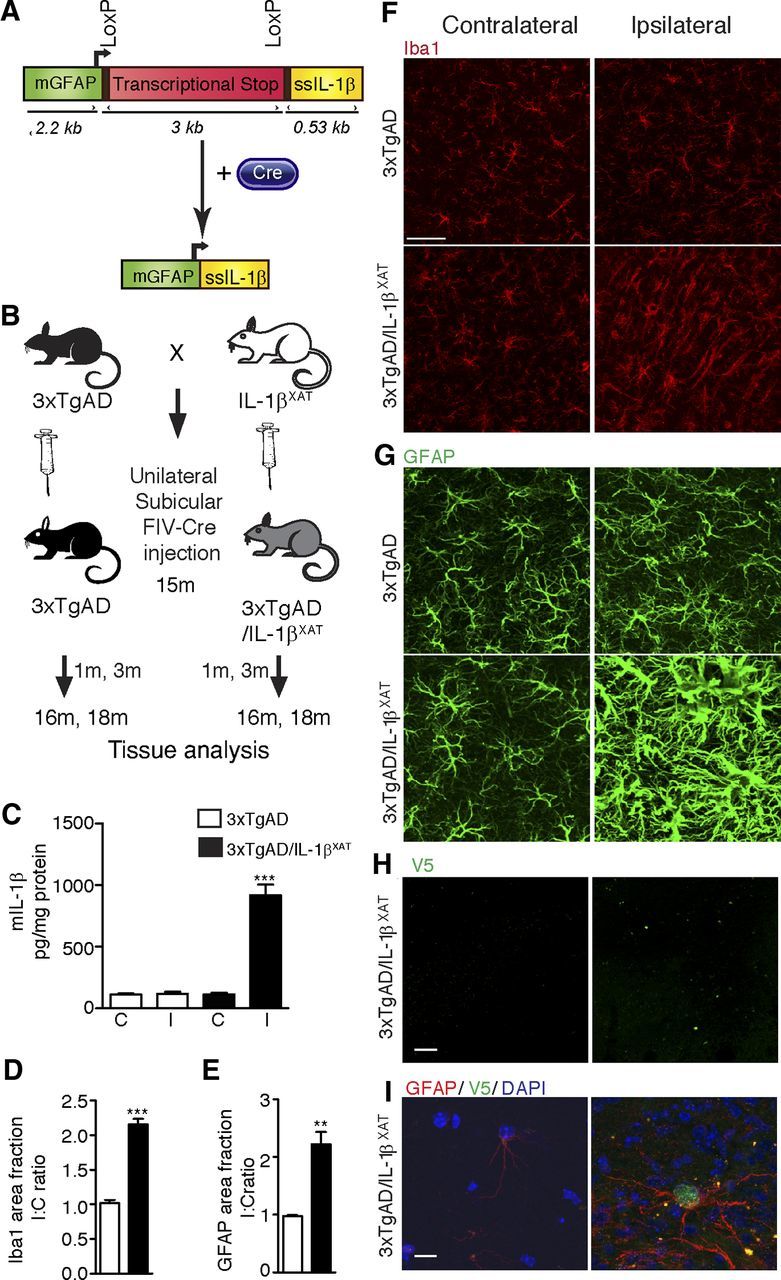
Research design and chronic neuroinflammatory phenotype in 3xTgAD/IL-1βXAT mice. A, The linear construct (∼10 kb) bears the murine GFAP promoter (mGFAP), a transcriptional stop signal flanked by LoxP sites, and the cDNA for hIL-1β gene fused to the signal sequence of mature hIL-1RA (ssIL-1β). After delivery of Cre, the transcriptional stop is excised out and the production of IL-1β is induced locally. B, 3xTgAD mice were bred to the IL-1βXAT mice. Fifteen-month-old 3xTgAD/IL-1βXAT and control littermates of the 3xTgAD genotype from the F1 generation were given a unilateral stereotactic injection of FIV–Cre in the subiculum. Groups of mice were killed 1 and 3 months after stereotactic surgery (at 16 and 18 months of age, respectively), and glial activation as well as amyloid and tau pathology were assessed. C, Murine IL-1β protein ELISA measurements. C, Contralateral; I, ipsilateral. Data were expressed as mean picograms mIL-1β per milligram protein ± SEM per group and analyzed by a one-way ANOVA test, followed by a Tukey's post hoc test. The level of mIL-1β in the ipsilateral hippocampus of 3xTgAD/IL-1βXAT mice is significantly greater than all other groups (***p < 0.0001), but the three other groups are not significantly different from each other (n = 3–9 per group). One month after transgene activation, elevation in markers of glial activation was observed in the ipsilateral subiculum of 3xTgAD/IL-1βXAT mice. D, Area fraction measurements of Iba1 immunohistochemical data. Data expressed as mean I/C ratio ± SEM per group and analyzed by unpaired Student's t test, ***p < 0.0001; n = 5–7 per group. E, Area fraction measurements of GFAP immunohistochemical data. Data expressed as mean I/C ratio ± SEM per group and analyzed by unpaired Student's t test, **p < 0.01; n = 3–4 per group. Representative confocal micrographs of the ipsilateral and contralateral subiculum immunostained with the microglial marker Iba1 (F) and the astrocytic marker GFAP (G) are shown. Scale bars, 30 μm. H, Representative photomicrographs from 16-month-old 3xTgAD/IL-1βXAT mice immunostained with an antibody against V5, a viral epitope tag present in the injected FIV–Cre construct. Scale bars, 200 μm. I, Representative high-power photomicrograph from the same experiment demonstrating the presence of V5 in the nucleus of a GFAP-positive astrocyte in the ipsilateral but not the contralateral subiculum. Scale bars, 10 μm.
Reduced amyloid load in 3xTgAD/IL-1β XAT mice after 1 and 3 months of transgene expression
We examined Aβ deposition in 3xTgAD/IL-1βXAT and 3xTgAD mice after 1 and 3 months of transgene expression, respectively. At both time points, 6E10 immunopositive plaques were significantly reduced in the ipsilateral subiculum in 3xTgAD/IL-1βXAT mice (Fig. 2A,B). The ratios between ipsilateral and contralateral measures declined sharply in the 3xTgAD/IL-1βXAT mice (0.35 ± 0.13 at 16 months and 0.27 ± 0.03 at 18 months) versus 3xTgAD mice (1.31 ± 0.21 at 16 months and 0.99 ± 0.06 at 18 months). Two-way ANOVA using genotype and time as parameters showed a highly significant effect of genotype (i.e., presence or absence of the IL-1β gene) on the I/C ratio (F(1,9) = 31.03, p = 0.0003, n = 3–5 mice per group). The effect of transgene expression duration on the I/C ratios was not significant (F(1,9) = 1.78, p = 0.2152, n = 3–5 mice per group). To further validate the immunohistochemical data, we measured levels of different Aβ isoforms by performing ELISA on soluble and insoluble fractions of hippocampal homogenates derived from 16-month-old 3xTgAD and 3xTgAD/IL-1βXAT mice. We found a ∼50% reduction in insoluble Aβ42 (I/C ratio = 0.46 ± 0.02 for 3xTgAD/IL-1βXAT mice vs 1.00 ± 0.03 for 3xTgAD mice) and Aβ40 (I/C ratio = 0.56 ± 0.02 for 3xTgAD/IL-1βXAT mice vs 1.00 ± 0.02 for 3xTgAD mice) (Fig. 2C,D). To investigate the role of sustained IL-1β overexpression on Aβ oligomers, we also measured levels of soluble Aβ aggregates or oligomers in hippocampal homogenates. We observed a decrease in Aβ oligomers in the ipsilateral hippocampus in the 3xTgAD/IL-1βXAT mice (I/C ratio = 0.67 ± 0.05 for 3xTgAD/IL-1βXAT mice vs 1.09 ± 0.09 or 3xTgAD mice; Fig. 2E). We also observed significant reductions of Aβ40 and Aβ42 in the soluble fractions of the hippocampal lysates (Fig. 2F,G).
Figure 2.
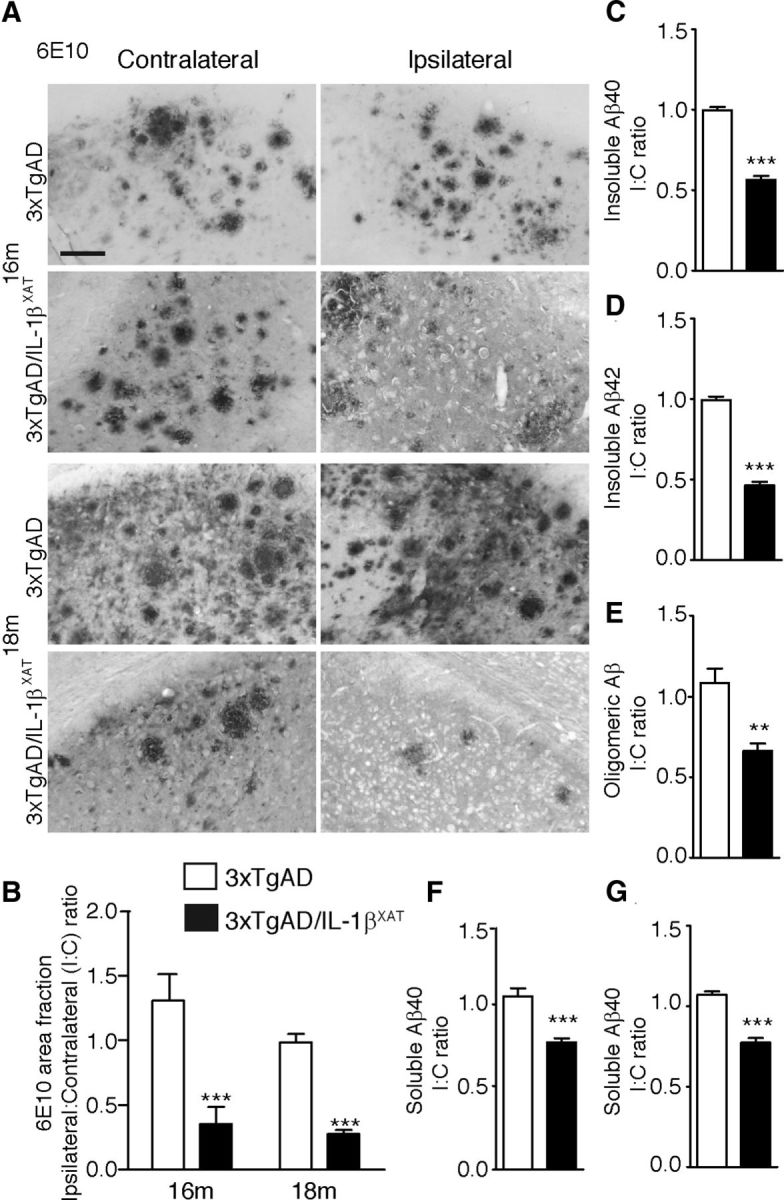
Sustained IL-1β expression ameliorates amyloid load in 3xTgAD/IL-1βXAT mice. A, Representative photomicrographs of contralateral and ipsilateral subicular sections from 3xTgAD and 3xTgAD/IL-1βXAT mice immunostained with the anti-amyloid antibody 6E10 at 16 and 18 months. Scale bars, 100 μm. B, Quantification of 6E10 immunopositive amyloid plaque area fraction in the ipsilateral and contralateral subiculum of 3xTgAD and 3xTgAD/IL-1βXAT mice at 16 and 18 months. Data are represented as the mean I/C ratio ± SEM for each group and was analyzed by a two-way ANOVA with Bonferroni's post hoc test; n = 3–5 mice per group. C–G, ELISA results obtained from whole ipsilateral and/or contralateral hippocampi of 3xTgAD and 3xTgAD/IL-1βXAT mice at 16 months. Mean I/C ratios ± SEM of Aβ40 and Aβ42 in the guanidinium-HCl (Insoluble) fraction (C, D), Tper (Soluble) fraction (F, G), and Oligomeric Aβ in the soluble fraction (E) are shown. In all measures, 3xTgAD/IL-1βXAT mice demonstrated a lower amyloid load in the ipsilateral subiculum/hippocampus at 16 and 18 months compared with 3xTgAD controls; n = 3–9 mice per group. Data were analyzed with unpaired Student's t tests; **p < 0.01, ***p < 0.0001.
Evidence of increased association and activation of microglia around amyloid plaques in 3xTgAD/IL-1βXAT mice
We reasoned that the observed reduction in amyloid load in this model might be attributable to reduced deposition of Aβ or its enhanced clearance and tested both. When tested in vivo in our model, immunoblots against full-length transgenic human APP failed to show any difference between 3xTgAD control and 3xTgAD/IL-1βXAT hippocampi (Fig. 3A,B), indicating that basal levels of APP expression are unaltered in both genotypes. BACE levels were also unchanged between the control and 3xTgAD/IL-1βXAT groups (Fig. 3A,C) as well as β-CTF (Fig. 3A,D), which suggests that gamma secretase activity is not significantly different in the 3xTgAD/IL-1βXAT mice from control mice.
Figure 3.
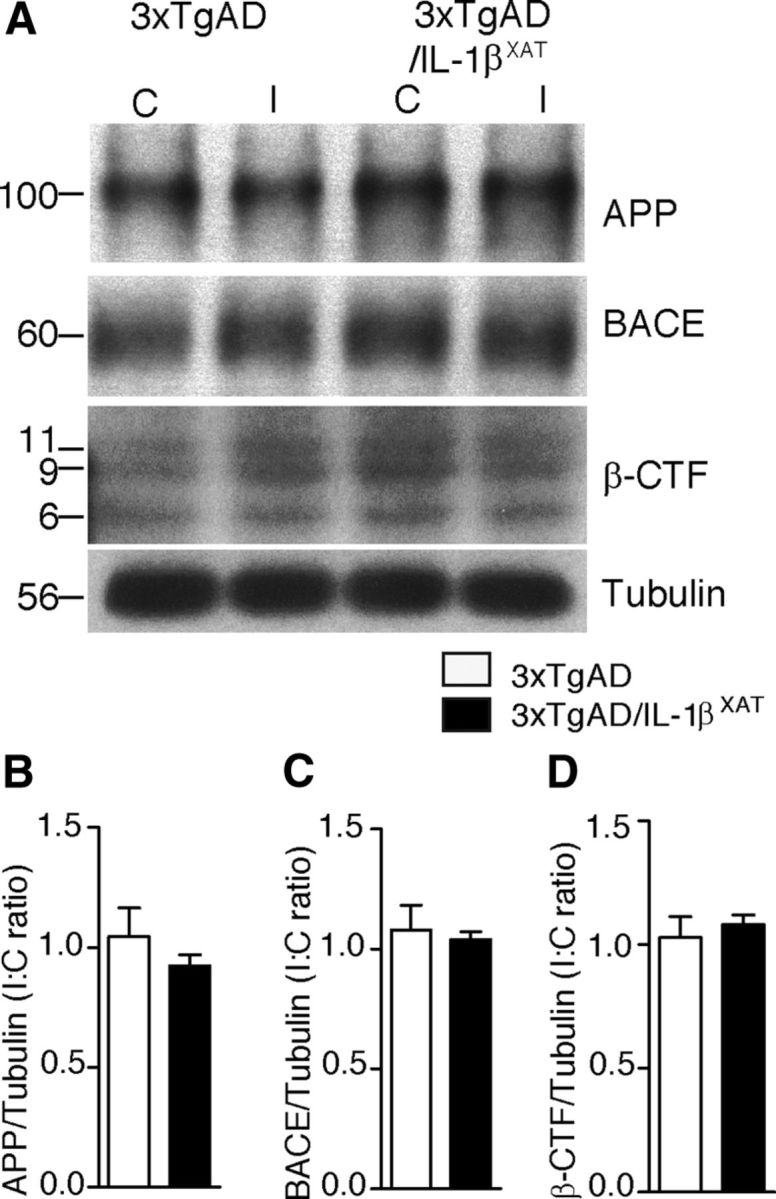
Sustained IL-1β expression does not alter expression of APP or its processing in 3xTgAD/IL-1βXAT mice. Homogenates from ipsilateral and contralateral hippocampi of 16-month-old 3xTgAD and 3xTgAD/IL-1βXAT mice were subjected to Western blot analysis with markers of APP expression and processing. Group means were not significantly different overall. A, Representative Western blot images of contralateral (C) and ipsilateral (I) hippocampi of 3xTgAD and 3xTgAD/IL-1βXAT mice probed with antibodies against APP, BACE, β-CTF, and tubulin at 16 months. Molecular weights are expressed in kilodaltons. Mean I/C ratio ± SEM of relative band intensities per group (normalized to tubulin band intensities) is shown for APP (B), BACE (C), and β-CTF (D); n = 3–9 mice per group.
Next, we investigated whether the reduced amyloid burden reflected an enhanced clearance of amyloid. We and others have reported a role of plaque-associated microglia in amyloid clearance in the past (Malm et al., 2005; Simard et al., 2006; Shaftel et al., 2007b). To analyze plaque-associated microglia, we performed confocal analysis of sections stained with Congo Red for fibrillar Aβ plaques, Iba-1 for microglia, and Hoechst as a nuclear stain. Ipsilateral subiculum showed markedly greater staining intensity for Iba-1 around Congo-Red-positive plaques in the 3xTgAD/IL-1βXAT animals (Fig. 4A–C). Counts of plaque-associated microglia showed 4.6 and 6.6 times more plaque-associated microglia in the ipsilateral subiculum of 3xTgAD/IL-1βXAT mice compared with control mice after 1 and 3 months of IL-1β overexpression by one-way ANOVA (F(2,7) = 21.27, p = 0.0011 at 16 and 18 months, respectively; Fig. 4D). Only Iba1 immunopositive microglial cells whose nuclei were in direct contact with plaques were counted in this analysis.
Figure 4.
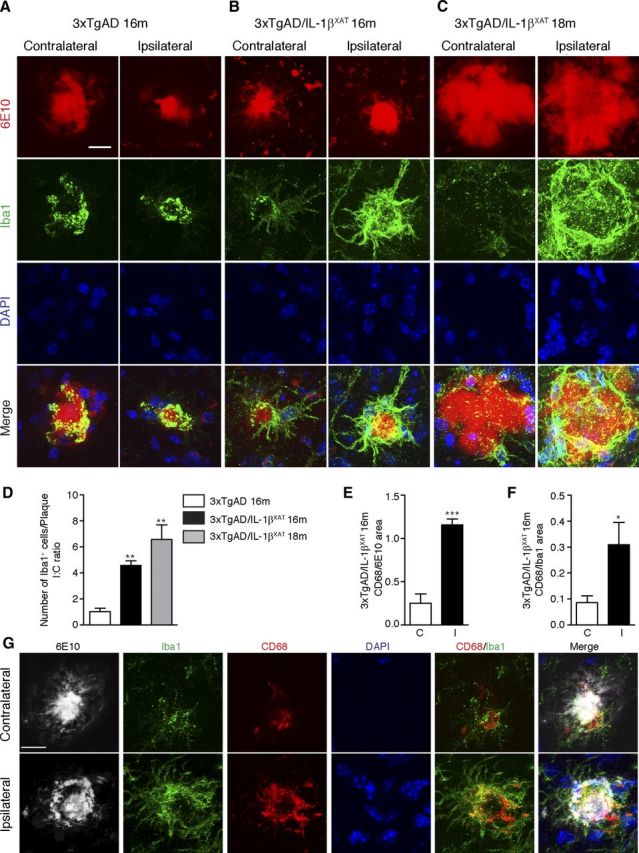
Sustained IL-1β overexpression enhances plaque-associated microglia in 3xTgAD/IL-1βXAT mice. Representative confocal micrographs of contralateral and ipsilateral subicular sections stained with Congo Red (fibrillar Aβ plaques), Iba-1 (microglia), and DAPI (nuclei) are shown. 3xTgAD/IL-1βXAT mice showed more plaque-associated microglia at 16 months (B) and 18 months (C) compared with their 3xTgAD counterparts (A) in the ipsilateral subiculum. Scale bars, 30 μm. On quantification, 3xTgAD/IL-1βXAT mice demonstrate approximately fourfold more plaque-associated microglia at 16 months and ∼6.5-fold more plaque associated microglia at 18 months in the ipsilateral subiculum compared with their 3xTgAD counterpart at 16 months (D). Data are shown as mean I/C ratio ± SEM of Iba1-positive cells per plaque; n = 3–4 mice per group. Data were analyzed with one-way ANOVA, followed by a Bonferroni's post hoc test; **p < 0.01. Quantitative results are shown for contralateral and ipsilateral hemispheres of 16-month-old 3xTgAD/IL-1βXAT mice brain sections stained with 6E10 (amyloid plaques), CD68 (activated microglia), Iba1 (microglia), and DAPI (nuclei). The ipsilateral subiculum demonstrates a 4.6-fold increase in CD68 area when normalized to plaque area (E). Data were expressed as mean CD68/6E10 area Fraction within a 500 pixel diameter around the plaques ± SEM for contralateral (C) and ipsilateral (I) subiculum. Analyzed by a paired Student's t test, ***p < 0.001; n = 4 mice. The ipsilateral subiculum also demonstrates a 3.5-fold increase in CD68 area when normalized to Iba1 area (F). Data were expressed as mean CD68/Iba1 area fraction within a 500 pixel diameter around the plaques ± SEM for contralateral (C) and ipsilateral (I) subiculum. Analyzed by a paired Student's t test, *p < 0.05; n = 4 mice. G, Representative confocal micrographs from the experiment are shown. Scale bars, 20 μm.
We then sought to investigate whether the plaque-associated microglia were more activated in the ipsilateral subiculum of the 3xTgAD/IL-1βXAT mice compared with the contralateral subiculum. To this end, we stained sections of the 16-month-old 3xTgAD/IL-1βXAT brain with 6E10, CD68, a marker of microglial activation, Iba1, and Hoechst. The ipsilateral subiculum, along with greater intensity of Iba1, also showed a greater extent of immunostaining for CD68 around the 6E10-positive amyloid plaques (Fig. 4G). After calculating the area fraction occupied by CD68 in the vicinity of amyloid plaques and normalizing it to either 6E10 area fraction (Fig. 4E) or Iba1 area fraction (Fig. 4F), we found an approximately fourfold increased presence of CD68 in plaque-associated microglia in the ipsilateral 3xTgAD/IL-1βXAT subiculum. This indicates that the plaque-associated microglia are more activated in the ipsilateral than the contralateral subiculum and consequently suggests that they might be better phagocytes of amyloid.
Sustained IL-1β overexpression exacerbates tau pathology after 1 and 3 months of transgene expression in 3xTgAD/IL-1βXAT mice
To characterize the effect of IL-1β overexpression on tau pathology, we performed immunostaining on sections nearest to the FIV–Cre injection site after 1 and 3 months of IL-1β overexpression (at 16 and 18 months of age). At 16 months, we observed a 2- to 2.5-fold increase in immunoreactivity at pT205 (part of the AT8 epitope) in the CA1 neurons and the stratum radiatum immediately adjacent to the ipsilateral subiculum in the 3xTgAD/IL-1βXAT mice. In contrast, control 3xTgAD mice did not show any significant difference in immunoreactivity in these areas between the contralateral and the ipsilateral hemispheres (Fig. 5A,D,E). Very similar results were seen with AT180; the increase in immunoreactivity in the ipsilateral CA1 fields and stratum radiatum was 4.3- and 4.0-fold more than the contralateral counterparts (Fig. 5B,F,G). In contrast, total human transgenic tau as detected by the HT7 antibody was not significantly different between ipsilateral and contralateral hemispheres in any genotype, suggesting that IL-1β overexpression did not alter the expression of the human tau transgene, despite changes in phosphorylation (Fig. 5C,H,I). To further validate the immunohistology data, we analyzed ipsilateral and contralateral hippocampi from 16-month-old 3xTgAD and 3xTgAD/IL-1βXAT mice with Western blot. We detected twofold to threefold increases in pT205, AT180, and PHF1 phospho-epitopes in the ipsilateral hemisphere of 3xTgAD/IL-1βXAT but not in control 3xTgAD mice. We did not find any changes in the levels of total tau (Fig. 5J–M). In 18-month-old animals, pS396 immunoreactivity (part of the PHF-1 epitope) similarly increased 2.5-fold in the CA1 neurons and their processes adjacent to the ipsilateral subiculum in 3xTgAD/IL-1βXAT animals but not control 3xTgAD animals (Fig. 6A,C). Again, quantification of HT7 immunoreactivity did not reveal any changes between ipsilateral and contralateral hemispheres of either genotype (Fig. 6B,D).
Figure 5.
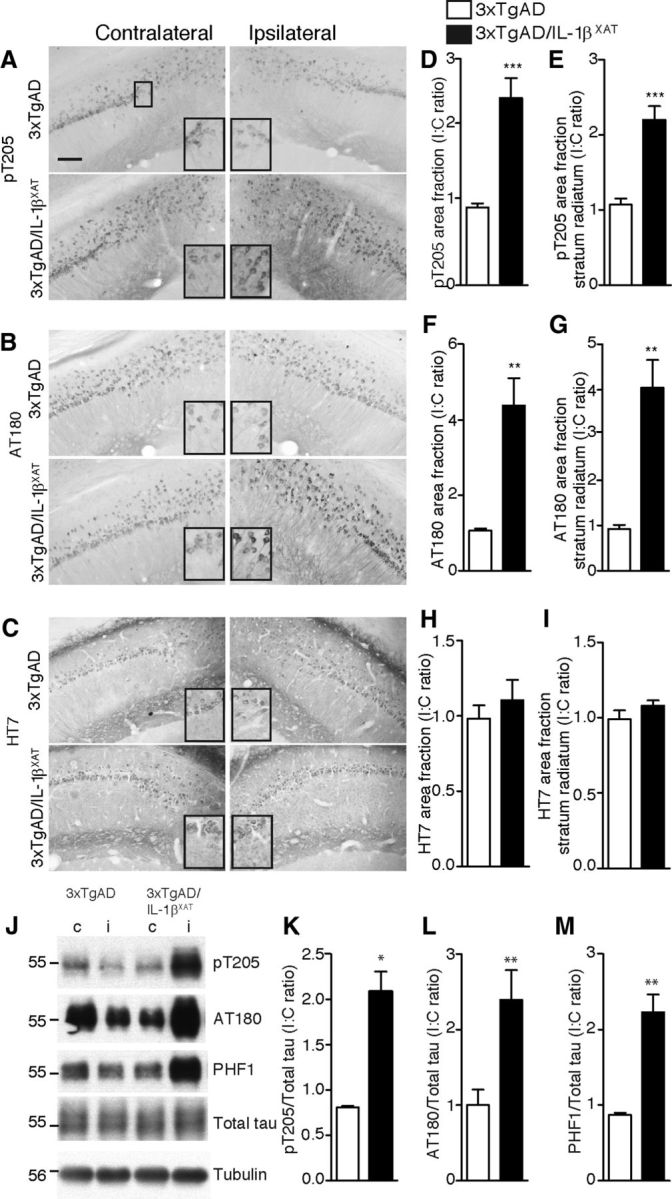
Sustained overexpression of IL-1β leads to enhanced tau phosphorylation in 16-month-old 3xTgAD/IL-1βXAT mice. Sections of the subiculum–CA1 junction of 16-month-old 3xTgAD and 3xTgAD/IL-1βXAT brains were probed with antibodies against phospho-tau and total tau. Representative images of section stained with phospho-tau antibodies pT205 (A) and AT180 (B) show increased immunostaining in the ipsilateral hemispheres of 3xTgAD/IL-1βXAT mice. HT7 immunostaining (C) failed to show any difference between the ipsilateral and contralateral hemispheres of either genotype. Scale bars, 100 μm. Insets show sections from the images as marked in the first panel of A magnified 2× digitally. Immunopositive area fractions were quantified in the entire field visualized (D, F, H) or in the stratum radiatum (E, G, I). All numerical data are represented as mean I/C ratio ± SEM per group. 3xTgAD/IL-1βXAT mice demonstrate an approximate twofold increase in pT205 immunostaining (D, E) and an approximate fourfold increase in AT180 immunostaining (F, G) but not in HT7 immunostaining (H, I) in the ipsilateral subiculum; n = 4–6 mice per group; data were analyzed with an unpaired Student's t test. Lysates from ipsilateral (i) and contralateral (c) hippocampi of 16-month-old 3xTgAD and 3xTgAD/IL-1βXAT mice were subjected to Western blot analysis with antibodies against phospho-tau and total tau. Representative images of Western blots with pT205, AT180, and PHF1, as well as total tau are shown (J). Molecular weights are expressed in kilodaltons. 3xTgAD/IL-1βXAT mice demonstrate an approximate twofold increase in tau phosphorylation at pT205 (K), AT180 (L), and PHF1 (M) epitopes. All band intensities were normalized to total tau; n = 3–9 mice per group. Data were analyzed with unpaired Student's t tests; *p ≤ 0.05, **p < 0.01, ***p < 0.001.
Figure 6.
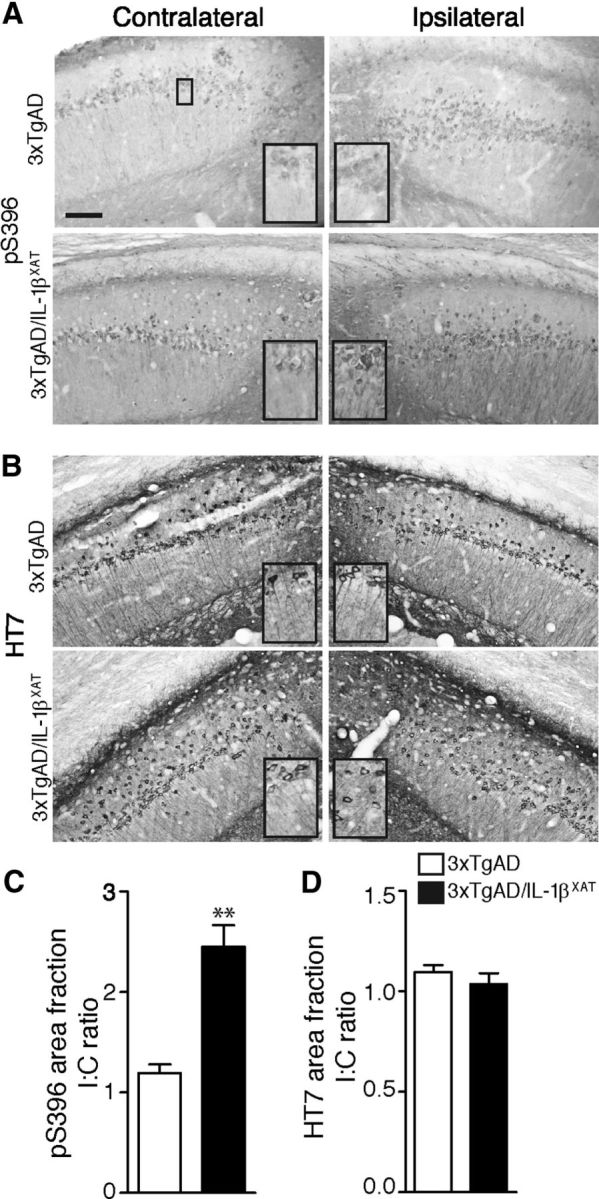
Sustained overexpression of IL-1β leads to enhanced tau phosphorylation in 18-month-old 3xTgAD/IL-1βXAT mice. Sections of the subiculum–CA1 junction of 18-month-old 3xTgAD and 3xTgAD/IL-1βXAT brains were probed with antibodies against phospho-tau and total tau. Representative images of section stained with the phospho-tau antibody pS396 (A) show increased immunostaining in the ipsilateral hemispheres of the 3xTgAD/IL-1βXAT mice. HT7 immunostaining (B) failed to show any difference between the ipsilateral and contralateral hemispheres of either genotype. Scale bars, 100 μm. Insets show sections from the images as marked in the first of Fig. 5A magnified 2× digitally. C, D, Immunopositive area fractions were quantified in the entire field visualized. 3xTgAD/IL-1βXAT mice demonstrate a an approximate twofold to threefold increase in pS396 immunostaining (C) but not in HT7 immunostaining (D). Numerical data are represented as mean I/C ratio ± SEM per group; n = 4–6 mice per group. Data were analyzed with an unpaired Student's t test; **p < 0.01.
Evidence of increased GSK3β and p38MAPK activity in 3xTgAD/IL-1βXAT mice
Once we obtained clear evidence of enhanced tau phosphorylation in the 3xTgAD/IL-1βXAT mice, we set out to investigate kinases that might be involved. Both GSK3β and p38MAPK have been studied for their ability to phosphorylate tau (Chun and Johnson, 2007). By Western blot analysis, we found significantly reduced phosphorylation of the Ser9 epitope of GSK3β, an important inhibitor of GSK3β activity (Dajani et al., 2001), in the ipsilateral 3xTgAD/IL-1βXAT hippocampus after 1 month of transgene expression (at 16 months of age; Fig. 7A,B). We also found evidence of increased phospho-p38 in the ipsilateral hippocampus of 16-month-old 3xTgAD/IL-1βXAT mice, suggesting that p38MAPK activity is also elevated in these animals compared with controls (Fig. 7A,C). The steady-state levels of GSK3β or p38 remained unaltered between ipsilateral and contralateral hemispheres across all genotypes.
Figure 7.

Sustained overexpression of IL-1β leads to activation of kinase pathways in 16-month-old 3xTgAD/IL-1βXAT mice. Lysates from ipsilateral and contralateral hippocampi of 16-month-old 3xTgAD and 3xTgAD/IL-1βXAT mice were subjected to Western blot analysis with markers of kinase activation. Representative image of Western blots (A) and densitometric quantification of the band intensities (B, C) demonstrated a decrease in pS9GSK3β in the ipsilateral hippocampus of the 3xTgAD/IL-1βXAT mice suggesting an increase in the activity of GSK3β and an increase in phospho-p38MAPK suggesting enhanced activity of p38MAPK, but the steady-state levels of both enzymes were not changed between groups. Molecular weights are expressed in kilodaltons. Band intensities of phospho-epitopes were normalized to the steady-state levels of GSK3β and p38MAPK, respectively. Numerical data are represented as mean I/C ratio ± SEM per group; n = 3–9 per group. Data were analyzed with unpaired Student's t tests; *p ≤ 0.05, ***p < 0.001.
Discussion
In this study, we present evidence of a differential regulation of amyloid and tau pathology by neuroinflammation in a mouse model of AD. After discovery of the association between plaques and microglia expressing immune markers in the late 1980s (McGeer et al., 1987; Griffin et al., 1989), a growing body of research has implicated neuroinflammation as a consistent component of AD pathology. As a prominent marker of microglia surrounding senile plaques, IL-1 was thought to be a major player in AD neuroinflammation. In vitro, IL-1β increases the expression of APP mRNA (Goldgaber et al., 1989; Gray and Patel, 1993), its translation (Rogers et al., 1999), and its processing to Aβ (Brugg et al., 1995; Liao et al., 2004). A single injection of recombinant IL-1β into rat brain led to increased expression of APP (Sheng et al., 1996), and IL-1RA knock-out mice were more vulnerable to Aβ oligomers (Craft et al., 2005). Based on these observations, a “cytokine cycle” was proposed, which implicates IL-1β as one of the major, if not causative, early event in amyloidogenesis. However, many of these studies were performed in vitro, mimicked an acute upsurge of IL-1β, or gave indirect evidence of the involvement of IL-1 signaling in AD pathogenesis. In AD, the increase of IL-1 is likely chronic and develops over a period of months and years, in which case, the role played by IL-1β might not be accurately understood from these previous studies. Our laboratory developed the first animal model of conditional overexpression of IL-1β to address the issue directly. Surprisingly, IL-1βXAT mice crossed with the APP/PS-1 mouse model of AD demonstrated reduced amyloid pathology after transgene activation at multiple time points and without apparent neuronal loss (Shaftel et al., 2007b; Matousek et al., 2012). We report a similar reduction of amyloid deposition with immunohistochemical and biochemical methods in the current study. The toxicity of Aβ oligomers has been studied across multiple systems and models (Larson and Lesné, 2012), and we report a reduction in oligomers in our model. The 3xTgAD/IL-1βXAT mice did not show any changes in basal levels of APP and BACE expression or levels of APP β-CTFs, which is indicative of unaltered secretase activity.
Our observation of a fourfold to sixfold increase in plaque-associated microglia in the subiculum of the 3xTgAD/IL-1βXAT mice is consistent with previous observations in IL-1βXAT mice crossed with APP/PS-1 mice (Shaftel et al., 2007b; Matousek et al., 2012). The increased overlap of microglia and plaques suggests that the decrease of Aβ observed in our model is a result of increased clearance by microglia, which is also supported by our observation of increased CD68 in plaque-associated microglia. Microglia, the primary immune effector cells of the brain, can phagocytose Aβ in vitro and in vivo (Fu et al., 2012; Liu et al., 2012). Several investigators have reported reduction of Aβ along with concomitant increases in microglial activation after modulation of innate immunity in vivo. For example, intracranial lipopolysaccharide (LPS) administration in mouse models of AD led to a reduction in amyloid pathology (DiCarlo et al., 2001; Herber et al., 2004), an effect shown to depend on microglial clearance of Aβ (Herber et al., 2007). Fractalkine receptor (CX3CR1) gene deletion, which alters the pattern of microglial activation markers, was associated with reduced fibrillar amyloid in APP/PS1 mice (Lee et al., 2010b). Interestingly, CX3CR1−/− animals also demonstrated an increase in IL-1β levels. It has been proposed that accumulation of Aβ plaques results from decreased clearance by dysfunctional microglia (Hickman et al., 2008) and that microglial-mediated Aβ clearance depends on their activation phenotype (Colton, 2009). Studies are underway in our laboratory to establish the specific phenotype of plaque-associated microglia in different murine models of AD overexpressing IL-1β.
The second major finding reported in this study is the exacerbation of tau pathology, demonstrated by elevated phospho-tau at multiple AD-relevant epitopes in 3xTgAD/IL-1βXAT mice. We therefore present evidence of a possible causative role for IL-1β-associated neuroinflammation on tau pathology. Although there have been many confounds over the nature of the relationship between amyloid and IL-1β, all evidence linking IL-1β or neuroinflammation to tau pathology have pointed toward a deleterious relationship. For example, IL-1 was shown to increase tau phosphorylation in vitro using neuronal–microglial coculture experiments (Li et al., 2003), and rat brains impregnated with slow-release IL-1 pellets showed increased tau phosphorylation (Sheng et al., 2000). Moreover, parenchymal LPS injections worsened tau pathology in a transgenic murine model of forebrain-specific P301L tau overexpression (Lee et al., 2010a) and led to exacerbated tau pathology in 3xTgAD mice (Kitazawa et al., 2005). Ablation of CX3CR1 in mice and resulting increases in microglial activation were associated with exacerbated tau pathology in hTau mice (Bhaskar et al., 2010). Finally, a recent study found attenuated tau pathology after administration of an antibody against IL-1R (Kitazawa et al., 2011). However, because the antibody was administered systemically, the influence of peripheral effects on study outcome cannot be ruled out. In light of such data, our hypothesis was that IL-1β-driven neuroinflammation would exacerbate tau pathology, and our results are consistent with this idea.
We also found evidence of increased GSK3β and p38MAPK activity with sustained IL-1β expression in 3xTgAD mice. An increase in phospho-p38 (Thr180/Tyr182) was observed in the hippocampi of 3xTgAD/IL-1βXAT mice, suggesting increased p38MAPK activity. The association of p38MAPK with tau phosphorylation under an inflammatory stimulus has been studied in vitro and in vivo (Sheng et al., 2001; Li et al., 2003). IL-1 signaling can itself upregulate p38MAPK activity (Allan et al., 2005), which can phosphorylate tau (Sheng et al., 2000). We also observed a decrease in the phospho-Ser9 epitope of GSK3β, which is indicative of increased GSK3β activity. GSK3β has been widely studied as a prominent tau kinase in vivo. GSK3β colocalizes with neurons bearing neurofibrillary tangles in transgenic mice (Ishizawa et al., 2003), and a regulated model of GSK3β overexpression demonstrated tau hyper-phosphorylation, somatodendritic mislocalization of tau, neurodegeneration, and gliosis (Lucas et al., 2001). Moreover, bigenic mice overexpressing both GSK3β and P301L tau conditionally show reversible AD-type pathology (Engel et al., 2006a), and treating these animals with lithium, a GSK3 inhibitor, reverted all tau pathology except mature tangles (Engel et al., 2006b). There is indirect evidence in the literature about the ability of IL-1 to regulate GSK-3β activity, either through activating the PI3K–Akt pathway in neurons or through disruption of the β-catenin pathway (Shaw et al., 1997; Auron, 1998; Davis et al., 2006; Peineau et al., 2007; Kitazawa et al., 2011; Ma et al., 2011). However, the precise signaling pathways that link IL-1β to tau pathology and the specific cell types they are activated in 3xTgAD/IL-1βXAT mice cannot be determined without conducting additional mechanistic studies.
We report a differential regulation of the key pathological features of AD by IL-1β. IL-1β-mediated microglial activation appears to have an adaptive role with respect to amyloid, but along with activation of pathways that likely clear amyloid, it upregulates multiple kinase pathways that lead to exacerbated tau pathology seen in our model (Fig. 8). Whether this is a direct effect of IL-1β on neurons is unknown. In addition, although we have focused on microglia, activated astrocytes and other cell types may also play a role in influencing AD pathology in our model. As already discussed, the reduction of Aβ pathology and aggravation of tau pathology after manipulation of innate immunity has been reported previously in different models overexpressing either amyloid or tau. However, we report amyloid and tau pathology being influenced in an opposing manner by CNS-specific sustained overexpression of IL-1β in a mouse model that captures both these pathological features. There is robust genetic and molecular evidence indicating that amyloid pathology lies upstream of tau pathology in AD, which is the basic assumption on which the amyloid hypothesis stands (Hardy and Selkoe, 2002; Selkoe, 2011). However, an important question remains whether resolution of amyloid pathology alone is enough to reduce tau pathology and influence disease outcome. Our results indicate that removal of amyloid via activation of innate immunity might not be sufficient to mitigate overall disease pathology and might even aggravate it. In a follow-up study of the phase 1 trial of ANI792, in which subjects with AD were inoculated with Aβ1–42 and QS21 adjuvant, brains of all examined patients showed advanced Braak stages of tau pathology (V and VI) despite near total removal of plaques (Holmes et al., 2008). Importantly, none of the patients showed any improvement in disease progression.
Figure 8.

Proposed role of IL-1β in our model. IL-1β transgene expression acts on the resident microglia and astrocytes, leading to their activation, which in turn triggers a positive feedback loop of IL-1β production and neuroinflammation. Microglial activation may aid in amyloid clearance. The local inflammatory milieu also leads to activation of kinase pathways that may directly or indirectly result in enhanced tau phosphorylation. Questions for future investigation are indicated.
In summary, the ability of IL-1-driven neuroinflammation to mediate opposite effects on amyloid load and tau phosphorylation becomes critically important in light of recent endeavors toward developing therapies targeting Aβ isoforms through activation of innate immunity or FcR-mediated microglial clearance. Countless studies have established tau toxicity as an inherent part of AD pathology, so any effect that therapies targeting Aβ might exert on tau needs to be addressed cautiously.
Footnotes
This work was supported by National Institutes of Health Grant RO1 AG030149 (M.K.O.). We thank Dr. Peter Davies (Albert Einstein School of Medicine, New York, NY) for the gift of the PHF-1 antibody and Dr. Salvatore Oddo (The Barshop Institute for Longevity and Aging Studies, The University of Texas Health Science Center at San Antonio, San Antonio, TX) for the 3xTgAD mice. We also thank Jack Walter, Mallory Olschowka, and Lee Trojanczyk for technical assistance, Dr. Linda Callahan for her assistance at the University of Rochester Medical Center Confocal Microscopy Core, and Jennie Miller for preparing the FIV–Cre virus used in this study.
The authors declare no competing financial interests.
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- Auron PE. The interleukin 1 receptor: ligand interactions and signal transduction. Cytokine Growth Factor Rev. 1998;9:221–237. doi: 10.1016/s1359-6101(98)00018-5. [DOI] [PubMed] [Google Scholar]
- Barger SW, Harmon AD. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature. 1997;388:878–881. doi: 10.1038/42257. [DOI] [PubMed] [Google Scholar]
- Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR. Evidence for glial-mediated inflammation in aged APPSW transgenic mice. Neurobiol Aging. 1999;20:581–589. doi: 10.1016/s0197-4580(99)00065-2. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68:19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugg B, Dubreuil YL, Huber G, Wollman EE, Delhaye-Bouchaud N, Mariani J. Inflammatory processes induce beta-amyloid precursor protein changes in mouse brain. Proc Natl Acad Sci U S A. 1995;92:3032–3035. doi: 10.1073/pnas.92.7.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Johnson GV. Glycogen synthase kinase 3β phosphorylates tau at both primed and unprimed sites. Differential impact on microtubule binding. J Biol Chem. 2003;278:187–193. doi: 10.1074/jbc.M206236200. [DOI] [PubMed] [Google Scholar]
- Chun W, Johnson GV. The role of tau phosphorylation and cleavage in neuronal cell death. Front Biosci. 2007;12:733–756. doi: 10.2741/2097. [DOI] [PubMed] [Google Scholar]
- Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft JM, Watterson DM, Hirsch E, Van Eldik LJ. Interleukin 1 receptor antagonist knockout mice show enhanced microglial activation and neuronal damage induced by intracerebroventricular infusion of human beta-amyloid. J Neuroinflammation. 2005;2:15. doi: 10.1186/1742-2094-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajani R, Fraser E, Roe SM, Young N, Good V, Dale TC, Pearl LH. Crystal structure of glycogen synthase kinase 3β: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell. 2001;105:721–732. doi: 10.1016/s0092-8674(01)00374-9. [DOI] [PubMed] [Google Scholar]
- Davis CN, Mann E, Behrens MM, Gaidarova S, Rebek M, Rebek J, Jr, Bartfai T. MyD88-dependent and -independent signaling by IL-1 in neurons probed by bifunctional Toll/IL-1 receptor domain/BB-loop mimetics. Proc Natl Acad Sci U S A. 2006;103:2953–2958. doi: 10.1073/pnas.0510802103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo G, Wilcock D, Henderson D, Gordon M, Morgan D. Intrahippocampal LPS injections reduce Aβ load in APP+PS1 transgenic mice. Neurobiol Aging. 2001;22:1007–1012. doi: 10.1016/s0197-4580(01)00292-5. [DOI] [PubMed] [Google Scholar]
- Engel T, Hernández F, Avila J, Lucas JJ. Full reversal of Alzheimer's disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J Neurosci. 2006a;26:5083–5090. doi: 10.1523/JNEUROSCI.0604-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel T, Goñi-Oliver P, Lucas JJ, Avila J, Hernández F. Chronic lithium administration to FTDP-17 tau and GSK-3β; overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J Neurochem. 2006b;99:1445–1455. doi: 10.1111/j.1471-4159.2006.04139.x. [DOI] [PubMed] [Google Scholar]
- Fu H, Liu B, Frost JL, Hong S, Jin M, Ostaszewski B, Shankar GM, Costantino IM, Carroll MC, Mayadas TN, Lemere CA. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Aβ by microglia. Glia. 2012;60:993–1003. doi: 10.1002/glia.22331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgaber D, Harris HW, Hla T, Maciag T, Donnelly RJ, Jacobsen JS, Vitek MP, Gajdusek DC. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc Natl Acad Sci U S A. 1989;86:7606–7610. doi: 10.1073/pnas.86.19.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray CW, Patel AJ. Regulation of β-amyloid precursor protein isoform mRNAs by transforming growth factor-β1 and interleukin-1β in astrocytes. Mol Brain Res. 1993;19:251–256. doi: 10.1016/0169-328x(93)90037-p. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, 3rd, Araoz C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A. 1989;86:7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer's disease: the potential role of a “cytokine cycle ” in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Heneka MT, O'Banion MK. Inflammatory processes in Alzheimer's disease. J Neuroimmunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Herber DL, Roth LM, Wilson D, Wilson N, Mason JE, Morgan D, Gordon MN. Time-dependent reduction in Aβ levels after intracranial LPS administration in APP transgenic mice. Exp Neurol. 2004;190:245–253. doi: 10.1016/j.expneurol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Herber DL, Mercer M, Roth LM, Symmonds K, Maloney J, Wilson N, Freeman MJ, Morgan D, Gordon MN. Microglial activation is required for Abeta clearance after intracranial injection of lipopolysaccharide in APP transgenic mice. J Neuroimmune Pharmacol. 2007;2:222–231. doi: 10.1007/s11481-007-9069-z. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer's disease mice. J Neuroscience. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of A[beta]42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Ishizawa T, Sahara N, Ishiguro K, Kersh J, McGowan E, Lewis J, Hutton M, Dickson DW, Yen SH. Co-localization of glycogen synthase kinase-3 with neurofibrillary tangles and granulovacuolar degeneration in transgenic mice. Am J Pathol. 2003;163:1057–1067. doi: 10.1016/S0002-9440(10)63465-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer's disease. J Neurosci. 2005;25:8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, Cribbs DH, LaFerla FM. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-Catenin pathway function in an Alzheimer's disease model. J Immunol. 2011;187:6539–6549. doi: 10.4049/jimmunol.1100620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YC, Shaftel SS, Miller JN, Tallents RH, Chang Y, Pinkert CA, Olschowka JA, Dickerson IM, Puzas JE, O'Banion MK, Kyrkanides S. Intraarticular induction of interleukin-1β expression in the adult mouse, with resultant temporomandibular joint pathologic changes, dysfunction, and pain. Arthritis Rheum. 2006;54:1184–1197. doi: 10.1002/art.21771. [DOI] [PubMed] [Google Scholar]
- Larson ME, Lesné SE. Soluble Aβ oligomer production and toxicity. J Neurochem. 2012;120:125–139. doi: 10.1111/j.1471-4159.2011.07478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DC, Rizer J, Selenica ML, Reid P, Kraft C, Johnson A, Blair L, Gordon MN, Dickey CA, Morgan D. LPS-induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation. 2010a;7:56. doi: 10.1186/1742-2094-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol. 2010b;177:2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao YF, Wang BJ, Cheng HT, Kuo LH, Wolfe MS. Tumor necrosis factor-α, interleukin-1β, and interferon-γ stimulate γ-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J Biol Chem. 2004;279:49523–49532. doi: 10.1074/jbc.M402034200. [DOI] [PubMed] [Google Scholar]
- Li Y, Liu L, Barger SW, Griffin WS. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci. 2003;23:1605–1611. doi: 10.1523/JNEUROSCI.23-05-01605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, Rübe CE, Walter J, Heneka MT, Hartmann T, Menger MD, Fassbender K. TLR2 is a primary receptor for Alzheimer's amyloid β peptide to trigger neuroinflammatory activation. J Immunol. 2012;188:1098–1107. doi: 10.4049/jimmunol.1101121. [DOI] [PubMed] [Google Scholar]
- Lucas JJ, Hernández F, Gómez-Ramos P, Morán MA, Hen R, Avila J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Tzavaras N, Tsokas P, Landau EM, Blitzer RD. Synaptic stimulation of mTOR is mediated by Wnt signaling and regulation of glycogen synthetase kinase-3. J Neurosci. 2011;31:17537–17546. doi: 10.1523/JNEUROSCI.4761-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm TM, Koistinaho M, Pärepalo M, Vatanen T, Ooka A, Karlsson S, Koistinaho J. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to β-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18:134–142. doi: 10.1016/j.nbd.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Mastrangelo MA, Bowers WJ. Detailed immunohistochemical characterization of temporal and spatial progression of Alzheimer's disease-related pathologies in male triple-transgenic mice. BMC Neurosci. 2008;9:81. doi: 10.1186/1471-2202-9-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matousek SB, Ghosh S, Shaftel SS, Kyrkanides S, Olschowka JA, O'Banion MK. Chronic IL-1β-mediated neuroinflammation mitigates amyloid pathology in a mouse model of Alzheimer's disease without inducing overt neurodegeneration. J Neuroimmune Pharmacol. 2012;7:156–164. doi: 10.1007/s11481-011-9331-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987;79:195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- Meda L, Baron P, Prat E, Scarpini E, Scarlato G, Cassatella MA, Rossi F. Proinflammatory profile of cytokine production by human monocytes and murine microglia stimulated with β-amyloid[25–35] J Neuroimmunol. 1999;93:45–52. doi: 10.1016/s0165-5728(98)00188-x. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular a[beta] and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT, Collingridge GL. LTP Inhibits LTD in the hippocampus via regulation of GSK3β. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Rogers JT, Leiter LM, McPhee J, Cahill CM, Zhan SS, Potter H, Nilsson LN. Translation of the Alzheimer amyloid precursor protein mRNA is up-regulated by Interleukin-1 through 5′-untranslated region sequences. J Biol Chem. 1999;274:6421–6431. doi: 10.1074/jbc.274.10.6421. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Resolving controversies on the path to Alzheimer's therapeutics. Nat Med. 2011;17:1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- Shaftel SS, Carlson TJ, Olschowka JA, Kyrkanides S, Matousek SB, O'Banion MK. Chronic Interleukin-1β expression in mouse brain leads to leukocyte infiltration and neutrophil-independent blood brain barrier permeability without overt neurodegeneration. J Neurosci. 2007a;27:9301–9309. doi: 10.1523/JNEUROSCI.1418-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O'Banion MK. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest. 2007b;117:1595–1604. doi: 10.1172/JCI31450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaftel SS, Griffin WS, O'Banion MK. The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflammation. 2008;5:7. doi: 10.1186/1742-2094-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw M, Cohen P, Alessi DR. Further evidence that the inhibition of glycogen synthase kinase-3β by IGF-1 is mediated by PDK1/PKB-induced phosphorylation of Ser-9 and not by dephosphorylation of Tyr-216. FEBS Lett. 1997;416:307–311. doi: 10.1016/s0014-5793(97)01235-0. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Ito K, Skinner RD, Mrak RE, Rovnaghi CR, Van Eldik LJ, Griffin WS. In vivo and in vitro evidence supporting a role for the inflammatory cytokine interleukin-1 as a driving force in Alzheimer pathogenesis. Neurobiol Aging. 1996;17:761–766. doi: 10.1016/0197-4580(96)00104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Mrak RE, Griffin WS. Neuritic plaque evolution in Alzheimer's disease is accompanied by transition of activated microglia from primed to enlarged to phagocytic forms. Acta Neuropathol. 1997a;94:1–5. doi: 10.1007/s004010050664. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Mrak RE, Griffin WS. Glial-neuronal interactions in Alzheimer disease: progressive association of IL-1alpha+ microglia and S100beta+ astrocytes with neurofibrillary tangle stages. J Neuropathol Exp Neurol. 1997b;56:285–290. [PubMed] [Google Scholar]
- Sheng JG, Zhu SG, Jones RA, Griffin WS, Mrak RE. Interleukin-1 promotes expression and phosphorylation of neurofilament and tau proteins in vivo. Exp Neurol. 2000;163:388–391. doi: 10.1006/exnr.2000.7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Jones RA, Zhou XQ, McGinness JM, Van Eldik LJ, Mrak RE, Griffin WST. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer's disease: potential significance for tau protein phosphorylation. Neurochem Intl. 2001;39:341–348. doi: 10.1016/s0197-0186(01)00041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]