

Abstract
Loss of the de novo DNA methyltransferases Dnmt3a and Dnmt3b in embryonic stem cells obstructs differentiation; however, the role of these enzymes in somatic stem cells is largely unknown. Using conditional ablation, we show that Dnmt3a loss progressively impairs hematopoietic stem cell (HSC) differentiation over serial transplantation, while simultaneously expanding HSC numbers in the bone marrow. Dnmt3a-null HSCs show both increased and decreased methylation at distinct loci, including substantial CpG island hypermethylation. Dnmt3a-null HSCs upregulate HSC multipotency genes and downregulate differentiation factors, and their progeny exhibit global hypomethylation and incomplete repression of HSC-specific genes. These data establish Dnmt3a as a critical participant in the epigenetic silencing of HSC regulatory genes, thereby enabling efficient differentiation.
Hematopoietic stem cells reside in the bone marrow and are defined by their capacity for the lifelong maintenance of blood and bone marrow, achieved through their differentiation into a myriad of cell types, as well as the regeneration of stem cells via self-renewal. The mechanisms that dictate a fate of differentiation versus self-renewal are still poorly understood. A number of transcription factors involved in the maintenance of HSC function have been identified; however, there is little insight into how epigenetic mechanisms orchestrate the activities of these factors to ensure blood homeostasis.
DNA methylation of CpG dinucleotides is a key epigenetic modification that influences tissue- and context-specific gene expression1. CpG methylation is catalyzed by a family of DNA methyltransferase enzymes comprising three members—Dnmt1, Dnmt3a and Dnmt3b2. Studies of mouse mutants have shown that these factors are essential for normal embryonic development, as their absence results in death from the ten-somite stage to 4 weeks after birth, depending on which Dnmt gene is inactivated and how this inactivation was achieved3. Whereas the Dnmt1 methyltransferase is thought to be principally involved in the maintenance of pre-existing methylation4, Dnmt3a and Dnmt3b act as de novo methyltransferases, modifying unmethylated DNA3. Embryonic stem (ES) cells lacking both Dnmt3a and Dnmt3b progressively lose differentiation potential with cell passage5, although the potential for self-renewal is maintained.
The role of DNA methylation in somatic stem cells has recently begun to surface. In neural progenitors, Dnmt3a has been shown to enable the expression of neurogenic genes through gene-body methylation6. In HSCs, loss of Dnmt1 leads to nearly immediate and complete loss of HSC activity in vivo7, and HSCs from mice with impaired Dnmt1 activity cannot suppress key myeloerythroid regulators and cannot differentiate into lymphoid progeny8. Loss of Dnmt3a or Dnmt3b alone was thought to have no impact on HSC function9; however, loss of both together was reported to abolish self-renewal but maintain the capacity for differentiation9. Given recent reports of DNMT3A mutations in over 20% of individuals with acute myeloid leukemia (AML)10–12 and around 10% of those with myelodysplastic syndrome (MDS)13, we re-evaluated the role of Dnmt3a in hematopoiesis.
RESULTS
Expression and function of Dnmt3a in HSCs
In the hematopoietic system, Dnmt3a expression was highly enriched in the most primitive long-term HSCs (LT-HSCs) compared to progenitors and differentiated cells (Fig. 1a). To investigate the function of Dnmt3a in hematopoiesis, we generated inducible conditional knockout mice by crossing mice carrying a loxP-flanked (floxed) copy of the gene (Dnmt3afl/fl mice)14 with mice carrying the Mx1-Cre driver. To examine the result of Dnmt3a loss in HSCs independent of possible effects on the niche, purified HSCs were transplanted into wild-type recipients before the induction of Dnmt3a deletion, with 250 Mx1-Cre+;Dnmt3afl/fl HSCs (side population+ c-Kit+, lineage−, Sca-1+) transplanted along with 250 × 103 whole bone marrow (WBM) cells from distinguishable wild-type mice. Four weeks after transplantation, Dnmt3a deletion was induced by injection with polyinosinic-polycytidylic acid (pIpC). Control mice throughout this study (unless otherwise specified) consisted of Dnmt3afl/fl littermates lacking Mx1-Cre, which were treated identically to experimental mice, including receiving injections of pIpC to control for interferon-mediated effects15. We cannot exclude the possibility of minor Dnmt3a-independent effects from the Mx1-Cre transgene. Analysis of pIpC-treated, donor-derived HSCs or bone marrow showed efficient Dnmt3a mRNA ablation and no detectable full-length or truncated protein (Supplementary Fig. 1).
Figure 1.

Dnmt3a is highly expressed in HSCs and its ablation has profound functional effects. (a) Real-time PCR analysis of Dnmt3a mRNA in LT-HSCs, short-term HSCs (ST-HSCs) and representative committed progenitors and differentiated cells. MPPs, multi-potential progenitors; CLPs, common lymphoid progenitors; CMPs, common myeloid progenitors; MEPs, megakaryocyte-erythroid progenitors; GMPs, granulocyte-macrophage progenitors (see Online Methods for purification schemes). Mean ± s.e.m. values are shown for three biological replicates. (b) Contribution of control (Dnmt3afl/fl;Mx1-Cre−) and Dnmt3a-null (Dnmt3afl/fl;Mx1-Cre+) HSCs to recipient mouse peripheral blood in secondary competitive transplants, measured at monthly intervals. Mean ± s.e.m. values are shown. (c) Lineage differentiation in secondary recipients of transplanted control and Dnmt3a-null HSCs. Shown are percentages of donor-derived (CD45.2+) myeloid cells (Gr1+ or Mac1+), B cells (B220+) and T cells (CD4+ or CD8+) in peripheral blood analyzed 16 weeks after transplantation. Differences that are significant between control and Dnmt3a-null HSCs are indicated. Mean ± s.e.m. values are shown (N = 15–37 mice). (d) Hoechst staining and flow cytometry analysis of the bone marrow of secondary recipient mice. Top, the boxed region shows the percentage of side population (SP) cells from mice transplanted with HSCs of the indicated genotypes. Bottom, SP cells were further gated using c-Kit+, lineage− and Sca-1+ (KLS) markers to reveal the proportion of test (CD45.2+) versus competitor (CD45.1+) HSCs. (e) Alternative HSC phenotype schemes for test cells gated first by KLS show similar expansion of the Dnmt3a-null HSC compartment. (f) Quantification of total HSC frequency in the bone marrow of secondary recipient mice by three phenotypic definitions. Hatched area indicates the proportion of CD45.2+ test cells. SLAM is CD150+, CD48−, KLS gating. Mean ± s.e.m. values are shown. (g) Analysis of progenitor frequencies in secondary recipient mice. Hatched area indicates the contribution of CD45.2+ test cells. Mean ± s.e.m. values are shown. **P < 0.001; ***P < 0.001.
Monthly analysis of test cell contribution to peripheral blood generation in primary recipients revealed no differences between mice transplanted with Dnmt3a-null or control HSCs (Supplementary Fig. 2). Although Dnmt3a was ablated, we reasoned that the DNA methylation already present might not be eliminated unless the HSCs divided. Thus, we forced stem cell turnover by transplanting the HSCs into secondary recipients. We purified Dnmt3a-null or control HSCs from primary recipients and transferred 250 of each to secondary recipients along with fresh wild-type competitor WBM cells. In this context, Dnmt3a-null HSCs exhibited a substantially greater contribution to peripheral blood production compared to control HSCs (Fig. 1b). The Dnmt3a-null HSCs retained multilineage engraftment, with a slight bias toward B-cell differentiation relative to controls (Fig. 1c).
Analysis of the bone marrow 18 weeks later revealed a striking increase in the number of phenotypically defined HSCs in the recipients of Dnmt3a-null HSCs. The approximate doubling of peripheral blood production was accompanied by a >7-fold increase in HSC numbers in mice transplanted with Dnmt3a-null cells (Fig. 1d,e). Whereas in recipients of control HSCs, ~35–60% of the regenerated HSCs were derived from the test cells, in recipients of Dnmt3a-null HSCs, over 90% of the HSC pool was comprised of HSCs derived from the transplanted Dnmt3a-null cells, regardless of the criteria used for HSC identification (Fig. 1f). The wild-type WBM competitor cells generated approximately 5,000 HSCs in the bone marrow of mice transplanted with either control or Dnmt3a-null HSCs. Whereas control HSCs generated 3,000–5,500 HSCs in each recipient per 250 input HSCs, transplantation of 250 purified Dnmt3a-null HSCs generated 40,000–85,000 HSCs per mouse.
This dominance of the HSC pool by cells derived from Dnmt3a-null HSCs was not matched by a similar contribution of these HSCs to the quantity or proportion of the immediate downstream progenitors (Fig. 1g and Supplementary Table 1), indicating that the effect of Dnmt3a loss was largely restricted to the most primitive HSCs. Expansion of Dnmt3a-null HSCs in vivo could not be attributed to enhanced proliferation (Fig. 2a,b and Supplementary Fig. 3) nor to exceptional resistance to apoptosis (Fig. 2c). Nevertheless, the function of Dnmt3a-null HSCs was not normal: compared to control mice, blood production was not proportional to the representation of cognate HSCs in the marrow, suggesting a possible differentiation defect.
Figure 2.
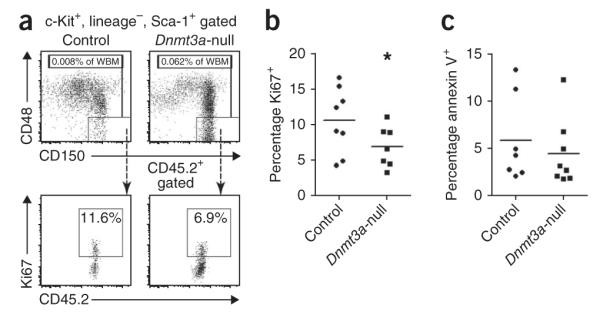
Cellular kinetics of Dnmt3a-null HSCs. (a) HSC gating scheme for the analysis of proliferation and apoptosis in secondary recipient mice transplanted with control or Dnmt3a-null HSCs. (b) Ki67 staining shows a significant reduction in the proliferative index of Dnmt3a-null HSCs relative to control HSCs; *P < 0.05. (c) Annexin V staining shows no difference in the apoptotic rate between control and Dnmt3a-null HSCs. Bars in b and c indicate the mean values for each genotype.
Dnmt3a loss impairs long-term HSC differentiation in vivo
The potential differentiation defect in Dnmt3a-null HSCs was reminiscent of ES cells deficient for both Dnmt3a and Dnmt3b. Self-renewal of ES cells lacking both of these genes is maintained, but these cells lose the ability to differentiate with serial passage5. To test whether HSCs lacking Dnmt3a would behave similarly, we purified 250 secondary HSCs and transplanted them into tertiary recipients, effectively passaging them in vivo. Although the differentiation capacity of both control and Dnmt3a-null HSCs declined, Dnmt3a-null HSCs continued to show modestly enhanced peripheral blood generation (Fig. 3a), with contribution to all lineages (Supplementary Fig. 2b). However, Dnmt3a-null HSCs again regenerated remarkable numbers of HSCs, with an average of 50-fold more HSCs derived from Dnmt3a-null cells than from their control counterparts (Fig. 3b). This trend continued in quaternary recipients (Fig. 3a–c). The effect of Dnmt3a loss on HSC activity was cell autonomous, as Dnmt3a-null HSCs showed higher in vitro colony-forming activity compared to control HSCs after each stage of transplantation (Supplementary Fig. 4a). PCR analysis of single HSC-derived colonies showed highly efficient Dnmt3a deletion (Supplementary Fig. 4b,c).
Figure 3.

Dnmt3a-null HSCs show inhibition of long-term differentiation in serial competitive transplantation of HSCs. (a) The proportion of peripheral blood generated from the test cells in recipient mice 16 weeks after transplantation. (b) Quantification of donor-derived HSCs in the bone marrow of recipient mice 18 weeks after transplantation, defined as CD45.2+, SPKLS cells. Data are representative of at least three individual transplantation experiments for each stage of serial transfer (N = 15–37 mice per group). Mean ± s.e.m. values are shown. (c) Flow cytometry data of quaternary recipient mice transplanted with control or Dnmt3a-null HSCs showing virtually all continuously amplified HSCs in bone marrow were derived from Dnmt3a-null HSCs (CD45.2+). (d) Differentiation and self-renewal quotients, calculated at the end of each round of transplantation with Dnmt3a-null and control HSCs. ***P < 0.001.
Together, these data suggested a crucial role for Dnmt3a in the choice between differentiation and self-renewal. Whereas Dnmt3a-null HSCs contributed more to blood production compared to control HSCs, the observed increase in contribution was not proportional to the robust expansion of HSCs derived from Dnmt3a-null cells in the bone marrow (Supplementary Fig. 5). To quantify these HSC characteristics, we calculated HSC amplification (the number of donor-derived HSCs recovered at the end of transplantation per original input HSC, or the ‘self-renewal quotient’) and differentiation capacity (16-week white blood cell count per microliter of blood multiplied by the percentage of test cell chimerism and divided by the number of donor-derived HSCs, or the ‘differentiation quotient’) (Fig. 3d and Supplementary Table 2). This analysis showed that control HSCs exhibit a gradual decline in both differentiation and self-renewal after each successive round of serial transplantation. In contrast, the in vivo differentiation capacity of Dnmt3a-null HSCs declined sharply, but their self-renewal potential amplified after secondary transplantation and was sustained, showing no decrease even after four rounds of transplantation. These data suggest that Dnmt3a loss particularly affects LT-HSCs, such that in the absence of Dnmt3a, cell division is more likely to result in self-renewal rather than differentiation.
Dnmt3a loss in HSCs results in both hyper- and hypomethylation
We began to investigate the mechanisms through which Dnmt3a enables HSC differentiation by examining DNA methylation alterations in Dnmt3a-null HSCs. Comparison of global 5-methylcytosine (5mC) levels by high-performance liquid chromatography–mass spectrometry (HPLC-MS) between transplanted control and Dnmt3a-null HSCs showed no overall difference in the level of 5mC in the whole genome (Fig. 4a). To investigate methylation differences in specific genomic regions, we performed reduced representation bisulfite sequencing (RRBS), which enables quantitative methylation analysis of relatively small cell numbers16,17, comparing transplanted control and Dnmt3a-null HSCs (Supplementary Table 3). For each genotype, two RRBS replicates with high correlation (~0.98; Supplementary Table 4) were analyzed (the final DNA methylation map is a composite of the data from the two biological replicates). The mean methylation ratio was not different between Dnmt3a-null and control HSCs, an observation in agreement with the HPLC-MS data.
Figure 4.

Dnmt3a loss in HSCs results in both hyper- and hypomethylation. (a) HPLC-MS analysis of global 5mc levels as a proportion of the total cytosine in purified HSCs from secondary recipient mice (N = 2). (b) RRBS analysis of tertiary recipient mice transplanted with control or Dnmt3a-null HSCs. Plots show the degree of differential methylation (between Dnmt3a-null and control HSCs) and its relationship to local CpG density (blue). Left, all hypomethylated (red, CpGs ≤33% methylated) and hypermethylated (green, CpGs ≥33% methylated) DMCs in Dnmt3a-null HSCs. Right, DMCs located within CGIs. (c) Independent bisulfite sequencing analysis of selected hypermethylated CGIs in Dnmt3a-null HSCs. (d) Bisulfite sequencing analysis of selected hypomethylated genes in Dnmt3a-null HSCs. Schematic diagrams for each gene are shown in c and d (not to scale). Exons are represented by vertical rectangles. White horizontal bars indicate CGIs, and black horizontal bars show the tested region. Open and filled circles represent unmethylated and methylated CpGs, respectively. Differences in methylation between control and Dnmt3a-null cells that were statistically significant are indicated; *P < 0.05, ***P < 0.001.
In the composite methylation map, ~1 million CpGs, termed covered CpGs, had at least tenfold coverage in both control and Dnmt3a-null samples. Although covered CpGs only make up 5% of the total 21.3 million CpGs in the mouse genome, ~5 of 6 CpG islands (CGIs) and two-thirds of promoters had more than three covered CpGs (Supplementary Fig. 6). This coverage allowed the examination of differential DNA methylation throughout various genomic features; for this analysis, we defined a change of ≥33% to indicate a differentially methylated CpG (DMC)16. Whereas some genomic features showed the expected loss of methylation (hypomethylation) following Dnmt3a ablation, some regions showed a notable increase in methylation (hypermethylation) (Fig. 4b). When all DMCs were considered, approximately 58% showed hypermethylation, and 42% were hypomethylated. CpG-rich and CpG-poor regions were affected differently. Within CGIs, nearly 95% of DMCs became hypermethylated in Dnmt3a-null HSCs (Fig. 4b). Regulatory regions (for example, promoters) lacking CGIs were 3–7 times more susceptible to alterations in DNA methylation than their CGI-containing counterparts (Supplementary Table 5). DMCs in CGI-associated promoters similarly tended to be hypermethylated, whereas DMCs in promoters without CGIs were in general equally likely to become hypo- or hypermethylated (Supplementary Fig. 7). This relationship between local CpG density and Dnmt3a-dependent methylation held true for many genomic features (Supplementary Fig. 7).
Because it seemed counterintuitive that loss of the Dnmt3a DNA methyltransferase enzyme would lead to hypermethylation of CGIs, we verified the hypermethylation of some of the gene-associated CGIs in Dnmt3a-null HSCs by independent bisulfite sequencing. This analysis confirmed hypermethylation of CGIs associated with Basp1, Pdxdc1 and Wbscr17 in Dnmt3a-null HSCs (Fig. 4c), validating the RRBS data. Although hypermethylation in Dnmt3a-null HSCs is paradoxical, the expression levels of Dnmt1 and Dnmt3b were unchanged (Supplementary Fig. 8), leaving the possibility that the aberrant activity of these enzymes in the absence of Dnmt3a could account for this phenomenon.
To examine whether particular gene functional categories were associated with changes in DNA methylation, we grouped DMCs into differentially methylated regions (DMRs) defined by at least three nearby DMCs of the same class, identifying 1,063 hypermethylated DMRs associated with 534 genes and 741 hypomethylated DMRs associated with 434 genes in Dnmt3a-null HSCs (Supplementary Table 6). Many of the hypomethylated genes are commonly associated with human hematopoietic malignancies, such as Prdm16, Stat1, Ccnd1, Myc, Mn1, Msi2, Men1, Erg and Runx1, and several were validated by targeted bisulfite sequencing (Fig. 4d). KEGG pathway and Oncomine concept analyses revealed a significant enrichment for hypomethylated genes in the categories of cancer-causal genes (P = 2.76 × 10−5) and gene sets overexpressed in leukemias such as AML (P = 5.25 × 10−4) and acute lymphoblastic leukemia (ALL, P = 2.67 × 10−6) (Supplementary Fig. 9).
Multipotency gene upregulation in Dnmt3a-null HSCs
The effects of these altered methylation patterns in Dnmt3a-null HSCs on gene expression were examined using global transcriptional profiling. Using stringent bioinformatic criteria, we identified 516 unique genes differentially expressed between transplanted Dnmt3a-null and control HSCs (225 downregulated and 291 upregulated in Dnmt3a-null HSCs; Supplementary Table 7). In the group of genes upregulated in Dnmt3a-null HSCs, there was an enrichment for genes highly expressed in normal HSCs, including 22 genes previously identified as ‘HSC fingerprint’ genes (expressed in mouse HSCs but not differentiated blood cells18), as well as other genes known to be crucial for HSC function, such as Gata3 (ref. 19), Runx1 (ref. 20), Pbx1 (ref. 21) and Cdkn1a22. Conversely, Dnmt3a-null HSCs showed lower expression of factors that are crucial for HSC differentiation, such as those encoded by Flk2 (ref. 23), Ikaros24, Sfpi1 (PU.1)25 and Mef2c26. Changes in expression for several genes from each category were validated by real-time PCR (Fig. 5a). The concomitant upregulation of HSC multipotency genes and downregulation of differentiation genes may contribute to the bias of Dnmt3a-null HSCs toward self-renewal.
Figure 5.

Dnmt3a loss in HSCs leads to higher expression of HSC multipotency genes. (a) Relative expression levels of select multipotency, HSC fingerprint18 and differentiation genes measured by real-time PCR analysis. Mean ± s.e.m. values are from three replicates in Dnmt3a-null HSCs normalized to the expression levels in control HSCs (dashed line). (b) Bisulfite sequencing analysis of multipotency and HSC fingerprint genes in control and Dnmt3a-null HSCs. Open horizontal bars indicate CGIs, and black horizontal bars show the tested region. Open and filled circles represent unmethylated and methylated CpGs, respectively. Differences in methylation between control and Dnmt3a-null HSCs that were statistically significant are indicated. (c) H3K4me3 ChIP analysis of DMRs in control and Dnmt3a-null HSCs. Mean ± s.e.m. values are shown (N = 3 replicate experiments). (d) Dnmt3a ChIP analysis of DMRs in wild-type hematopoietic progenitors (KLS cells, N = 2 replicate experiments) reveals Dnmt3a binding to CGIs in Runx1 and Gata3 but not in Nr4a2. Mean ± s.e.m. values are shown. *P < 0.05, **P < 0.01, ***P < 0.001.
Basal gene expression broadly correlated with global methylation patterns in control and Dnmt3a-null HSCs, in that expressed genes tended to have lower overall levels of methylation compared to transcriptionally silent genes (Supplementary Fig. 10a). However, there was little correlation between changes in methylation and differential gene expression in the mutant HSCs (Supplementary Fig. 10b). Other studies have similarly observed a poor correlation in various human disease states10,27. Although the relationship between DNA methylation and gene expression is also dependent on other epigenetic factors and the presence of required transcription factors at specific developmental stages, this finding suggests that the dysregulation of a limited number of key direct targets of Dnmt3a produces a wide array of indirect effects.
We next examined specific genes, linking methylation and gene expression changes to probe the mechanisms behind the loss of differentiation capacity in Dnmt3a-null HSCs. The upregulation of several genes in Dnmt3a-null HSCs relative to control cells might contribute to this phenotype (inhibition of differentiation, enhanced self-renewal, lower proliferative index), including Runx1 (4.3-fold increase), Gata3 (2.3-fold increase) and Nr4a2 (1.9-fold increase). Overexpression of both Gata3 (ref. 19) and Runx1 (ref. 28) in normal HSCs leads to the inhibition of differentiation, and overexpression of Nr4a2 forces HSCs into quiescence29. According to the RRBS data, the Runx1 and Gata3 loci contained some of the largest hypomethylated DMRs in Dnmt3a-null HSCs. Independent bisulfite sequencing of these regions showed notable hypomethylation of the associated CGIs (Fig. 5b). These genes fit into the classic regulatory model, in which decreased methylation leads to increased transcription. The increased expression of Nr4a2 in Dnmt3a-null HSCs relative to control HSCs could not be attributed to hypomethylation, as there was only a marginal decrease in methylation in the CGI in exon 3 of Nr4a2 (Fig. 5b). Notably, the Gata3 and Runx1 CGIs that were methylated in control HSCs were associated with the gene body rather than with promoter regions; the genes were still highly expressed and their promoters unmethylated in both control and Dnmt3a-null HSCs (Supplementary Fig. 11). These data, although on a limited number of genes, support the view that Dnmt3a can affect non-promoter DNA methylation6.
A recent study of Dnmt3a-null neural progenitors correlated Dnmt3a target genes with high levels of trimethylation of lysine 4 on histone H3 (H3K4me3), a histone mark indicative of transcriptionally permissive chromatin states6. Consistent with their higher expression level, Runx1, Gata3 and Nr4a2 all showed increased H3K4me3 in Dnmt3a-null HSCs in the hypomethylated regions when compared with control cells (Fig. 5c). To determine whether these genes are direct targets of the Dnmt3a enzyme, we performed chromatin immunoprecipitation (ChIP) with Dnmt3a in wild-type hematopoietic stem and progenitor cells (c-Kit+, lineage−, Sca-1+). Strong binding of Dnmt3a was detected for Runx1 and Gata3 but not for Nr4a2 (Fig. 5d). The data from this small group of genes suggest that the loss of Dnmt3a in HSCs leads to the upregulation of HSC multipotency genes by two distinct mechanisms: (i) regulation of direct targets, which are characterized by Dnmt3a binding in wild-type HSCs and hypomethylation in Dnmt3a-null HSCs (for example, Runx1 and Gata3), and (ii) regulation of indirect targets, which show no significant methylation changes in Dnmt3a-null HSCs but have altered histone modifications. The second class of genes (for example, Nr4a2) may be regulated by factors encoded by direct Dnmt3a targets, like Runx1. Thus, in HSCs, Dnmt3a may have direct and indirect effects on gene expression.
Dnmt3a represses the stem cell program during HSC differentiation
If Dnmt3a is important for the differentiation of HSCs, we hypothesized that the impact of its loss may also be evident in differentiated Dnmt3a-null progeny. To test this idea, we examined DNA methylation in a representative cell lineage, purifying B cells from secondary transplant recipient mice in which Dnmt3a was ablated but differentiation was still effective. In contrast to our observations in HSCs, the overall level of 5mC was reduced in Dnmt3a-null B cells relative to control B cells (Fig. 6a). Because we could obtain relatively large numbers of differentiated cells, we used digital restriction enzyme analysis of methylation (DREAM) sequencing30 to identify at high resolution and independent of bisulfite treatment loci differentially methylated in control and Dnmt3a-null B cells. Analyzing CpGs with >20 reads, differences in methylation of >25% could be detected with a false discovery rate (FDR) <0.07% (Supplementary Table 8). Of the 20,365 CpGs with sufficient reads, 282 sites (1.4%) showed >25% hypomethylation in Dnmt3a-null B cells, whereas only 10 sites (0.05%) showed hypermethylation (hypomethylation was above the expected FDR, hypermethylation was not). Unlike in HSCs, Dnmt3a-null B cells showed a net reduction in global methylation and almost all DMCs were hypomethylated (Fig. 6b).
Figure 6.
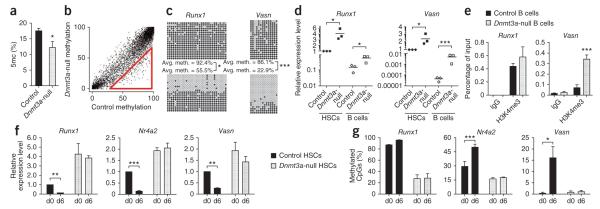
Dnmt3a is required to suppress the stem cell program in HSCs to permit differentiation. (a) HPLC-MS analysis of global 5mc levels as a proportion of the total cytosine in B cells from secondary recipient mice. Mean ± s.e.m. values are shown (N = 7 mice). (b) DREAM analysis of B cells in secondary recipient mice. SmaI sites with at least 20 sequence tags in control B cells are plotted showing the methylation ratio between the genotypes. The red triangle indicates sites of hypomethylation in Dnmt3a-null B cells in 1.4% of all CpGs (FDR = 0.07%). (c) Bisulfite sequencing across the Vasn and Runx1 CGIs in control (top) and Dnmt3a-null (bottom) B cells. Differences in methylation between control and Dnmt3a-null cells that were statistically significant are indicated. (d) Cognate gene expression for cells analyzed in c. Diamonds indicate control cells, and squares indicate Dntm3a-null cells. HSCs are represented by filled symbols and B cells by open symbols. Bars indicate the average gene expression for each cell population. Differences in expression between control and Dnmt3a-null cells that were statistically significant are indicated. (e) H3K4me3 ChIP analysis for Runx1 and Vasn in control and Dnmt3a-null B cells. Mean ± s.e.m. values are shown (N = 4 replicate experiments). (f) Expression of Dnmt3a-responsive genes at day 0 (d0) and day 6 (d6) after 5-FU exposure measured by real-time PCR. Expression levels are relative to normalized expression of that gene in d0 control HSCs. Mean ± s.e.m. values are shown, and statistically significant differences in expression at the two time points are indicated. (g) Proportion of methylated CpGs as detected by bisulfite sequencing across CGIs. Mean ± s.e.m. values are shown for three biological replicates, and statistically significant differences in methylation at the two time points are indicated. *P < 0.05, **P < 0.01, ***P < 0.001.
Several of the hypomethylated sites in Dnmt3a-null B cells were associated with genes that in the hematopoietic system are normally expressed primarily in HSCs, including Runx1 and Vasn. These genes were almost completely methylated in control B cells (Fig. 6c) but were largely unmethylated in Dnmt3a-null B cells, which correlated with incomplete gene silencing in the knockout cells (Fig. 6d). As in the Dnmt3a-null HSCs, these genes also exhibited higher H3K4me3 levels than seen in control cells (Fig. 6e).
In addition to the hypomethylation of Runx1 and Vasn in B cells, there was a striking enrichment for HSC fingerprint genes, including Mycn, Ptpn14, Prdm16, Pdk4, Vwf and Vldlr (Supplementary Table 8). These same genes were upregulated in Dnmt3a-null HSCs (Supplementary Table 7), and several continued to be expressed in Dnmt3a-null B cells, as determined by real-time PCR (data not shown). This pattern of hypomethylation and residual expression of stem cell–specific genes in B cells suggests that a major role of Dnmt3a in HSCs is to methylate and repress the stem cell program, silencing HSC genes and permitting differentiation. The persistently higher expression of HSC multipotency genes in Dnmt3a-null HSCs may also reinforce the stem cell program, contributing to preferential self-renewal.
To test our hypothesis that Dnmt3a-mediated silencing of the stem cell program permits differentiation, we analyzed the behavior of candidate Dnmt3a-regulated genes following artificial stimulation of differentiation using 5-flurouracil (5-FU), which kills cycling hematopoietic cells, thus forcing HSCs to proliferate and differentiate to replenish the hematopoietic compartment. Six days after 5-FU exposure, HSCs are undergoing maximal proliferation, a state characterized by markedly reduced expression of the regulators of HSC quiescence, such as Nr4a2 and Pbx1 (ref. 31). At this time, control HSCs showed a significant decrease in the expression of Runx1, Nr4a2 and Vasn when compared to cells at day 0 of 5-FU exposure (Fig. 6e), coupled with increases in their methylation levels (Fig. 6f), indicating that the silencing of these genes is concomitant with HSC proliferation and differentiation. In contrast, in Dnmt3a-null HSCs, there was no difference in the methylation or expression levels of these genes at day 0 and day 6 after 5-FU exposure (Fig. 6f,g).
Exogenous Dnmt3a partially restores Dnmt3a-null HSC function
Finally, we considered whether the aberrant preference toward self-renewal could be partially corrected in Dnmt3a-null HSCs that had already lost substantial differentiation capacity. Dnmt3a-null HSCs that had been serially transplanted into two mice were transduced with retroviruses coexpressing Dnmt3a and GFP (MSCV-Dnmt3a-IRES-GFP, abbreviated to MSCV-Dnmt3a) or expressing GFP alone (MSCV-GFP) and were transplanted into tertiary recipients. At long intervals after transplantation (>18 weeks), the frequency of HSCs transduced with MSCV-GFP remained at levels near those for Dnmt3a-null HSCs transplanted into tertiary recipients, but the frequency of MSCV-Dnmt3a-transduced HSCs was substantially reduced, although not quite to the levels seen with control HSCs transplanted into tertiary recipients (Fig. 7a and Supplementary Fig. 12a). Moreover, when GFP+ HSCs from these recipient mice were purified and cultured in vitro, the colony-forming potential of the Dnmt3a-null HSCs transduced with MSCV-Dnmt3a was indistinguishable from that of control HSCs purified from tertiary recipient mice (Fig. 7b), whereas Dnmt3a-null HSCs transduced with MSCV-GFP generated higher colony numbers. Restoration of Dnmt3a expression in Dnmt3a-null HSCs was also able to restore the methylation patterns of Vasn and Runx1 in B cells (Fig. 7c) and limit their expression in HSCs and B cells (Supplementary Fig. 12b,c). Transduction of control HSCs with MSCV-Dnmt3a resulted in a reduction in colony-forming capacity compared to control HSCs transduced with MSCV-GFP alone (Supplementary Fig. 12d), also suggesting a possible gene dosage effect.
Figure 7.

Exogenous Dnmt3a partially restores function and methylation patterns. (a) Analysis of HSC (CD150+, CD48−, KLS gated) frequency 18-weeks after transplantation. Sca-1+ cells from secondary recipient mice transplanted with Dnmt3a-null HSCs were transduced with either MSCV-Dnmt3a or control (MSCV-GFP) retroviruses and transplanted into tertiary recipients. Also shown are HSC frequencies in tertiary recipients transplanted with non-transduced control or Dnmt3a-null HSCs. Mean ± s.e.m. values are shown (N = 7–12 mice), and statistically significant differences are indicated. (b) Colony-forming capacity of Dnmt3a-null HSCs transduced with MSCV-GFP or MSCV-Dnmt3a. Also shown are colony formation from non-transduced control and Dnmt3a-null HSCs after the third serial transplantation. Mean ± s.e.m. values are shown (N = 4 replicate plates), and statistically significant differences are indicated. (c) Bisulfite sequencing in B cells across Vasn and Runx1 CGIs after forced exogenous Dnmt3a expression in Dnmt3a-null HSCs. Statistically significant differences in methylation are indicated. *P < 0.05, **P < 0.01, ***P < 0.001.
DISCUSSION
Loss of de novo DNA methyltransferases in ES cells leads to a progressive block in differentiation potential. Here we show that this principle also holds true for at least one somatic stem cell, the HSC, although loss of only Dnmt3a is sufficient to cause this phenotype. A previous study, which reported a contrary result9, likely missed the phenotype due to less efficient expression of the Cre driver from a retroviral vector and a lack of serial transplantation. The phenotype of Dnmt3a-null HSCs is markedly distinct from that of Dnmt1-null HSCs, which show premature HSC exhaustion and lymphoid-deficient differentiation7,8, indicating distinct roles for the different DNA methyltransferases in HSCs.
We show that Dnmt3a has a specific role in permitting HSC differentiation, as in its absence, phenotypically normal stem cells accumulate but progressively lose their differentiation capacity. In the differentiated progeny of Dnmt3a-null HSCs, there is a reduction in global DNA methylation, along with the presence of hypomethylated sites that correlates with increased expression of genes normally restricted to stem cells (HSC multipotency genes). Dnmt3a-null HSCs also downregulate early differentiation factors and are thus retained in the stem cell state by biasing cell divisions toward self-renewal as opposed to differentiation (Fig. 8). Histone modifications are thought to have a primary and dynamic role in the epigenetic regulation of gene expression during development32,33, with the more stable DNA methylation events having a secondary or ‘lock-down’ function. However, our data suggest that DNA methylation also has a critical role, at least at some loci, in cell type–specific epigenetic instruction.
Figure 8.
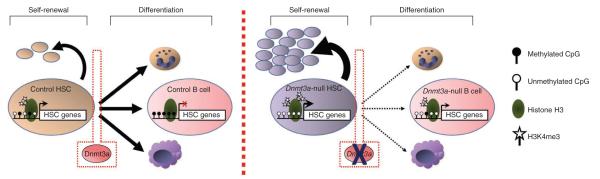
Model for Dnmt3a action in HSCs. HSC-specific genes are mostly unmethylated and expressed in normal HSCs (left). Upon receiving a signal to differentiate, Dnmt3a methylates and silences these regions to permit lineage commitment. This is associated with a loss of H3K4me3 and gene repression in B cells. Dnmt3a-null HSCs (right) cannot silence HSC genes, so upon receiving a stimulus to differentiate, HSC-specific genes remain expressed due to a lack of methylation and elevated H3K4me3. Upon cell division, the HSC self-renewal pathway remains active in Dnmt3a-null HSCs, resulting in their accumulation in the bone marrow. Of the few Dnmt3a-null HSCs that do differentiate, their progeny show incomplete methylation and partial repression of HSC genes.
At the level of DNA methylation, the Dnmt3a-null HSC phenotype is paradoxical. Despite the dramatic expansion of stem cells, there is no overall change in total methylcytosine level. We see a modest number of differentially methylated regions (1,804 DMRs) that surprisingly represent both increases and decreases in methylation in Dnmt3a-null HSCs, although our methylation profiling strategy interrogates only a fraction of the genome. It is possible that there are relatively few direct targets of Dnmt3a in HSCs, but their dysregulation in Dnmt3a-null HSCs causes widespread indirect changes. Consistent with this hypothesis, we show that Runx1 is a direct target of Dnmt3a in HSCs, and the encoded Runx1 protein has over 5,000 DNA binding sites in hematopoietic stem and progenitor cells34. However, it is unlikely that the phenotype of Dnmt3a-null HSCs can be attributed to a single gene but rather a constellation of changes mediated by DNA methylation, histone modification and gene expression.
The lack of correlation between methylation and differential gene expression in Dnmt3a-null HSCs is consistent with other studies10 and is indicative of the complex and still obscure role of DNA methylation. Accumulating evidence suggests that DNA methylation outside of promoter regions may have a major role in gene regulation6. The bias of the RRBS strategy and the fraction of the genome sampled may limit our ability to observe DNA methylation changes in the regions that would more readily explain the Dnmt3a-null HSC phenotype. Alternatively, other roles of Dnmt3a may contribute to the phenotype. For example, loss of Dnmt3a may affect the location of 5-hydroxymethylcytosine35, the role of which remains to be elucidated. Telomere integrity may also be affected, as Dnmt3a-null ES cells have elongated telomeres36, and telomere length can influence the replicative lifespan of HSCs37. Finally, Dnmt3a may interact with DNA within a protein complex in HSCs. The function of other proteins in this complex might be compromised in the absence of Dnmt3a.
Despite our currently incomplete understanding, we do observe that alterations in DNA methylation in Dnmt3a-null HSCs are broadly reminiscent of those associated with human malignancies. Hypermethylated CGIs and hypomethylated proto-oncogenes are a hallmark of many cancers38–40, including AML, in which DNMT3A mutations have recently been reported10–12. In this context, it may be surprising that we have not yet observed overt leukemia in mice transplanted with Dnmt3a-null HSCs, even more than a year after quaternary transplantation. Although we see dramatic expansion of the stem cell pool, these cells do not overwhelm the majority of hematopoietic elements in the bone marrow. Hematopoiesis in the transplant-recipient mice is derived mainly from the normal, competitor bone marrow cells. Thus, Dnmt3a deficiency alone appears insufficient to generate AML in this model, and second hits are likely to be required. Furthermore, the Dnmt3a-null status achieved here is distinct from DNMT3A mutation in humans with AML. The distinction could lead to different mechanisms of oncogenic transformation. Nevertheless, this mouse model should serve as a powerful tool to explore the relationship between Dnmt3a loss and the cooperating mutations found in humans with AML.
Although a direct link between mutations in DNMT3A, epigenetic changes and AML oncogenesis remains to be established, this study provides the first mechanistic insight into how Dnmt3a controls normal HSC function. We also show that Dnmt3a loss in HSCs leads to the inhibition of differentiation and the hypomethylation of genes with a causal role in cancer. Although hypomethylation per se may not be oncogenic in stem cells, the inability to shut down stem cell genes during differentiation may drive transformation with a secondary oncogenic event.
URLS
Genome browser tracks and additional data related to this paper are available at http://dldcc-web.brc.bcm.edu/lilab/HSC/; Bioconductor software, http://www.bioconductor.org/; R statistical software, http://cran.r-project.org/.
ONLINE METHODS
Mice and transplantation
All animal procedures were approved by the institutional animal care and use committee of the Baylor College of Medicine and were conducted in accordance with institutional guidelines. All mice were in the C57BL/6 background, distinguished by the presence of CD45.1 or CD45.2 alleles. Dnmt3afl/fl mice were obtained from the Beaudet laboratory and crossed to Mx1-Cre mice. For bone marrow transplantation, recipient C57BL/6 CD45.1+ mice were transplanted by retro-orbital injection following a split dose of 10.5 Gy of irradiation. Donor HSCs (250, CD45.2+) were competed against 2.5 × 105 unfractionated WBM cells with the opposite CD45 allele (matched to the recipient). Deletion of floxed alleles in donor HSCs was carried out 5–6 weeks after transplantation in primary recipients by six intraperitoneal injections (300 μg per mouse) of pIpC (Sigma) in PBS every other day. For serial HSC transplantation, WBM from transplanted recipients was isolated 18 weeks after transplantation, and donor HSCs were re-purified using CD45.2+, side populationKLS gating. Of these purified donor HSCs, 250 were competed against 2.5 × 105 fresh CD45.1+ WBM cells in new lethally irradiated recipients.
HSC purification and flow cytometry
WBM was isolated from femurs, tibiae and iliac crests. Side population staining was performed with Hoechst 33342 (Sigma) as previously described41. Magnetic enrichment was carried out on an AutoMACS using microbeads conjugated to antibodies recognizing biotin or mouse CD117 (all from Miltenyi Biotec). For the analysis of transplant-receiving mice, WBM was stained with antibodies to discriminate cells derived from competitors and donors (CD45.1+ and CD45.2+, respectively) and to isolate the cell type of interest (Supplementary Table 1), as previously described42. All antibodies were obtained from Becton Dickinson or eBioscience and used at 1:100 dilutions. Cell sorting was performed on a MoFlo cell sorter (Dako North America) and analysis performed on an LSR II (Becton Dickinson).
Plasmids and retroviral overexpression
Full-length mouse Dnmt3a cDNA (BC007466; Open Biosystems) was subcloned into MSCV-RFB-IRES-GFP vector using Gateway recombination. Viruses were packaged by co-transfection with pCL-Eco into 293T cells. The supernatants containing viruses were collected 48 h after transfection, and titers were determined using 3T3 cells. For transduction of hematopoietic progenitors, donor mice were treated with 5-FU (150 mg/kg; American Pharmaceutical Partners) 6 d before the harvest of bone marrow. WBM was enriched for Sca-1+ cells using magnetic enrichment and adjusted to a concentration of 5 × 105 cells/ml in transduction medium containing Stempro 34 (Gibco), nutrient supplement, penicillin/streptomycin, l-glutamine (2 mM), mouse stem cell factor (mSCF, 10 ng/ml; R&D Systems), mouse thromboprotein (mTPO, 100 ng/ml; R&D Systems) and polybrene (8 μg/ml; Sigma). The suspension was spin-infected at 250g at room temperature for 2 h, and cells were incubated for an additional 3 h at 37 °C.
Peripheral blood analysis
Mice were bled retro-orbitally, the red blood cells were lysed and each sample was incubated with the following antibodies on ice for 20 min: CD45.1-FITC, CD45.2-APC, CD4–Pacific Blue, CD8–Pacific Blue, B220–Pacific Blue, B220-PeCy7, Mac1-PeCy7 and Gr-1–PeCy7, as previously described42. Cells were then spun down and resuspended in a propidium iodide solution, and analysis was performed on live cells with an LSR II. Peripheral blood counts were performed with a Hemavet 950 (Drew Scientific).
Methocult assays and genotyping
Single HSCs were sorted into 96-well plates containing Methocult 3434 medium (Stem Cell Technologies) supplemented with penicillin/streptomycin and cultured in vitro at 37 °C for 2 weeks. Genomic DNA was prepared from Methocult colonies with the Spin Doctor Genomic DNA Isolation kit (Gerard Biotec). PCR screening of floxed allele deletion was performed as previously described9.
BrdU, Ki67 and propidium iodide staining
BrdU analysis of HSCs was performed as described15,43, with 1 mg BrdU per 6 g of mouse weight injected intraperitoneally 12 h before analysis. WBM was stained with antibodies to identify stem and progenitor cell compartments and then prepared for analysis of BrdU incorporation using the FITC-BrdU Flow kit (BD Pharmingen). For Ki67 analysis, WBM was prepared as above, with the substitution of antibody to human Ki67 (BD Pharmingen). For cell cycle analysis using propidium iodide, HSCs were sorted into deionized water containing 0.1% sodium citrate and 50 mg/ml propidium iodide and analyzed with a FACScan flow cytometer (BD Biosciences).
Annexin V staining
WBM was stained with antibodies to identify stem and progenitor cell compartments and then washed twice with cold PBS and incubated at room temperature for 15 min in 1× binding buffer (10 mM HEPES, 140 mM NaCl and 2.5 mM CaCl2) containing Annexin V–APC (BD Pharmingen) and propidium iodide. Cells were analyzed by flow cytometry within 1 h of staining.
Quantitative real-time PCR
RNA was isolated using the RNAqueous kit (Ambion) and reverse transcribed with random hexamer primers using SuperScript II (Invitrogen). cDNA input was standardized, and RT-PCR was performed in Taqman Master Mix (Applied Biosystems) with 18 S rRNA probe (VIC-MGB) and a gene-specific probe (FAM-MGB) for 60 cycles with an AbiPrism 7900HT (all from Applied Biosystems). Samples were normalized to 18S rRNA levels and fold change determined by the ΔΔCT method.
Western blotting
Western blots were prepared, probed with rabbit antibody to Dnmt3a (sc-20703, Santa Cruz Biotechnology) and mouse antibody to β-actin (sc-47778, Santa Cruz Biotechnology) and visualized using enhanced chemiluminescence (ECL) detection reagents according to the manufacturer’s instructions (RPN2132, GE Healthcare).
Microarrays
HSCs (CD45.2+, side populationKLS, CD150+) were sorted from three biological replicate pools of secondary recipient mice for each genotype. Re-sorting of initial samples confirmed the purity of each sort was >98%. RNA was isolated with the RNAqueous kit as above and linearly amplified in two rounds of T7 in vitro transcription (MessageAmp, Ambion), with labeled biotin-UTP and biotin-CTP (Enzo Biotech) incorporated during the second amplification step. Labeled RNA (20 μg) was hybridized to Affymetrix MOE430.2 chips. Raw images and intensity files were generated with GCOS 1.0 software (Affymetrix). Microarray chips had to pass quality control tests, including having a scale factor of <5, a 50-to-30 probe ratio of <20, a replicate correlation coefficient of >0.96 in unbiased global clustering analysis and limited RNA degradation as determined using the 50-to-30 signal intensity ratios from chip probes. Normalization and model-based expression measurements were performed with GC-RMA software (see URLs). Differentially expressed genes were defined as those having a fold change of >1.50 between samples that was accompanied by an adjusted P value of <0.05 (Benjamini Hochberg FDR). Final gene lists were further filtered to remove sex chromosome genes, as the genders of donor HSCs were not matched, and genes significantly different between control HSCs and non-transplanted, wild-type HSCs44, to remove any genes that might be differently expressed as a result of serial transplantation.
Digital restriction enzyme analysis of methylation (DREAM)
Genomic DNA (5 μg) was digested with 5 μl of FastDigest SmaI endonuclease (Fermentas) for 3 h at 37 °C. Subsequently, 50 U (5 μl) of XmaI endonuclease (NEB) was added, and the digestion was continued for an additional 16 h. The digested DNA was purified using QIAquick PCR purification kit (Qiagen). The 3′ recessed ends of the DNA created by XmaI digestion were filled in with 3′-dA tails added by Klenow DNA polymerase lacking 3′-to-5′ exonuclease activity (NEB) and dCTP, dGTP and dATP mix (0.4 mM of each). Illumina paired-end sequencing adaptors were ligated using Rapid T4 DNA ligase (Enzymatics). The ligation mix was size selected by electrophoresis in 2% agarose. A slice corresponding to a 250- to 500-bp window, according to the DNA ladder, was cut out, and DNA was extracted from agarose. Eluted DNA was amplified with Illumina paired-end PCR primers using iProof High-Fidelity DNA Polymerase (Bio-Rad) and 18 cycles of amplification. The resulting sequencing library was cleaned with AMPure magnetic beads (Agencourt) and sequenced on an Illumina Genome Analyzer II. Sequencing reads were mapped to SmaI sites in the mouse genome (mm9), and signatures corresponding to methylated and unmethylated CpGs were enumerated for each SmaI site. Methylation frequencies for individual SmaI sites were then calculated. The methylation ratio is the ratio of the number of tags starting with CCGGG divided by the total number of tags mapped to a given SmaI site. We used at least 20 sequencing reads to analyze methylation levels at individual SmaI sites. Based on technical replicate experiments (data not shown), we could distinguish differences in methylation of >25% with an FDR of 0.07%.
High-performance liquid chromatography–mass spectrometry (HPLC-MS)
DNA hydrolysis was performed as previously described45. Briefly, 1 μg of genomic DNA was denatured by heating at 100 °C. Nuclease P1 (5 U; Sigma) was added and the mixture incubated at 37 °C for 2 h. A 1/10 volume of 1 M ammonium bicarbonate and 0.002 U of venom phosphodiesterase 1 (Sigma) were added to the mixture and the incubation continued for 2 h at 37 °C. Next, 0.5 U of alkaline phosphatase (Roche) was added and the mixture incubated for 1 h at 37 °C. Before injection into an Agilent Zorbax Eclipse XDB-C18 4.6 × 50-mm column (3.5-μm particle size), the reactions were diluted with water to dilute out the salts and enzymes. Liquid chromatography separation was performed at a flow rate of 220 μl min−1. Quantification was performed using a LC-ESI-MS/MS system in the multiple reaction–monitoring (MRM) mode as described46.
Chromatin immunoprecipitation and real-time PCR
ChIP was performed as described previously46. FACS-sorted cells were used as a source of chromatin. Antibodies used included IgG control (sc-2027; Santa Cruz Biotechnology) and antibodies to H3K4me3 (07-473; Millipore) and Dnmt3a (64B1446; Abcam). The immunoprecipitated DNA was quantified by using real-time PCR with SYBR Green dye on an ABI Prism 7000 system (Applied Biosystems). All PCR signals from non-immunoprecipitated samples were referenced to their respective input to normalize for differences in cell number and for primer efficiency. The sequences for the primers used in this study are provided in Supplementary Table 9.
Reduced representation bisulfite sequencing (RRBS)
Genomic DNA (50–100 ng) was digested with 10 U MspI (NEB), which cuts at CCGG sites in a methylation-insensitive manner47. Digested fragments were end repaired, A tailed and ligated to Illumina adaptors. End repair and 3′ adenylation was performed in a 50-μl reaction volume containing 4 mM 5′ methylated dCTP, 4 mM dGTP, 40 mM dATP and 10 U of 3′-to-5′ exo- Klenow DNA polymerase and incubated at 30 °C for 20 min followed by 20 min at 37 °C. Adaptor ligation was performed in 50-μl reactions containing 300 mM pre-methylated adaptors and 1,000 U of T4 DNA polymerase and incubated at 16 °C overnight. Adaptor-ligated DNA was subjected to size selection on a 3% NuSieve 3:1 agarose gel. DNA marker lanes were excised from the gel and stained with SYBR Gold (Invitrogen). Slices of 160–350 bp were excised from the unstained gel and purified using MinElute spin columns (Qiagen). Size-selected fragments were bisulfite treated using the EpiTect Bisulfite Kit (Qiagen), with the minor modification that five additional cycles (5 min at 95 °C followed by 90 min at 60 °C) were included. After bisulfite conversion, DNA was eluted in 40 μl of elution buffer, and 0.8 μl of DNA was used for analytical PCR to determine the minimum number of PCR cycles required to obtain enough material for sequencing. Final PCR products were purified on MinElute columns, assessed on 4–20% polyacrylamide Criterion Tris-buffered EDTA (TBE) gels (Bio-Rad) and quantified using a Qubit fluorometer (Invitrogen). The libraries were sequenced on the Illumina Genome Analyzer II.
Sequences were identified by standard Illumina base-calling software. After qualify control filtering16,17, the RRBS reads were mapped to the mouse genome (mm9) using RRBSMAP48 software. Three biological samples were subjected to RRBS for each genotype, and the two replicates with the highest correlation were chosen for analysis (the final DNA methylation map is the composite of two replicates). The P value and difference of methylation ratios for each covered CpG were calculated using Fisher’s exact test. DMCs were defined such that the adjusted P value is <0.05 and the methylation ratio difference is ≥0.33. Candidate DMRs were first selected as regions with at least three consecutive DMCs in the same methylation class (for example, all hypomethylated DMCs or all hypermethylated DMCs), and the distance between any two consecutive DMCs had to be <300 bp. We assigned a DMR to a RefSeq gene as long as the DMR was located between 1 kb upstream of the transcription start site and 1 kb downstream of the transcription termination site of the gene.
Bisulfite PCR sequencing
Bisulfite PCR was performed as described previously49. Genomic DNA (2 μg) was denatured with 0.2 M NaOH at 37 °C for 10 min followed by incubation with 30 μl of freshly prepared 10 mM hydroquinone and 520 μl of 3 M sodium bisulfite (pH 5.0) at 50 °C for 16 h. DNA was purified on a Wizard Miniprep Column (Promega), desulfonated with 0.3 M NaOH at 25 °C for 5 min, precipitated with ammonium acetate and ethanol and dissolved in 50 μl of Tris-EDTA buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). Bisulfite-treated DNA (40–80 ng) was amplified with gene-specific primers in a two-step PCR. PCR products from the second step were cloned into the pCR4-TOPO vector (Invitrogen), transformed into competent bacteria and sequenced. Sequences for the primers used in this study are provided in Supplementary Table 9.
Statistics
Student’s t tests and one-way ANOVA were used for statistical comparisons where appropriate.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank all members of the Goodell laboratory for scientific advice, A. Rosen, Y. Zheng and L. Yang for mouse management and technical support, C. Threeton for flow cytometry, L. White (microarray core) and A. Gnirke for assistance with the RRBS technique. G.A.C. was supported by a grant from the US National Institutes of Health (NIH K99 DK084259-01A1) and is an American Society of Hematology Scholar. This work was also supported by NIH grants (AG036562, HL086223, CA100632, CA129831, P50CA126752, DK092883, CA125123 and DK58192), the Ellison foundation, the American Heart Association (CPRIT grant RP110028) and the Functional Genomics Core (P30 DK056338).
Footnotes
Accession numbers. Raw data have been submitted to the NCBI GEO database (GSE27322).
Note: Supplementary information is available on the Nature Genetics website.
AUTHOR CONTRIBUTIONS G.A.C. designed and performed experiments, analyzed data and wrote the manuscript. Experiments were also designed by J.S.B., J.-P.J.I., L.A.G., H.G., C.B., W.L. and M.A.G. and were performed by M.J., M.L., A.V. and J.J. Data were additionally analyzed and interpreted by M.J., D.S., M.L., C.B., A.V., J.J., S.L., Y.L., A.M., J.-P.J.I., L.A.G., W.L. and M.A.G. D.S., C.B., Y.X., S.L. and Y.L. developed critical software. The manuscript was written or edited by G.J.D., W.L., L.A.G., J.-P.J.I., J.S.B., C.B. and M.A.G.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Attwood JT, Yung RL, Richardson BC. DNA methylation and the regulation of gene transcription. Cell. Mol. Life Sci. 2002;59:241–257. doi: 10.1007/s00018-002-8420-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998;19:219–220. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 3.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 4.Lei H, et al. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 1996;122:3195–3205. doi: 10.1242/dev.122.10.3195. [DOI] [PubMed] [Google Scholar]
- 5.Chen T, Ueda Y, Dodge JE, Wang Z, Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 2003;23:5594–5605. doi: 10.1128/MCB.23.16.5594-5605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu H, et al. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science. 2010;329:444–448. doi: 10.1126/science.1190485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trowbridge JJ, Snow JW, Kim J, Orkin SH. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell. 2009;5:442–449. doi: 10.1016/j.stem.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bröske AM, et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat. Genet. 2009;41:1207–1215. doi: 10.1038/ng.463. [DOI] [PubMed] [Google Scholar]
- 9.Tadokoro Y, Ema H, Okano M, Li E, Nakauchi H. De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J. Exp. Med. 2007;204:715–722. doi: 10.1084/jem.20060750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ley TJ, et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan XJ, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 2011;43:309–315. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- 12.Yamashita Y, et al. Array-based genomic resequencing of human leukemia. Oncogene. 2010;29:3723–3731. doi: 10.1038/onc.2010.117. [DOI] [PubMed] [Google Scholar]
- 13.Walter MJ, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011;25:1153–1158. doi: 10.1038/leu.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaneda M, et al. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- 15.Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature. 2010;465:793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu H, et al. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat. Methods. 2010;7:133–136. doi: 10.1038/nmeth.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meissner A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chambers SM, et al. Hematopoietic fingerprints: an expression database of stem cells and their progeny. Cell Stem Cell. 2007;1:578–591. doi: 10.1016/j.stem.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen D, Zhang G. Enforced expression of the GATA-3 transcription factor affects cell fate decisions in hematopoiesis. Exp. Hematol. 2001;29:971–980. doi: 10.1016/s0301-472x(01)00670-1. [DOI] [PubMed] [Google Scholar]
- 20.Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 21.Ficara F, Murphy MJ, Lin M, Cleary ML. Pbx1 regulates self-renewal of long-term hematopoietic stem cells by maintaining their quiescence. Cell Stem Cell. 2008;2:484–496. doi: 10.1016/j.stem.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng T, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 23.Mackarehtschian K, et al. Targeted disruption of the flk2/flt3 gene leads to deficiencies in primitive hematopoietic progenitors. Immunity. 1995;3:147–161. doi: 10.1016/1074-7613(95)90167-1. [DOI] [PubMed] [Google Scholar]
- 24.Georgopoulos K, et al. The ikaros gene is required for the development of all lymphoid lineages. Cell. 1994;79:143–156. doi: 10.1016/0092-8674(94)90407-3. [DOI] [PubMed] [Google Scholar]
- 25.Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–1577. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- 26.Stehling-Sun S, Dade J, Nutt SL, DeKoter RP, Camargo FD. Regulation of lymphoid versus myeloid fate ‘choice’ by the transcription factor Mef2c. Nat. Immunol. 2009;10:289–296. doi: 10.1038/ni.1694. [DOI] [PubMed] [Google Scholar]
- 27.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Challen GA, Goodell MA. Runx1 isoforms show differential expression patterns during hematopoietic development but have similar functional effects in adult hematopoietic stem cells. Exp. Hematol. 2010;38:403–416. doi: 10.1016/j.exphem.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sirin O, Lukov GL, Mao R, Conneely OM, Goodell MA. The orphan nuclear receptor Nurr1 restricts the proliferation of haematopoietic stem cells. Nat. Cell Biol. 2010;12:1213–1219. doi: 10.1038/ncb2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jelinek J, et al. Digital restriction enzyme analysis of methylation (DREAM) by next generation sequencing yields high resolution maps of DNA methylation. Blood. 2009;114:567. [Google Scholar]
- 31.Venezia TA, et al. Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol. 2004;2:e301. doi: 10.1371/journal.pbio.0020301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 33.Fahrner JA, Eguchi S, Herman JG, Baylin SB. Dependence of histone modifications and gene expression on DNA hypermethylation in cancer. Cancer Res. 2002;62:7213–7218. [PubMed] [Google Scholar]
- 34.Wilson NK, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010;7:532–544. doi: 10.1016/j.stem.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 35.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalo S, et al. DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat. Cell Biol. 2006;8:416–424. doi: 10.1038/ncb1386. [DOI] [PubMed] [Google Scholar]
- 37.Allsopp RC, Morin GB, DePinho R, Harley CB, Weissman IL. Telomerase is required to slow telomere shortening and extend replicative lifespan of HSCs during serial transplantation. Blood. 2003;102:517–520. doi: 10.1182/blood-2002-07-2334. [DOI] [PubMed] [Google Scholar]
- 38.Ehrlich M, et al. Hypomethylation and hypermethylation of DNA in Wilms tumors. Oncogene. 2002;21:6694–6702. doi: 10.1038/sj.onc.1205890. [DOI] [PubMed] [Google Scholar]
- 39.Frigola J, et al. Differential DNA hypermethylation and hypomethylation signatures in colorectal cancer. Hum. Mol. Genet. 2005;14:319–326. doi: 10.1093/hmg/ddi028. [DOI] [PubMed] [Google Scholar]
- 40.Cho YH, et al. Aberrant promoter hypermethylation and genomic hypomethylation in tumor, adjacent normal tissues and blood from breast cancer patients. [PMC free article] [PubMed]
- 41.Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 1996;183:1797–1806. doi: 10.1084/jem.183.4.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Challen GA, Boles N, Lin KK, Goodell MA. Mouse hematopoietic stem cell identification and analysis. Cytometry A. 2009;75:14–24. doi: 10.1002/cyto.a.20674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng CG, Weksberg DC, Taylor GA, Sher A, Goodell MA. The p47 GTPase Lrg-47 (Irgm1) links host defense and hematopoietic stem cell proliferation. Cell Stem Cell. 2008;2:83–89. doi: 10.1016/j.stem.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Challen GA, Boles NC, Chambers SM, Goodell MA. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-β1. Cell Stem Cell. 2010;6:265–278. doi: 10.1016/j.stem.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song L, James SR, Kazim L, Karpf AR. Specific method for the determination of genomic DNA methylation by liquid chromatography–electrospray ionization tandem mass spectrometry. Anal. Chem. 2005;77:504–510. doi: 10.1021/ac0489420. [DOI] [PubMed] [Google Scholar]
- 46.Dahl JA, Collas P. A rapid micro chromatin immunoprecipitation assay (microChIP) Nat. Protoc. 2008;3:1032–1045. doi: 10.1038/nprot.2008.68. [DOI] [PubMed] [Google Scholar]
- 47.Smith ZD, Gu H, Bock C, Gnirke A, Meissner A. High-throughput bisulfite sequencing in mammalian genomes. Methods. 2009;48:226–232. doi: 10.1016/j.ymeth.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics. 2009;10:232. doi: 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kroeger H, et al. Aberrant CpG island methylation in acute myeloid leukemia is accentuated at relapse. Blood. 2008;112:1366–1373. doi: 10.1182/blood-2007-11-126227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.