

Abstract
Since the hallmark dermatologic features of Birt–Hogg–Dubé (BHD) syndrome were first described by three Canadian physicians in 1977, the clinical manifestations of BHD have been expanded to include hamartomas of the hair follicle, lung cysts, increased risk for spontaneous pneumothorax and kidney neoplasia. Twenty-five years later the causative gene FLCN was identified, and the mutation spectrum has now been defined to include mainly protein truncating mutations, but also rare missense mutations and large gene deletions/duplication. Second “hit” FLCN mutations in BHD kidney tumors and loss of tumorigenic potential of the FLCN-null UOK257 tumor cell line when FLCN is re-expressed underscore a tumor suppressor role for FLCN. The identification of novel FLCN interacting proteins FNIP1 and FNIP2/L and their interaction with 5′-AMP activated protein kinase (AMPK) has provided a link between FLCN and the AMPK-mTOR axis and suggested molecular targets for therapeutic intervention to treat BHD kidney cancer and fibrofolliculomas. The generation of FLCN-null cell lines and in vivo animal models in which FLCN (or FNIP1) has been inactivated have provided critical reagents to facilitate mechanistic studies of FLCN function. Research efforts utilizing these critical FLCN-deficient cell lines and mice have begun to uncover important signaling pathways in which FLCN and its protein partners may play a role, including TGF-β signaling, TFE3 transcriptional regulation, PGC1-α driven mitochondrial biogenesis, apoptotic response to cell stress, and vesicular transport. As the mechanisms by which FLCN inactivation leads to BHD manifestations are clarified, we can begin to develop therapeutic agents that target the pathways dysregulated in FLCN-deficient fibrofolliculomas and kidney tumors, providing improved prognosis and quality of life for BHD patients.
Keywords: Birt–Hogg–Dubé syndrome, BHD, FLCN, Folliculin, FNIP1, FNIP2, Tumor suppressor, Inherited kidney cancer, Fibrofolliculoma
Introduction
Thirty-five years ago three Canadian physicians Arthur Birt, Georgina Hogg and James Dubé described the presence of “hereditary pilar hamartomas” that were inherited as an autosomal dominant trait in 15 of 37 individuals over 25 years of age in a 70-member kindred [1]. These benign skin tumors were named fibrofolliculomas and were found in association with trichodiscomas and common achrocordons in these individuals (Fig. 1). This triad of dermatologic lesions became the classic hallmark feature of Birt–Hogg–Dubé (BHD) syndrome. A report in 1996 [2] by Chung and coworkers followed by further documentation of cases by Toro and colleagues several years later [3] expanded the BHD phenotype to include the presence of multiple lung cysts and a predisposition to develop spontaneous pneumothorax (Fig. 1). In a study of 33 BHD families, Zbar and coworkers estimated that the odds for developing pneumothorax after adjusting for age were 50.3 times greater in a person with BHD than in his unaffected siblings [4].
Fig. 1.
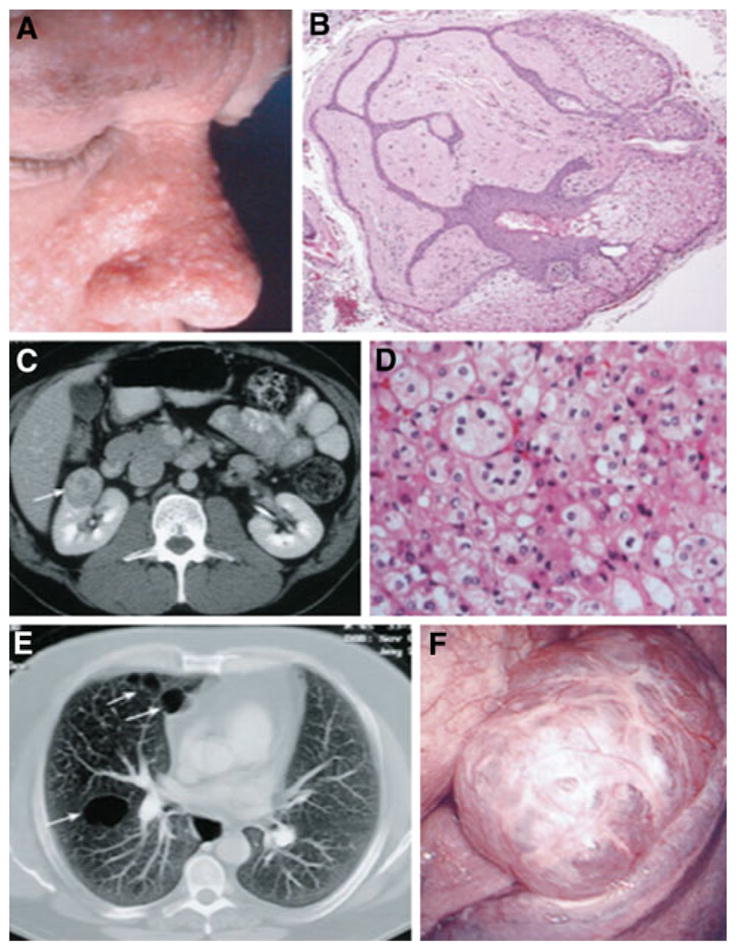
Clinical manifestations of BHD. a Fibrofolliculomas on the face of a BHD patient. b Photomicrograph of a fibrofolliculoma showing the anastomosing epithelial strands and dense connective tissue. c Abdominal CT scan showing renal tumor in BHD patient. d Photomicrograph of the chromophobe renal carcinoma shown in C, ×200. e Thoracic CT showing lung cysts in a BHD patient. f Numerous purple blebs on pleural surface of lower left lung lobe from a BHD patient seen by video-assisted thoracoscopy. Used with permission from Ref. [4]
Kidney neoplasia is confirmed as part of the BHD phenotype but association with colon neoplasia remains controversial
Although a case of bilateral multifocal chromophobe renal carcinoma in a BHD patient was presented by Roth and coworkers in 1993, it was not known whether this was a feature of the disease or an incidental finding [5]. A clear association of renal neoplasia with BHD skin lesions was confirmed, however, when 7 individuals in 3 familial kidney cancer families at the National Institutes of Health (NIH), USA, were identified in which fibrofolliculomas cosegregated with renal oncocytomas or papillary renal tumors [3]. In an expanded study of BHD families from the NIH, Zbar and colleagues estimated that the odds of a person affected with BHD developing renal neoplasia were 6.9 times greater than a person not affected with BHD [4], thereby underscoring the importance of life long screening of BHD patients and at-risk family members for early detection and treatment of asymptomatic renal tumors. A detailed analysis of the pathology of BHD renal tumors by Pavlovich et al. [6] revealed that, unlike other inherited renal cancer syndromes that present with a single histologic subtype of renal tumor (i.e., von Hippel-Lindau disease, hereditary papillary renal carcinoma, hereditary leiomyoma renal cell carcinoma), BHD is associated with a variety of renal tumor histologies (clear cell, papillary, chromophobe, oncocytoma), most frequently hybrid oncocytic tumors comprised of features of both chromophobe renal carcinoma and renal oncocytoma (Fig. 1).
The association of colon polyps and colon carcinoma with BHD has been controversial. Early reports suggested a possible link between intestinal or colonic polyposis and BHD skin lesions [7–9] but a more comprehensive risk assessment in a larger cohort reviewed at the NIH [4] did not find an increased risk for colonic polyps or colon cancer in individuals with BHD over that of the general population. The question of whether colon neoplasia is part of the BHD phenotype remains open since cases of colon polyps and/or colon cancer in BHD patients continue to be reported [10, 11].
Germline mutations in FLCN on chromosome 17p11.2 predispose to BHD syndrome
Using genetic linkage analysis in the family described by Birt, Hogg and Dubé, Schmidt and coworkers were able to localize the disease locus for BHD to chromosome 17p11.2 [12] and in 2002 Nickerson et al. [13] identified germline mutations in a novel gene, FLCN, in a panel of BHD family probands. To date over 100 unique FLCN mutations in the coding region of the gene have been catalogued [14]. The majority of these mutations are protein truncating (frame-shift, nonsense, insertion/deletion, splice site) predicted to inactivate the encoded protein folliculin with an insertion/ deletion mutational “hot spot” identified in a tract of cytosines in exon 11 [13–19]. However, several missense mutations have been reported that may compromise protein stability [20]. The recent discovery of large gene deletions, some of which encompass the putative FLCN promoter [21, 22], and a large FLCN gene duplication [22] suggests that folliculin function may be compromised by complete loss of or partial reduction in activity or by abrogating promoter-driven expression of FLCN. No clear genotype-phenotype associations between FLCN mutation type or location and skin, lung or kidney manifestations have been identified, but a significantly lower frequency of kidney neoplasia was seen in patients with a deleted cytosine in the exon 11 mutational “hot spot” compared with an inserted cytosine at that location [15]. Interestingly a significantly higher risk of colon neoplasia was observed in patients with the “hot spot” mutation in exon 11 compared to those with a substitution mutation in the FLCN gene [11].
FLCN is a tumor suppressor gene
Ten years after the FLCN gene discovery was published, the function of folliculin remains elusive despite intense efforts of many research groups to characterize this novel protein. Seminal work by Vocke and coworkers, in which a somatic “second hit” FLCN mutation or loss of heterozygosity on chromosome 17p was identified in the majority of BHD-associated renal tumors, has confirmed a tumor suppressor role for folliculin [23]. (Interestingly FLCN haploinsufficiency appears to be sufficient for the development of fibrofolliculomas in BHD patients [24].) Re-expression of FLCN in a FLCN-null kidney cancer cell line established from a BHD patient renal tumor abrogated the tumor-promoting potential of these cells in nude mice [25, 26], further supporting folliculin as a tumor suppressor. Knockdown of FLCN in the ACHN renal tumor cell line led to formation of significantly larger tumors when injected into athymic nude mice, and re-expression of FLCN in VHL-deficient 786-O cells decreased tumor growth in mice, further underscoring the tumor suppressor properties of FLCN [27].
Folliculin functional studies identify interacting protein partners FNIP1 and FNIP2
Since protein–protein interactions often provide clues to the function of those proteins, Baba et al. [28] used folliculin coimmunoprecipitation experiments to identify the first folliculin-interacting protein, FNIP1, a highly conserved novel protein that interacts with the carboxy-terminus of folliculin but whose function is as yet unknown. Subsequent FNIP1 coimmunoprecipitation experiments demonstrated that FNIP1 interacts with 5′-AMP activated protein kinase (AMPK) in a FLCN-independent manner. AMPK is a well-known sensor of energy and nutrient levels in cells and responds to these stresses by negatively regulating mammalian target of rapamycin (mTOR), the master controller of protein synthesis and cell growth [29]. Baba and coworkers [30] showed that FNIP1 could be phosphorylated by AMPK, which may be important for its protein expression or stability. FLCN was also phosphorylated directly or indirectly by AMPK and by mTOR since inhibitors of these kinases reduced levels of phosphorylated FLCN, and FLCN phosphorylation was enhanced when in complex with FNIP1. Wang and colleagues identified Serine 62 as a FLCN residue indirectly phosphorylated by AMPK (but not mTOR) and also showed that FLCN phosphorylation was enhanced by binding with FNIP1 and FNIP2 (see below) suggesting that Serine 62 phosphorylation may serve to regulate FLCN/FNIP1/FNIP2/AMPK complex formation (Fig. 2).
Fig. 2.

FLCN, FNIP1 and FNIP2 interact with AMPK and modulate mTOR signaling. Tumor suppressors are red. FLCN interactors are green. Potential functional interactions of FLCN/FNIP1/FNIP2 are in yellow boxes. Arrow indicates activation. Horizontal line with vertical end indicates inhibition. Question mark indicates conflicting data supporting both activation and inhibition.
Recently in an effort to understand the functional significance of the FLCN/FNIP1 interaction, two groups independently generated Fnip1 knockout mice that developed striking defects in B cell development. Baba et al. [31] demonstrated developmental arrest at the pro-B stage in Fnip1−/− and tamoxifen-inducible Fnip1 conditional knockout mice that was a consequence of mTOR-independent caspase-induced pre-B cell apoptotic death. Fnip1−/− mice developed by Park et al. [32] displayed normal pre-B cell signaling but dysregulated AMPK-mTOR signaling, leading to enhanced apoptosis in response to metabolic stress and complete block in B cell development. Both in vivo models support an essential role for FNIP1 in B cell differentiation.
A second FLCN interactor discovered by Hasumi and coworkers using bioinformatics searches displayed 49 % identity to FNIP1 and was named FNIP2 [33] (also reported as FNIPL [34] and MAPO1[35]). FNIP2 along with FNIP1 forms complexes with FLCN, AMPK and both FNIPs. Tissue expression patterns of FNIP1 and FNIP2 suggest that they may be tissue-specific for certain tissues and redundant in other tissues, and interestingly, their expression in sporadic tumors found most often in BHD patients (oncocytoma and chromophobe renal tumors) was elevated relative to normal kidney[33]. Recent work from Lim and colleagues supports a role for MAPO1 (FNIP2) in association with FLCN to activate AMPK and control the induction of apoptosis in response to O6-methylguanine, which is produced in cells as a consequence of exposure to alkylating agents and can contribute to mutation and cancer [35].
FLCN modulates mTOR in a context dependent manner in FLCN deficient mice
Animal models of human disease provide important research tools for elucidating the details of dysregulated biochemical pathways and can serve as prototypes for testing potential therapeutic agents. A number of genetically engineered animal models in which the FLCN gene is inactivated have been generated to date. Baba and coworkers achieved kidney-specific inactivation of Flcn in the mouse by crossing Flcn floxed mice with KSP(cadherin 16) Cre transgenic mice[36] producing a highly cystic kidney phenotype that resulted in renal failure and death in the first month of life. A second kidney-targeted Flcn-knockout mouse model generated by Chen et al. [37] developed polycystic kidneys and cystic renal carcinoma with a similar shortened lifespan. Both animal models displayed activated mTOR and its downstream pathway effectors in the Flcn-deficient kidneys. Encouragingly, they partially responded to rapamycin therapy, which inhibits mTOR, with an extended life span and reduction in size and cyst development in the Flcn-deficient kidneys suggesting that rapamycin may provide a potential therapeutic intervention for BHD patients. In another heterozygous Flcn knockout mouse model developed by Hasumi et al. [38] spontaneous solid renal tumors developed after the age of 10 months arising from kidney cells that had lost the wild type copy of Flcn, thus supporting FLCN inactivation as the initiating step for kidney tumorigenesis in BHD. These mouse tumors histologically resembled the human BHD-associated renal tumors and developed in association with hyperplastic and complex renal cysts that exhibited different latency periods, thereby more closely mimicking the human BHD syndrome than the kidney-directed Flcn-knockout mouse models. Importantly, these Flcn+/− kidney tumors displayed activation of mTOR, increased levels of proteins involved in both complex 1 (mTORC1) and complex 2 (mTORC2), and activation and elevated levels of AKT. These findings were confirmed in human BHD-associated renal tumors and suggest that a dual kinase mTOR inhibitor may be more effective than rapamycin alone in treating BHD-associated renal tumors.
However, conflicting data have been published regarding the role of FLCN in modulating the mTOR pathway. In studies of adenomas and cysts that developed in a heterozygous Flcn mouse model with an in-frame insertion in the Flcn gene, Hudon et al. [27] observed elevated levels of the mTOR downstream effector phospho-S6 in large multilocular polycystic kidneys that lacked Flcn expression, but reported decreased phospho-S6 in single small cysts within the mouse kidneys. They reported decreased phospho-S6 in mouse xenograft tumors that developed from intraperitoneal injections of ACHN kidney cancer cells in which FLCN expression was inactivated by siRNA, but saw no differences in S6 activation when these cells were grown in vitro regardless of culture conditions. Similarly, Hartman et al. [39] generated a heterozygous Flcn mouse model with targeted disruption of the Flcn gene in which solid tumors developed after 12 months with features resembling chromophobe renal tumors and oncocytic cysts. Very low or no phospho-S6 expression was detected in these kidney lesions by immunohistochemistry, which was supported by their in vitro results using several cultured cell lines in which FLCN expression was knocked down. Furthermore, in experiments with yeast that were deleted in the yeast homolog, bhd, this group found that FLCN inactivation led to downregulated yeast Tor2 [40]. Taken together the opposing results supporting activation of mTOR in response to FLCN inactivation on the one hand, and suppression of mTOR under conditions of FLCN knockdown on the other hand have led to the hypothesis that modulation of mTOR activity by FLCN may be context dependent [27, 39].
FLCN may function in multiple signaling pathways as a tumor suppressor
Now, a decade after the FLCN gene discovery, we are experiencing an explosion of new and exciting research that promises to reveal the details of how folliculin acts as a tumor suppressor. Based on recently published work from a number of laboratories, folliculin may play a role in regulating cell growth through other signaling pathways in addition to the AMPK-mTOR axis (Fig. 2). Using gene expression micro-array analysis in FLCN-expressing, FLCN-mutant and FLCN-null renal tumor cells, Hong et al. [26] found reduced expression of genes involved in TGF-β signaling in FLCN-deficient cell lines compared with FLCN-replete lines, and confirmed decreased TGF-β signaling in BHD-associated renal tumors lacking FLCN expression. In further support of a role for folliculin in TGF-β signaling, Cash and coworkers reported resistance to cell-intrinsic apoptosis in FLCN−/− embryonic stem cells, BHD-associated human renal tumors and FLCN-deficient mouse kidney tumors due to reduced transcription of the pro-apoptotic gene Bim. They confirmed the cause as generalized loss of TGF-β driven transcription and hypo-acetylation of Histone H3 associated with TGF-β target gene promoters including Bim [41]. Since the histone deacetylase (HDAC) inhibitor, trichostatin A, was able to rescue the apoptotic resistance through restored Bim levels and subsequent cell death, HDAC inhibitors might be of potential therapeutic use in treating BHD tumors.
Other research from Hong et al. [42] supports a connection between FLCN and the transcription factor TFE3, a member of the MiTF/TFE transcription factor subfamily, known to be involved in TFE3-fusions that lead to translocation renal carcinomas with early onset and aggressive phenotypes. They identified an upregulated TFE3 target gene, GPNMB, in an expression microarray analysis of FLCN-null renal tumor cells, confirmed GPNMB expression in BHD-associated renal tumors, and showed that re-expression of FLCN in the tumor cell line abrogated GPNMB expression. Importantly they demonstrated decreased post-translational modification of TFE3 and nuclear localization in FLCN-deficient renal tumor cells, FLCN−/ − mouse embryonic fibroblasts and FLCN−/ − mouse kidney tumors, and demonstrated TFE3 nuclear localization in BHD-associated renal tumors. Conversely, increased TFE3 post-translational modification (phosphorylation) in FLCN-expressing cells and sequestration of TFE3 in the cytoplasm of FLCN-expressing normal kidney tissues was observed. Although the mechanism remains to be elucidated, FLCN may act to suppress TFE3 transcription through post-translational modification and play a role in TFE3-associated kidney cancer.
Clues to folliculin function may come from the characteristics of the renal oncocytomas, chromophobe and hybrid oncocytic renal tumors that develop in BHD patients, which contain large numbers of mitochondria. Klomp and coworkers performed gene expression profiling of a set of BHD-associated renal tumors compared with sporadic counterpart tumors, and identified a BHD-specific mitochondrial gene expression phenotype involving a deregulated PGC1-α (peroxisome proliferator-activated receptor γ coactivator 1α)-TFAM (transcription factor A, mitochondrial) axis. This includes TFAM-target genes involved in mitochondrial gene transcription and replication of the mitochondrial genome, and genes, in addition to TFAM, that are upregulated by PGC-1α and important for mitochondrial biogenesis [43]. They found a clear correlation between reduced FLCN expression and over expression of the PGC-1α target gene set in a variety of tumor types. These results suggest that a FLCN-PGC-1α-TFAM pathway most likely exists which is deregulated in response to loss or reduction of FLCN expression, implicating FLCN in efficient mitochondrial function.
Recently the crystal structure of the FLCN carboxy-terminal domain was determined by Nookala and colleagues that revealed a distant relationship to differentially expressed in normal cells and neoplasia (DENN) domain proteins, a family of proteins that serve as guanine exchange factors (GEF) for Rab GTPases to facilitate their role in vesicle membrane transport [44]. FLCN carboxy-terminal domain was found to be structurally similar to DENN1B-S protein whose cognate Rab GTPase is Rab35. Indeed, FLCN displayed GEF activity towards Rab35 in vitro suggesting that FLCN may act as a Rab GEF and play a role in vesicle membrane trafficking.
Successful therapeutic agents to treat BHD patients will specifically target growth promoting pathways dysregulated by loss of FLCN
The research findings reported to date have provided some insight into the mechanisms by which FLCN deficiency leads to the development of renal tumors and fibrofolliculomas in the setting of BHD syndrome, and suggest several molecular targets for therapeutic intervention to benefit BHD patients. Based upon the partial response of the kidney-specific FLCN-deficient mouse model to rapamycin, targeting the mTOR pathway may be a reasonable approach to treat BHD renal tumors, perhaps employing a dual kinase mTORC1/mTORC2 inhibitor in tumors in which both complex 1 and complex 2 are activated. However, careful consideration must be given to the conflicting data suggesting that FLCN deficiency may result in mTOR activation or inhibition depending upon context, before initiating a clinical trial with mTOR inhibitors.
A recent report from Preston et al. [45] presented evidence that UOK257 FLCN-null renal tumor cells have increased HIF activity and HIF-target gene expression which may drive their higher dependency on glucose metabolism (‘Warburg effect’). Targeting glycolysis with 2-deoxyglucose reduced cell proliferation of UOK257 cells, suggesting that inhibitors of glucose metabolism might be effective therapeutic agents against BHD renal tumors that display this same phenotype. In an interesting study by Lu and colleagues, selective growth-inhibiting sensitivity was induced by mithramycin in UOK257 FLCN-null renal tumor cells over UOK257-FLCN restored cells which was potentiated by a low dose of rapamycin suggesting that mithramycin might warrant further investigation as a potential therapeutic agent to treat BHD renal tumors [46].
Since the first description in 1977 of the hallmark dermatologic features of Birt–Hogg–Dubé syndrome, we have made substantial progress in defining the clinical manifestations of BHD in skin, lungs and kidneys, identifying FLCN as the responsible gene and characterizing the spectrum of germline mutations, discovering novel FLCN interacting proteins FNIP1 and FNIP2/L, and generating FLCN-null cell lines and in vivo animal models in which FLCN (or FNIP1) is inactivated. Through research efforts utilizing these critical FLCN-deficient cell lines and mice, we have begun to uncover important signaling pathways in which FLCN and its protein partners play a role. As the mechanistic details by which FLCN inactivation leads to BHD are clarified, we can begin to intelligently design therapeutic agents that target the pathways dysregulated in FLCN-deficient fibrofolliculomas and kidney tumors, which will provide improved prognosis and quality of life for BHD patients.
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, Frederick National Lab, Center for Cancer Research. This project has been funded in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN2612008 00001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, not does mention of trade names, commercial products or organizations imply endorsement by the U.S. Government.
Footnotes
Conflict of interest: The author declares no conflict of interest.
References
- 1.Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibro-folliculomas with trichodiscomas and acrochrochordons. Arch Dermatol. 1977;113:1674–1677. [PubMed] [Google Scholar]
- 2.Chung JY, Ramos-Caro FA, Beers B, Ford MJ, Flowers F. Multiple lipomas, angiolipomas, and parathyroid adenomas in a patient with Birt–Hogg–Dube syndrome. Int J Dermatol. 1996;35:365–367. doi: 10.1111/j.1365-4362.1996.tb03642.x. [DOI] [PubMed] [Google Scholar]
- 3.Toro JR, Glenn G, Duray P, Darling T, Weirich G, Zbar B, Linehan M, Turner ML. Birt–Hogg–Dubé syndrome: a novel marker of kidney neoplasia. Arch Dermatol. 1999;135:1195–1202. doi: 10.1001/archderm.135.10.1195. [DOI] [PubMed] [Google Scholar]
- 4.Zbar B, Alvord WG, Glenn G, Turner M, Pavlovich CP, Schmidt L, Walther M, Choyke P, Weirich G, Hewitt SM, Duray P, Gabril F, Greenberg C, Merino MJ, Toro J, Linehan WM. Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt–Hogg–Dubé syndrome. Cancer Epidemiol Biomarkers Prev. 2002;11:393–400. [PubMed] [Google Scholar]
- 5.Roth JS, Rabinowitz AD, Benson M, Grossman ME. Bilateral renal cell carcinoma in the Birt–Hogg–Dubé syndrome. J Am Acad Dermatol. 1993;29:1055–1056. doi: 10.1016/s0190-9622(08)82049-x. [DOI] [PubMed] [Google Scholar]
- 6.Pavlovich CP, McClellan MW, Eyler RA, Hewitt SM, Zbar B, Linehan WM, Merino MJ. Renal tumors in the Birt–Hogg–Dubé syndrome. Am J Surg Pathol. 2002;26:1542–1552. doi: 10.1097/00000478-200212000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Hornstein OP, Knickenberg M. Perifollicular fibromatosis cutis with polyps of the colon. Arch Dermatol Res. 1975;253:161–175. doi: 10.1007/BF00582068. [DOI] [PubMed] [Google Scholar]
- 8.Rongioletti F, Hazini R, Gianotti G, Rebora A. Fibrofolliculomas, trichodiscomas and achrocordons (Birt–Hogg–Dubé) associated with intestinal polyposis. Clin Exp Dermatol. 1989;14:72–74. doi: 10.1111/j.1365-2230.1989.tb00890.x. [DOI] [PubMed] [Google Scholar]
- 9.Le Guyadec T, Dufau JP, Poulain JF, Vaylet F, Grossin M, Lanternier G. Multiple trichodiscomas associated with colonic polyposis. Ann Dermatol Venereol. 1998;125:717–719. [PubMed] [Google Scholar]
- 10.Khoo SK, Giraud S, Kahnoski K, Chen J, Motorna O, Nickolov R, Binet O, Lambert D, Friedel J, Levy R, Ferlicot S, Wolken-stein P, Hammel P, Bergerheim U, Hedblad MA, Bradley M, Teh BT, Nordenskjold M, Richard S. Clinical and genetic studies of Birt–Hogg–Dube syndrome. J Med Genet. 2002;39:906e12. doi: 10.1136/jmg.39.12.906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nahorski MS, Lim DH, Martin L, Gille JJ, McKay K, Rehal PK, Ploeger HM, van Steensel M, Tomlinson IP, Latif F, Menko FH, Maher ER. Investigation of the Birt–Hogg–Dube tumour suppressor gene (FLCN) in familial and sporadic colorectal cancer. J Med Genet. 2010;47:385–390. doi: 10.1136/jmg.2009.073304. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt LS, Warren MB, Nickerson ML, Weirich G, Matrosova V, Toro JR, Turner ML, Duray P, Merino M, Hewitt S, Pavlovich CP, Glenn G, Greenberg CR, Linehan WM, Zbar B. Birt–Hogg–Dubé syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet. 2001;69:876–882. doi: 10.1086/323744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn G, Turner ML, Duray P, Merino M, Choyke P, Pavlovich CP, Sharma N, Walther M, Munroe D, Hill R, Maher E, Greenberg C, Lerman MI, Linehan WM, Zbar B, Schmidt LS. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt–Hogg–Dubé syndrome. Cancer Cell. 2002;2:157–164. doi: 10.1016/s1535-6108(02)00104-6. [DOI] [PubMed] [Google Scholar]
- 14.Lim DH, Rehal PK, Nahorski MS, Macdonald F, Claessens T, Van Geel M, Gijezen L, Gille JJ, Giraud S, Richard S, van Steensel M, Menko FH, Maher ER. A new locus-specific database (LSDB) for mutations in the folliculin (FLCN) gene. Hum Mutat. 2010;31:E1043–E1051. doi: 10.1002/humu.21130. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt LS, Nickerson ML, Warren MB, Glenn GM, Toro JR, Merino MJ, Turner ML, Choyke PL, Sharma N, Peterson J, Morrison P, Maher ER, Walther MM, Zbar B, Linehan WM. Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt–Hogg–Dubé syndrome. Am J Hum Genet. 2005;76:1023–1033. doi: 10.1086/430842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leter EM, Koopmans AK, Gille JJ, van Os TA, Vittoz GG, David EF, Jaspars EH, Postmus PE, van Moorselaar RJ, Craanen ME, Starink TM, Menko FH. Birt–Hogg–Dubé syndrome: clinical and genetic studies of 20 families. J Invest Dermatol. 2008;128:45–49. doi: 10.1038/sj.jid.5700959. [DOI] [PubMed] [Google Scholar]
- 17.Toro JR, Wei MH, Glenn GM, Weinreich M, Toure O, Vocke C, Turner M, Choyke P, Merino MJ, Pinto PA, Steinberg SM, Schmidt LS, Linehan WM. BHD mutations, clinical and molecular genetic investigations of Birt–Hogg–Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45:321–331. doi: 10.1136/jmg.2007.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kluger N, Giraud S, Coupier I, Avril MF, Dereure O, Guillot B, Richard S, Bessis D. Birt–Hogg–Dubé syndrome: clinical and genetic studies of 10 French families. Br J Dermatol. 2010;162:527–537. doi: 10.1111/j.1365-2133.2009.09517.x. [DOI] [PubMed] [Google Scholar]
- 19.Houweling AC, Gijezen LM, Jonker MA, van Doorn MB, Oldenburg RA, van Spaendonck-Zwarts KY, Leter EM, van Os TA, van Grieken NC, Jaspars EH, de Jong MM, Bongers EM, Johannesma PC, Postmus PE, van Moorselaar RJ, van Waesberghe JH, Starink TM, van Steensel MA, Gille JJ, Menko FH. Renal cancer and pneumothorax risk in Birt–Hogg–Dubé syndrome; an analysis of 115 FLCN mutation carriers from 35 BHD families. Br J Cancer. 2011;105:1912–1919. doi: 10.1038/bjc.2011.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nahorski MS, Reiman A, Lim DH, Nookala RK, Seabra L, Lu X, Fenton J, Boora U, Nordenskjöld M, Latif F, Hurst LD, Maher ER. Birt Hogg-Dubé syndrome-associated FLCN mutations disrupt protein stability. Hum Mutat. 2011;32:921–929. doi: 10.1002/humu.21519. [DOI] [PubMed] [Google Scholar]
- 21.Kunogi M, Kurihara M, Ikegami TS, Kobayashi T, Shindo N, Kumasaka T, Gunji Y, Kikkawa M, Iwakami S, Hino O, Takahashi K, Seyama K. Clinical and genetic spectrum of Birt–Hogg–Dube syndrome patients in whom pneumothorax and/ or multiple lung cysts are the presenting feature. J Med Genet. 2010;47:281–287. doi: 10.1136/jmg.2009.070565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benhammou JN, Vocke CD, Santani A, Schmidt LS, Baba M, Seyama K, Wu X, Korolevich S, Nathanson KL, Stolle CA, Linehan WM. Identification of intragenic deletions and duplication in the FLCN gene in Birt–Hogg–Dubé syndrome. Genes Chromosom Cancer. 2011;50:466–477. doi: 10.1002/gcc.20872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vocke CD, Yang Y, Pavlovich CP, Schmidt LS, Nickerson ML, Torres-Cabala CA, Merino MJ, Walther MM, Zbar B, Linehan WM. High frequency of somatic frameshift BHD gene mutations in Birt–Hogg–Dubé-associated renal tumors. J Natl Cancer Inst. 2005;97:931–935. doi: 10.1093/jnci/dji154. [DOI] [PubMed] [Google Scholar]
- 24.van Steensel MA, Verstraeten VL, Frank J, Kelleners-Smeets NW, Poblete-Gutiérrez P, Marcus-Soekarman D, Bladergroen RS, Steijlen PM, van Geel M. Novel mutations in the BHD gene and absence of loss of heterozygosity in fibrofolliculomas of Birt–Hogg–Dubé patients. J Invest Dermatol. 2007;127:588–593. doi: 10.1038/sj.jid.5700592. [DOI] [PubMed] [Google Scholar]
- 25.Yang Y, Padilla-Nash HM, Vira MA, Abu-Asab MS, Val D, Worrell R, Tsokos M, Merino MJ, Pavlovich CP, Ried T, Linehan WM, Vocke CD. The UOK 257 cell line: a novel model for studies of the human Birt–Hogg–Dubé gene pathway. Cancer Genet Cytogenet. 2008;180:100–109. doi: 10.1016/j.cancergencyto.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong SB, Oh H, Valera VA, Stull J, Ngo DT, Baba M, Merino MJ, Linehan WM, Schmidt LS. Tumor suppressor FLCN inhibits tumorigenesis of a FLCN-null renal cancer cell line and regulates expression of key molecules in TGF-beta signaling. Mol Cancer. 2010;9:160. doi: 10.1186/1476-4598-9-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hudon V, Sabourin S, Dydensborg AB, Kottis V, Ghazi A, Paquet M, Crosby K, Pomerleau V, Uetani N, Pause A. Renal tumour suppressor function of the Birt–Hogg–Dubé syndrome gene product folliculin. J Med Genet. 2010;47:182–189. doi: 10.1136/jmg.2009.072009. [DOI] [PubMed] [Google Scholar]
- 28.Baba M, Hong SB, Sharma N, Warren MB, Nickerson ML, Iwamatsu A, Esposito D, Gillette WK, Hopkins RF, 3rd, Hartley JL, Furihata M, Oishi S, Zhen W, Burke TR, Jr, Linehan WM, Schmidt LS, Zbar B. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci USA. 2006;103:15552–15557. doi: 10.1073/pnas.0603781103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 2009;196:65–80. doi: 10.1111/j.1748-1716.2009.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L, Kobayashi T, Piao X, Shiono M, Takagi Y, Mineki R, Taka H, Zhang D, Abe M, Sun G, Hagiwara Y, Okimoto K, Matsumoto I, Kouchi M, Hino O. Serine 62 is a phosphorylation site in folliculin, the Birt–Hogg–Dubé gene product. FEBS Lett. 2010;584:39–43. doi: 10.1016/j.febslet.2009.11.033. [DOI] [PubMed] [Google Scholar]
- 31.Baba M, Keller JR, Sun HW, Resch W, Kuchen S, Suh HC, Hasumi H, Hasumi Y, Kieffer-Kwon KR, Gonzalez CG, Hughes RM, Klein ME, Oh HF, Bible P, Southon E, Tessarollo L, Schmidt LS, Linehan WM, Casellas R. The Folliculin-FNIP1 pathway deleted in human Birt–Hogg–Dube syndrome is required for mouse B cell development. Blood. 2012 doi: 10.1182/blood-2012-02-410407. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park H, Staehling K, Tsang M, Appleby MW, Brunkow ME, Margineantu D, Hockenbery DM, Habib T, Liggitt HD, Carlson G, Iritani BM. Disruption of Fnip1 reveals a metabolic checkpoint controlling B lymphocyte development. Immunity. 2012;36:769–781. doi: 10.1016/j.immuni.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hasumi H, Baba M, Hong SB, Hasumi Y, Huang Y, Yao M, Valera VA, Linehan WM, Schmidt LS. Identification and characterization of a novel folliculin-interacting protein FNIP2. Gene. 2008;415(1–2):60–67. doi: 10.1016/j.gene.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takagi Y, Kobayashi T, Shiono M, Wang L, Piao X, Sun G, Zhang D, Abe M, Hagiwara Y, Takahashi K, Hino O. Interaction of folliculin (Birt-Hogg-Dubé gene product) with a novel Fnip1-like (FnipL/Fnip2) protein. Oncogene. 2008;27:5339–5347. doi: 10.1038/onc.2008.261. [DOI] [PubMed] [Google Scholar]
- 35.Lim TH, Fujikane R, Sano S, Sakagami R, Nakatsu Y, Tsuzuki T, Sekiguchi M, Hidaka M. Activation of AMP-activated protein kinase by MAPO1 and FLCN induces apoptosis triggered by alkylated base mismatch in DNA. DNA Repair (Amst) 2012;11:259–266. doi: 10.1016/j.dnarep.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 36.Baba M, Furihata M, Hong SB, Tessarollo L, Haines DC, Southon E, Patel V, Igarashi P, Alvord WG, Leighty R, Yao M, Bernardo M, Ileva L, Choyke P, Warren MB, Zbar B, Linehan WM, Schmidt LS. Kidney-targeted Birt–Hogg–Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J Natl Cancer Inst. 2008;100:140–154. doi: 10.1093/jnci/djm288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen J, Futami K, Petillo D, Peng J, Wang P, Knol J, Li Y, Khoo SK, Huang D, Qian CN, Zhao P, Dykema K, Zhang R, Cao B, Yang XJ, Furge K, Williams BO, Teh BT. Deficiency of FLCN in mouse kidney led to development of polycystic kidneys and renal neoplasia. PLoS One. 2008;3:e3581. doi: 10.1371/journal.pone.0003581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hasumi Y, Baba M, Ajima R, Hasumi H, Valera VA, Klein ME, Haines DC, Merino MJ, Hong SB, Yamaguchi TP, Schmidt LS, Linehan WM. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc Natl Acad Sci USA. 2009;106:18722–18727. doi: 10.1073/pnas.0908853106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartman TR, Nicolas E, Klein-Szanto A, Al-Saleem T, Cash TP, Simon MC, Henske EP. The role of the Birt–Hogg–Dubé protein in mTOR activation and renal tumorigenesis. Oncogene. 2009;28:1594–1604. doi: 10.1038/onc.2009.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Slegtenhorst M, Khabibullin D, Hartman TR, Nicolas E, Kruger WD, Henske EP. The Birt–Hogg–Dube and tuberous sclerosis complex homologs have opposing roles in amino acid homeostasis in Schizosaccharomyces pombe. J Biol Chem. 2007;282:24583–24590. doi: 10.1074/jbc.M700857200. [DOI] [PubMed] [Google Scholar]
- 41.Cash TP, Gruber JJ, Hartman TR, Henske EP, Simon MC. Loss of the Birt–Hogg–Dubé tumor suppressor results in apoptotic resistance due to aberrant TGFβ-mediated transcription. Oncogene. 2011;30:2534–2546. doi: 10.1038/onc.2010.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hong SB, Oh H, Valera VA, Baba M, Schmidt LS, Linehan WM. Inactivation of the FLCN tumor suppressor gene induces TFE3 transcriptional activity by increasing its nuclear localization. PLoS One. 2010;5:e15793. doi: 10.1371/journal.pone.0015793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klomp JA, Petillo D, Niemi NM, Dykema KJ, Chen J, Yang XJ, Sääf A, Zickert P, Aly M, Bergerheim U, Nordenskjöld M, Gad S, Giraud S, Denoux Y, Yonneau L, Méjean A, Vasiliu V, Richard S, MacKeigan JP, Teh BT, Furge KA. Birt–Hogg–Dubé renal tumors are genetically distinct from other renal neo-plasias and are associated with up-regulation of mitochondrial gene expression. BMC Med Genomics. 2010;3:59. doi: 10.1186/1755-8794-3-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nookala RK, Langemeyer L, Pacitto A, Ochoa-Montaño B, Donaldson JC, Blaszczyk BK, Chirgadze DY, Barr FA, Bazan JF, Blundell TL. Crystal structure of folliculin reveals a hid-DENN function in genetically inherited renal cancer. Open Biol. 2012;2:120071. doi: 10.1098/rsob.120071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Preston RS, Philp A, Claessens T, Gijezen L, Dydensborg AB, Dunlop EA, Harper KT, Brinkhuizen T, Menko FH, Davies DM, Land SC, Pause A, Baar K, van Steensel MA, Tee AR. Absence of the Birt–Hogg–Dubé gene product is associated with increased hypoxia-inducible factor transcriptional activity and a loss of metabolic flexibility. Oncogene. 2011;30:1159–1173. doi: 10.1038/onc.2010.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu X, Wei W, Fenton J, Nahorski MS, Rabai E, Reiman A, Seabra L, Nagy Z, Latif F, Maher ER. Therapeutic targeting the loss of the Birt–Hogg–Dube suppressor gene. Mol Cancer Ther. 2011;10:80–89. doi: 10.1158/1535-7163.MCT-10-0628. [DOI] [PubMed] [Google Scholar]