

Abstract
Fluorescence lifetime-resolved imaging microscopy (FLIM) has been used to monitor the enzymatic activity of a proteolytic enzyme, Membrane Type 1 Matrix Metalloproteinase (MT1-MMP), with a recently developed FRET-based biosensor in vitro and in live HeLa and HT1080 cells. MT1-MMP is a collagenaise that is involved in the destruction of extra-cellular matrix (ECM) proteins, as well as in various cellular functions including migration. The increased expression of MT1-MMP has been positively correlated with the invasive potential of tumor cells. However, the precise spatiotemporal activation patterns of MT1-MMP in live cells are still not well-established. The activity of MT1-MMP was examined with our biosensor in live cells. Imaging of live cells was performed with full-field frequency-domain FLIM. Image analysis was carried out both with polar plots and phase differential enhancement. Phase differential enhancement, which is similar to phase suppression, is shown to facilitate the differentiation between different conformations of the MT1-MMP biosensor in live cells when the lifetime differences are small. FLIM carried out in differential enhancement or phase suppression modes, requires only two acquired phase images, and permits rapid imaging of the activity of MT1-MMP in live cells.
Keywords: Fluorescence Lifetime Imaging Microscopy, FLIM, Förster resonance energy transfer, FRET, Frequency-domain, Fluorescent proteins, Biosensors, Phase suppression, Phase differential enhancement, Membrane proteinase
Introduction
Finding spatial and temporal correlations for signaling cascades regulating cell migration is crucial for understanding the mechanisms by which metastatic cancer cells invade neighboring tissues. Interestingly, the family of matrix metalloproteinases (MMPs) have been shown to be up-regulated and play important roles in many types of human cancers during metastasis [1–3]. Of these MMPs, a proteolytic enzyme called Membrane Type 1 Matrix Metal-loproteinase (MT1-MMP) has generated particular interest because of its involvement not only in the destruction of ECM (extracellular matrix) proteins but also in the transmission of signaling cascades to facilitate invasion [4–9]. However, our understanding of the spatiotemporal activity of MT1-MMP in live cells is still limited.
The function of MT1-MMP during metastatic events stems from its effect on, and relationships to, other proteins and enzymes in various signal transduction cascades. However, many unanswered questions exist about MT1-MMP's associated functions with other enzymes and proteins in cells. Kinases such as Src which are responsible the turnover and development of adhesion sites, have been implicated as possibly interacting with MT1-MMP to modulate its function [4, 10]. In addition, MT1-MMP's role regulating migration is not established; it has been linked to its internalization at the membrane [6], its ability to control the shedding of other membrane proteins such as CD44 [11] and syndecan-I [12] and possibly its processing of the products of ECM destruction [13]. Given the complexity of MT1-MMP's function and regulation in live cells, understanding the spatiotemporal activation patterns of MT1-MMP is crucial for developing a complete model of its biochemical functions in cells that direct events necessary for metastasis and invasion.
In order to visualize the enzymatic activity of MT1-MMP in live cells and in real-time, biosensors based on Förster resonance energy transfer (FRET) have been developed [14]. Most FRET biosensors involve molecules (e.g. proteins), which exist in two different conformations or extents of association; these molecules are labeled with donor and acceptor chromophores (often fluorescent proteins). Whenever donor and acceptor chromophores are sufficiently close and have mutually favorable orientations, energy is transferred non-radiatively from an excited donor molecule to a nearby acceptor molecule [15–18].
Our biosensor reports on the activity of MT1-MMP. The current version of the MT1-MMP biosensor [19] consists of a substrate specific to the catalytic domain of MT1-MMP, which is inserted between the enhanced cyan fluorescent protein (ECFP) and the yellow fluorescent protein for FRET (YPet) [20]. Once the substrate of the biosensor is cleaved by the active MT1-MMP, the donor and acceptor are permanently separated resulting in a complete loss of FRET. Changes in fluorescence emission from either the donor or the acceptor as a result of FRET are used to monitor the conformation of the MT1-MMP biosensor and consequently the activity of MT1-MMP.
Because of the quantitative relationship between the efficiency of FRET and the distance between the donor and acceptor, measurements of the FRET efficiency can accurately report on a biosensor's conformation. Intensity-based FRET techniques have been used in previous studies of the MT1-MMP biosensor; these methods rely on ratiometric assessments of variations in the donor's and acceptor's fluorescence to infer the biosensor's conformation [19, 21, 22]. These intensity-based measurements require controls that are often difficult to perform reliably. Other steady-state fluorescence methods for quantifying FRET efficiencies in live samples include acceptor and donor photobleaching, sensitized emission and polarization-based approaches [18, 23–25].
An alternative measurement is FLIM, which is a concentration independent measurement and is consequently not subject to the controls that are often difficult to carry out reliably with steady-state measurements of FRET in live cell systems. Genetically-encoded biosensors based on FRET in conjunction with FLIM have been increasingly applied to visualize molecular activity in live cells. Our homodyne, full-field, frequency-domain FLIM instrument provides rapid image acquisition for mapping FRET efficiencies, making it well-suited for studying biosensors in live cells [26, 27].
Fluorescence lifetimes characterize the average time that a molecule remains in the excited state prior to returning to the ground state [26, 28, 29]. Lifetimes can be measured either in the time domain, where the time-dependent fluorescence excited by a very short pulse of light is analyzed, or in the frequency domain, where fluorescence is examined during excitation with a repetitive modulation of the light intensity with harmonic components. We use the latter method with a single modulation frequency to achieve rapid data acquisition.
To increase contrast between different conformations of the biosensor when imaging with frequency-domain FLIM, we have applied a novel phase differential enhancement analysis, which is related to phase suppression methods [30–35]. One of the first applications of phase suppression was by Veselova et al. [36, 37] to separate and distinguish spectra of two fluorescence components in the same sample with different lifetimes. By adjusting the phase of the detector, they could block the fluorescence signal of one component with a certain lifetime, and observe only the spectrum of the other component. Later, this technique has been applied by several investigators for separating spectra from fluorophores with different lifetimes and to eliminate background [38–42].
In the work presented here, experiments with the MT1-MMP biosensor were performed in both HeLa and HT1080 cells, where the conformation of the intracellular MT1-MMP biosensor could be varied. Guided by initial experiments establishing the FRET efficiency of our system, a correspondence was established with live cells between the biosensor's conformation and the phase differential enhancement experiments. The results show that the contrast between the two states of our FLIM-based biosensor improves when the data are analyzed with phase differential enhancement compared to more typical evaluations of the time-dependent fluorescence emission of the donors and acceptors.
Materials and Methods
Measurements of Lifetimes in the Frequency Domain
The intensity of the excitation light illuminating the sample is repetitively modulated at frequency f=1/T, where T is the repetition period [28, 29, 33, 34, 43]. Each frequency component of a Fourier series expansion of the repetitive excitation intensity can be analyzed separately and independently. The fundamental sinusoidal component of the repetitive time-dependent excitation light at a radial frequency ωE = 2πf can be written as,
| (1) |
Eω/E0 is the modulation depth of the excitation light, and φE is the phase of the excitation light.
The fluorescence intensity of the sample, S(t), is modulated at the same frequency as the excitation, and is delayed in phase and de-modulated relative to the excitation light.
| (2) |
The relative shift in phase and de-modulation have associated lifetimes, (τφ) and (τM), defined according to Eqs. (3) and (4).
| (3) |
| (4) |
φ ≡φF − φE is the phase delay of the fluorescence relative to that of the excitation. M is the modulation ratio (fractional depth of the modulation depth of the sample relative to that of the excitation). For a single lifetime component τφ= τM; otherwise τφ < τM.
When samples have more than one lifetime component (see Appendix A), such as the ECFP studied here, S(t) is a sum (or more generally an integral if there is a distribution of lifetimes), where each lifetime component contributes a unique modulation ratio (weighted by its fractional intensity) and phase delay to the sum (or integral) [26, 28, 29, 44]. However, whether there is a single or many lifetime components, the measured fluorescence S(t) of Eq. (2) is always described in terms of a sinusoid at the frequency ωE with a particular modulation ratio and a single phase delay germane to the sample. Methods and techniques exist to extract multiple component lifetimes from complex samples in the frequency domain using a single or multiple modulation frequencies [45–48].
Polar Plot Representation of Lifetimes in the Frequency Domain
The interpretation of lifetime-resolved experiments in the frequency domain can be carried out graphically on a polar plot [44, 49, 50]. Equations (5) and (6) define the Cartesian coordinates of a vector [x,y] on a polar plot. The vector [x,y] can also be expressed in polar coordinates, where each point [x,y] = [M, φ] on a polar plot corresponds to a polar vector with its magnitude equal to the measured modulation ratio M = (x2 + y2)½, and a polar angle relative to the x-axis equal to the measured phase delay φ = arctan(y/x),
| (5) |
| (6) |
Each FLIM measurement (at every pixel of an image), whether the measured fluorescence has a single lifetime or many lifetimes, is represented uniquely by a vector on a polar plot. The [x, y] = [M, φ] vectors from fluorescence measurements of single component fluorophores lie exactly on a universal semi-circle. This semi-circle is centered at [x,y] = [0.5,0] with a radius of 0.5 (see e.g. Figs. 1, 3 and 5; [44, 49-51]). For a multi-component fluorescence signal, each individual component, i, contributes fractionally to the measured phase delay and modulation ratio that define the measured vector on the polar plot. The contribution of each lifetime component to the vector on the polar plot is weighted by its corresponding fractional contribution to the steady-state intensity (fractional intensity contribution). Therefore, polar plot vector [x, y] = [M, φ] of a measured fluorescence signal with multi-component lifetimes is a weighted linear combination of a set of vectors of the single component lifetimes [x, y] = [M, φ]= Σi αi[x, y]i= Σiα i[M,φ]i; αi is the fractional intensity contribution of component i, and Σiαi = 1. The end points of all measured polar vectors that describe multi-component systems (and which represent fluorescence that is directly excited with light) are located inside this semicircle on the polar plot. See Appendix B for a more thorough discussion.
Fig. 1.
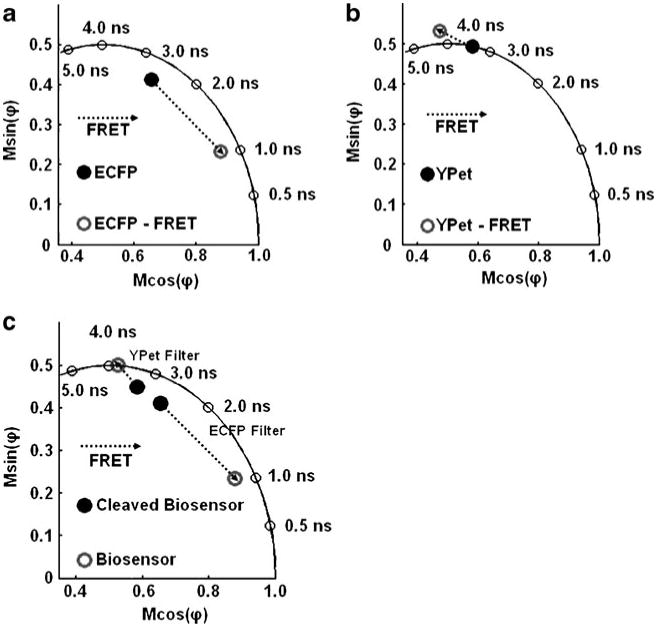
Time-resolved in-vitro analysis of the MT1-MMP biosensor: a The detected phase delay and modulation ratio of the purified MT1-MMP biosensor before and after cleavage were detected through the ECFP channel and are plotted on the polar plot. b The phase delay and modulation ratio of YPet on the MT1-MMP biosensor before (measured) and after (simulated assuming that only the fluorescence of the acceptor excited by FRET is observed—see text) cleavage, detected through the YPet channel. c The trajectories of the phase delay and modulation ratio of the MT1-MMP biosensor before and after cleavage, visualized through the channels specific for ECFP and YPet as indicated
Fig. 3.
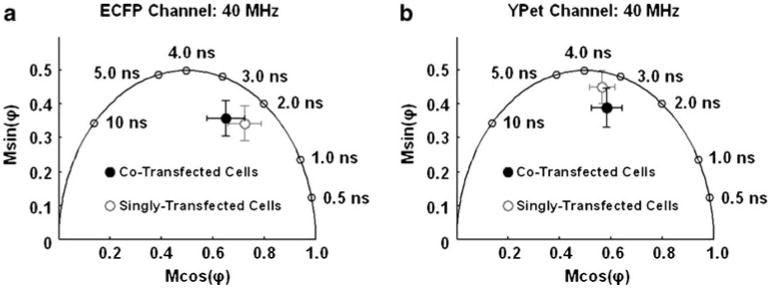
The polar plots describing the donor and acceptor on the MT1-MMP biosensor measured in HeLa cells: a As visualized through the ECFP channel, the average position on the polar plot of the HeLa cells (open circles, n=5) singly transfected with the MT1-MMP biosensor and the average position of the HeLa cells transfected with the MT1-MMP biosensor and MT1-MMP enzyme (filled circles, n=5) are shown. Error bars in x and y are the standard deviations in each coordinate axis. b The average polar positions of the same cells described in (a) are plotted here having been imaged through the YPet channel
Fig. 5.
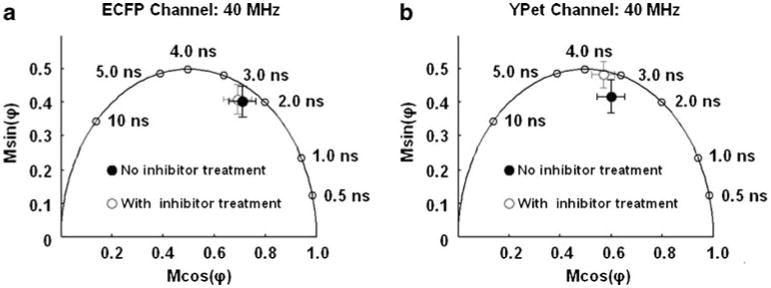
Variations in polar coordinate of ECFP and YPet on the MT1-MMP biosensor transfected into HT1080 cells: a The average position on the polar plot describing a set (n=5) of HT1080 cells transfected with the MT1-MMP biosensor and measured in the ECFP channel is shown as an open circle. The average position on the polar plot calculated from the data collected through the ECFP channel describing a set of HT1080 cells (n=5) incubated with the GM6001 inhibitor is indicated. Error bars are standard deviations in x and y. b The presented data depict the same cells as in (a), but visualized through the YPet channel
Polar plots are particularly advantageous for presenting and analyzing FLIM data from live cell imaging because the fluorescence decay kinetics can be uniquely characterized in the frequency domain by a single measured point. It is important to emphasize that the phase delay and modulation ratio are directly measured parameters and a polar plot representation does not assume any model of lifetimes or their distribution; in this sense, the polar plot is a model free description of the fluorescence measurement.
Homodyne Detection of Fluorescence Lifetimes
Our full-field frequency-domain FLIM is carried out by repetitively modulating the gain of the detector (image intensifier) at high frequencies [26, 43, 52] with the same modulation frequency as the excitation light (homodyne). The modulation of the excitation light and the detector gain are phase locked (synchronized). The high frequency fluorescence signal is thereby transformed to a DC signal that depends on the phase difference between the modulation of the intensifier amplification and of the excitation light. All information encoded in the original fluorescence signal is retained throughout the homodyne procedure. See Appendix for a more thorough description.
For a normal FLIM measurement, a series of images (phase images) is acquired at successive phase differences between the excitation light and the detector amplification (detector phases) over a full period, 0 to 2π. The series of phase recordings at each pixel of the resultant images is analyzed as a sinusoidal curve, from which the phase delay and modulation ratio of the sample's fluorescence can be determined at every pixel (see the previous section). The number of selected phases depends on the purpose of the experiment.
Phase Suppression with Full-Field Homodyne FLIM
Complete Phase Suppression of One Component
Each fluorescence component (say the ith component) contributes to the fluorescence signal with a unique phase delay φF,i − φE and modulation ratio, Mi. By taking two phase images where cos(φG − φE + φF,i) = 0 and subtracting the two images (the phases of these two selected homodyne signals, (φG − φE + φF,i) , differ by π), the sinusoidal signal from the ith fluorescence component with a certain phase delay (for instance, φF,i − φE) can be completely suppressed, leaving only contributions in the difference image from fluorescence components with different phase delays. Phase suppression works in homodyne detection because the sinusoidal contributions of fluorophores with different phase delays (different lifetimes) are shifted in phase relative to one another (Supp. Fig. 1B and C). Any other fluorescent species (for instance, species j with φF,j≠i≠ φF,i) in the sample with a different characteristic phase delay such that cos(φG − φE + φF,j≠i) ≠ 0 for the two detector phases selected previously for the suppression of species i, will accordingly have a non-zero intensity after the subtraction of the two phase images. Thus if there are only two components, i and j and if component i has been suppressed, only component j will be visible in the difference image.
When phase suppression is carried out at every pixel of an image containing multiple fluorophores, the resulting difference intensity images map the location of different fluorophores. Furthermore, we know the lifetime of the fluorescence at all pixels (or objects in the image), because the phase reading for the component which is fully suppressed is 90° from the phase setting corresponding to that lifetime. In addition, once the two detector phases have been selected for the suppression, image acquisition is limited to only two exposures at two different detector phases; thus the data acquisition by phase suppression is more rapid than measuring at multiple detector phases required to determine the actual values of the modulation ratio and phase delay.
Maximizing Differences between Two Phase Images: Phase Differential Enhancement
Rather than complete suppression of a particular component, two detector phases of the homodyne FLIM instrument can be chosen to provide the largest difference in intensity between two fluorescing species when the phase suppression strategy is applied. In this paper, this approach will be referred to as phase differential enhancement. For example, after the subtraction of two phase images, the user may be left with a positive intensity corresponding to a specific fluorescing species and a negative intensity corresponding to another unique fluorescing species. Hence, the absolute intensity difference would have a value different from zero. That is, phase differential enhancement does not suppress the intensity of a specific lifetime component, as discussed for phase suppression, but enhances the AC phase difference between components with similar lifetimes. This approach is shown in this work to be very useful for the MT1-MMP biosensor.
Selecting the Modulation Frequency for Phase Differential Enhancement
When applying phase differential enhancement, it is beneficial to maximize the difference in phase delay between the two species being studied. The difference in the phase delay between distinct fluorescent species can be maximized by measuring with an optimal modulation frequency. The function (φ(τ) = φF(τ) − φE), Eq. (2), can be used to determine the optimal modulation frequency for maximum difference in phase delay (and therefore, maximum intensity differences between two species with phase differential enhancement) by taking its derivative as a function of lifetime.
| (7) |
When is evaluated at a particular lifetime value and plotted as a function of frequency, the maximum corresponds to the frequency that provides the greatest change in phase delay (φ(τ)) for a small change in the lifetime (τ). Typical lifetimes for the fluorophores used in this paper are 2 ns ECFP and 4 ns for YPet. According to this analysis, ∼80 MHz is the optimal frequency for distinguishing changes in the phase delays for ECFP, and ∼40 MHz is the optimal frequency for detecting the corresponding changes in phase delay for YPet on the MT1-MMP biosensor (Supp. Fig. 2).
Measuring FRET Efficiency
FRET efficiency (E) can be quantified directly by measuring the donor's lifetime without an acceptor (τD) and with an acceptor (τDA)
| (8) |
However, typical of many cyan variants, the chromophore of ECFP exhibits two lifetimes. Both lifetimes are quenched by energy transfer. Usually an average lifetime is used to quantify FRET efficiency [28, 29, 53]. The average lifetime is defined as the weighted sum of all component lifetimes (τi) each lifetime multiplied by its corresponding species fraction (ai),
| (9) |
The FRET efficiency of a multi-component system can then be approximated by the following equation where 〈τ〉DA and 〈τ〉D are the average lifetimes of donor in the presence of the acceptor and free donor,
| (10) |
This assumes that there is a complete labeling of donors and acceptors in the sample. Corrections must be made for imperfect labeling to compute FRET efficiencies.
The extent of energy transfer in a system can also be determined by measuring the phase delay and modulation ratio of the acceptor's fluorescence. Although the acceptor's lifetime does not vary as a result of FRET, the measured phase delay and modulation ratio of the acceptor's fluorescence will be altered from that when the acceptor is directly excited. This is because the excitation of the acceptor by FRET from the donor follows the time course of the donor decay [51]. Analysis shows that the measured phase delay of an acceptor undergoing FRET (φAD) is the sum of the phase delay of the donor undergoing FRET (φDA) and the phase delay of the acceptor that would be measured if the acceptor was directly excited (φA). In addition, the measured modulation ratio of the emission of an acceptor that is excited by FRET (MAD) is the product of the modulation ratio of the donor during FRET (MDA) and the modulation ratio of the directly excited acceptor (MA).
| (11) |
| (12) |
The above equations have been derived previously by convoluting the acceptor's fundamental fluorescence response with the intensity-modulated emission of a donor fluorophore [51, 53] and by examining the measured rate of de-excitation of the acceptor in the presence of the donor with kinetic models [54, 55].
In-Vitro Characterization
The ECFP/YPet MT1-MMP biosensor (protein) was expressed with an N-terminal His6 tag in Escherichia coli and purified by Nickel chelation [56]. Unless otherwise stated, a 4 μM solution of the biosensor was measured before and after digestion with trypsin for the in-vitro characterization. The biosensor (protein) was suspended in proteolytic assay buffer described previously [21]. Trypsin was used for this characterization because it can completely cleave the biosensor. The specificity of the substrate in the biosensor for the catalytic domain of MT1-MMP has been described previously [19].
Steady-state spectra were acquired on a PC1 fluorometer (ISS Inc., Champaign, IL) at an excitation wavelength of 440 nm. Spectra were collected with the polarizer orientations at magic angle conditions and corrected to account for the volume of trypsin added in the cleavage assays, the variations in lamp intensity and the spectral sensitivity of the emission channel. The corrections, integrations of the spectra and the extraction of the spectra of ECFP and YPet were carried out in Excel (Microsoft, Redmond, WA) and MATLAB (MathWorks, Natick, MA). A set of reference spectra for both ECFP and YPet (Roger Tsien Lab1), allowed the isolation of each component spectrum from the measured spectra of the biosensor (containing both components) by linear component analysis. The extracted spectra of ECFP, YPet and the biosensor were fit to Gaussian functions for numerical integration.
Lifetime measurements were carried out on our homodyne frequency-domain FLIM instrument at a modulation frequency of 40 MHz [27]. The excitation source was a 440 nm laser (Crystal Laser, Reno, NV) and the emission was collected through filters (Omega Optical, Brattleboro, VT) for ECFP (460–500 nm) or YPet (510–565 nm). The sample was imaged on an inverted microscope in solution on a glass-bottom dish using a 40× air objective (Leica, Wetzlar, Germany). The lifetime of YPet was measured at a modulation frequency of 40 MHz while being directly excited by the 488 nm line of an Ar+ laser (Uniphase, San Jose, CA). The reference solution for all frequency-domain FLIM measurements was fluorescein at a concentration of 50 μM in 0.1 N NaOH (lifetime of 4.3 ns [57]).
Multi-frequency lifetime measurements were conducted to determine the average lifetime of ECFP attached to the MT1-MMP biosensor. Measurements were carried out on a K2 frequency-domain lifetime fluorometer (ISS Inc, Champaign, IL), using magic angle polarizer conditions. A 440 nm laser (Crystal Laser, Reno, NV) was the excitation source. A set of 20 modulation frequencies was chosen between 10 MHz and 100 MHz. The emission filter was a 488±10 nm bandpass filter (Chroma, Bellows Falls, VT). The concentration of samples, typically within the range of 0.5–2 μM, was adjusted to avoid saturation of the detector and re-absorption. The lifetime components and fractional contributions to the steady-state intensity were computed by a least squares fitting algorithm in the Vinci Analysis Software (ISS Inc, Champaign, IL). The fluorescein fluorophore used as a lifetime standard for these experiments was dissolved in 0.1 N NaOH at an optical density equal to 0.02 at an excitation wavelength of 490 nm (lifetime=4.3 ns [57]) was the reference fluorophore.
Live Cell Imaging
Transfection of the HeLa and HT1080 cells was performed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to manufacturer's instructions. Cells were plated on glass-bottomed dishes and starved with 0.5% Fetal Bovine Serum (FBS) (Atlanta Biologicals, Lawrenceville, GA) for 36–48 h prior to imaging. For the MT1-MMP inhibition experiment, HT1080 cells maintained in DMEM (Invitrogen, Carlsbad, CA) with 0.5% FBS were pre-treated with GM6001 (Merck, Germany) at a concentration of 20 μM for approximately 18 h before imaging to inhibit the MT1-MMP activity. All images were collected on Leica DMIRB microscope operating with an oil immersion 100X objective (Leica, Germany) at room temperature.
For FLIM imaging, the excitation source was a 440 nm laser (Crystal Laser, Reno, NV) with its intensity modulated at frequencies of either 40 MHz or 80 MHz. Fluorescence was collected through filters (Omega Optical, Brattleboro, VT) specific either for ECFP (460–500 nm) or YPet (510–565 nm).
The magnitude of the separation between the points on the polar plot representing the intact and cleaved biosensor depends on the choice of modulation frequency. A frequency of 40 MHz provided the necessary resolution for the concomitant detection of ECFP and YPet for both conformations of the MT1-MMP biosensor in the live cells. Therefore, the supporting in-vitro measurements were also performed at a modulation frequency of 40 MHz.
Cell images were analyzed by MATLAB (Mathworks, Natick, MA). In brief, the images were de-noised with a custom de-noising algorithm developed for the full-field FLIM instrument [57]. The boundary of the cell was then determined by segmenting the intensity images by applying Otsu's method [58]. Because of slight variations in background and signal levels among different cells, a different threshold was applied to each cell for precise detection of cell boundaries.
Results
In-Vitro Characterization
Initial experiments established the FRET efficiency, spectral features and the frequency-domain lifetime characteristics (phase delay and modulation ratio) of the MT1-MMP biosensor in vitro.
Lifetime data in the spectral region of donor (ECFP) fluorescence were collected in a cuvette. The lifetime of ECFP decreased from 2.84 ns for pure ECFP to 0.70 ns for ECFP with FRET, indicating an energy transfer efficiency of approximately 0.75 (see Table 1).
Table 1.
The multi-frequency lifetime measurements were quantified after fitting the data to two lifetime components to characterize ECFP's chromophore. The average lifetimes are listed in the table, reflecting an average FRET efficiency near 0.75
| a1 | τ1 (ns) | a2 | τ2 (ns) | ‹τ› (ns) | |
|---|---|---|---|---|---|
| ECFP-RET | 0.92 | 0.42 | 0.08 | 3.99 | 0.71 |
| ECFP | 0.44 | 1.24 | 0.56 | 4.08 | 2.83 |
Separately, homodyne FLIM measurements of ECFP's fluorescence on the MT1-MMP biosensor before and after cleavage of the biosensor were carried out in solution at a modulation frequency of 40 MHz. A shortening in the overall lifetime of the ECFP as a result of energy transfer is also seen in the polar plot in Fig. 1a. The locations of the points on the polar plot recorded with homodyne FLIM system are consistent with the total phase delay and modulation ratio measured on the multi-frequency cuvette-based instrument at a similar modulation frequency.
With FLIM, in the spectral region corresponding to the acceptor (YPet), we have also measured lifetime-resolved signals of the biosensor before and after it was cleaved (Fig. 1c); both the donor and acceptor emit at these wavelengths (emission between 510 nm and 565 nm; excitation wavelength of 440 nm). A phase delay and modulation ratio corresponding to about 3.8 ns was recorded for the intact biosensor; these data (Fig. 1c) are located near the semicircle. After the biosensor was cleaved, the phase delay and modulation ratio corresponded to a multi-component lifetime. Multiple lifetimes are observed partly because the absorbance spectrum of YPet (and many of the YFP variants) has a tail extending into the low wavelength region indicating that YPet is simultaneously directly excited in addition to ECFP. Therefore, the phase delay and modulation ratio corresponding to an approximate lifetime of 3.8 ns for the intact biosensor (undergoing FRET) contains intensity contributions from directly excited YPet, YPet's emission excited by energy transfer as well as some emission from ECFP. Likewise, the phase delay and modulation ratio measured for the cleaved biosensor at emission wavelengths characterizing mostly YPet contain intensity contributions from both the unpaired ECFP and YPet.
Phase and modulation data collected for both conformations of the biosensor at emission wavelengths between 510 nm and 565 nm were further analyzed to check for consistency with the acceptor lifetime FRET theory. To extend this analysis of the acceptor's lifetime, the lifetime of pure YPet was also measured by directly exciting the MT1-MMP biosensor undergoing FRET with a longer wavelength (488 nm) and was found to be a single lifetime at 3.45 ns (filled circle in Fig. 1b). Using the spectra in Fig. 2, fractional contributions of each species to the total steady-state intensity were computed for each conformation of the biosensor (intact and cleaved) at emission wavelengths characteristic of YPet. The corresponding vector on the polar plot for YPet undergoing energy transfer could then be simulated and plotted (empty circle in Fig. 1b).
Fig. 2.
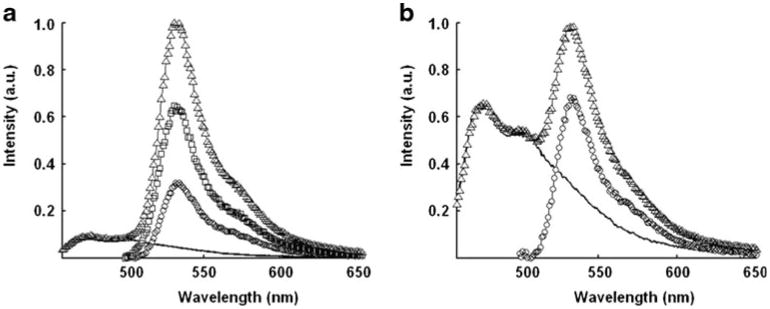
The analysis of the steady-state spectra of the MT1-MMP biosensor in vitro: The spectra represent the contributions of three species: 1) the direct excitation of ECFP's emission (−), 2) YPet's emission due to its direct excitation (○), and 3) YPet's emission due to FRET (□) to (a) the spectrum of the intact (Δ) and to (b) the spectrum of the cleaved biosensor (Δ). Of course, the cleaved biosensor lacks the contribution from YPet's emission due to FRET
As can be seen, the magnitude of the phase shift between the polar coordinates of YPet during FRET (empty circle in Fig. 1b) and ECFP during FRET (empty circle in Fig. 1a) is close to the phase delay of pure YPet (filled circle in Fig. 1b). The total modulation ratio (length of the polar vector) of YPet during FRET is 0.72 which is approximately the product of the modulation depth of pure YPet 0.76 and the modulation depth of ECFP during FRET 0.91.
The trajectories on the polar plots of the FLIM data describing the biosensor show two unique tracks for the donor and acceptor when measured using their respective filters (Fig. 1c). These trajectories are basic information, from which we interpret the measured variations in the phase delay and modulation ratio of the biosensor within live cells.
Visualizing Exogenous MT1-MMP Activity in Live Cells
Analysis on the Polar Plot
A series of HeLa cells transfected with the MT1-MMP biosensor either alone (n=5) or together with the MT1-MMP enzyme (n=5) were imaged with the FLIM system at a modulation frequency of 40 MHz. Since HeLa cells are deficient in endogenous MT1-MMP, the biosensors remained intact in cells expressing only the intact biosensor. However, when HeLa cells expressed both the biosensor and the MT1-MMP enzyme, a large fraction of intracellular biosensors were cleaved
As shown in Fig. 3a, there was a change in both the phase delay and the modulation ratio collected through the filter for ECFP (ECFP channel) between the HeLa cells expressing the intact biosensor only and the HeLa cells expressing both the MT1-MMP biosensor and the MT1-MMP enzyme. When seen on a polar plot, the trend of this change in the two phase delays and modulation ratios paralleled that of the in-vitro experiments, indicating cleavage of the biosensor in the co-transfected HeLa cells. Similarly, phase delays and modulation ratios collected through the filter for YPet (YPet channel) indicated that the polar coordinate describing the cells expressing both biosensor and MT1-MMP enzyme moved farther into the semi-circle, as shown on the polar plot (Fig. 3b). However, the overall change in the polar coordinate of each species (through either channel) was small and their associated distributions demonstrated significant overlap on the polar plot (Fig. 3a-b).
Phase Differential Enhancement Applied to the ECFP Channel
To increase the discrimination of the FRET-FLIM experiment, the individual intensity images collected at each of the eight detector phases used in our homodyne detection were analyzed for both singly transfected HeLa cells (n=5) and co-transfected HeLa cells (n=5). HeLa cells examined for analysis with phase differential enhancement were measured at a modulation frequency of 80 MHz through the ECFP channel (Supp. Fig. 3).
For each cell, the phase-selected intensity images taken through the ECFP channel at eight different detector phases (phase images) were used to compute an image of the DC offset at each pixel of the image. The eight phase images were then divided in a pixel-wise manner by this DC offset image. For all cells (singly transfected and co-transfected), the average intensity of each phase image normalized by the DC offset was computed for each detector phase setting (Fig. 4a). The standard deviations for each of the points plotted in Fig. 4a were all less than two percent of their corresponding intensity values in the plot. The fluorescein reference standard was measured separately before and after FLIM imaging of the singly and co-transfected cells.
Fig. 4.
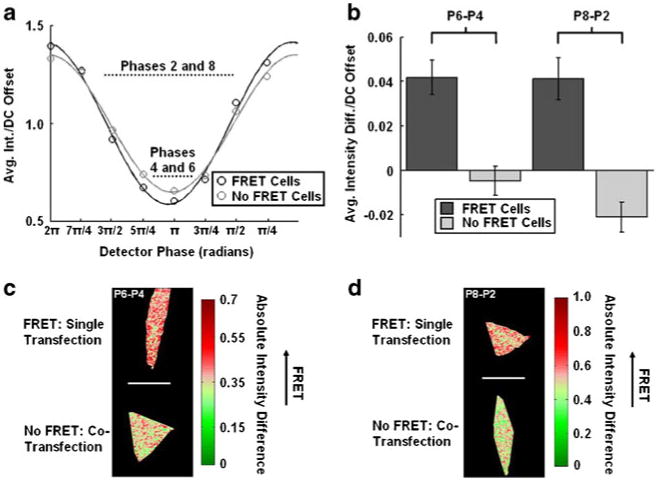
Phase differential enhancement applied to the ECFP channel characterizing the MT1-MMP biosensor in HeLa cells: a The average intensity of each phase image measured at the eight phases applied to our detector describing the MT1-MMP biosensor through the ECFP channel in singly- (intact biosensor with FRET, black, n=5) and co-transfected (cleaved biosensor without FRET, light gray, n=5) HeLa cells are shown. Also displayed are the fits corresponding to each curve used for the homodyne detection of each set of HeLa cells. The data points were normalized to their respective DC offsets for plotting and analysis. b The bar graphs show the average intensity differences/DC offset of the MT1-MMP biosensor in singly- (intact biosensor with FRET) and co-transfected (cleaved biosensor without FRET) HeLa cells after the phase image #6 or #8 was subtracted from those of #4 (P6-P4) or #2 (P8-P2), respectively. The error bars represent standard errors. c-d The representative cell images resulted from the phase differential enhancement in P6-P4 and P8-P2 groups
An inspection of the plot of the average intensities for each phase image normalized to their respective DC offset for each set of HeLa cell (singly transfected and co-transfected cells) shows that phase differential enhancement can further distinguish the two sets of cells (Fig. 4a). Specifically, subtracting the phase-6 image from the phase-4 image resulted in approximately an order of magnitude difference in the resulting intensity differences between singly- and co-transfected HeLa cells (Fig. 4b).
Similarly, subtraction of phase-8 and phase-2 images shown in Fig. 4a also provided a large difference in intensity between the two sets of cells. However, in this case, both sets of HeLa cells (singly transfected and co-transfected) still retain significant non-zero intensities after the subtraction (Fig. 4b). After subtraction of the intensity images corresponding to phase 8 and phase 2, the intensity difference of the co-transfected cells is negative, whereas the singly transfected HeLa cells maintain a consistently positive intensity difference. Although in these examples, one set of cells was not completely suppressed, the absolute difference in intensity between the two sets of cells after the subtraction was sufficient to distinguish clearly two different sets of cells (Fig. 4b). By subtracting these two phase images that maximize differences, the normalized intensity difference images of cells containing intact biosensor had nearly three times the intensity differences of the cells containing cleaved biosensors (Fig. 4b and d).
The resulting images from the phase differential enhancement strategies (Fig. 4c and d) indicate that there is a nearly homogenous distribution of biosensors in both sets of HeLa cells (singly- and co-transfected).
Visualizing Endogenous MT1-MMP Activity in Live Cells
Analysis on the Polar Plot
Variations in the conformation of the MT1-MMP biosensor were also studied in cancerous HT1080 cells by examining corresponding changes in the phase delay and modulation ratio of both ECFP and YPet. HT1080 cells were incubated with or without GM6001, which blocks MT1-MMP activity and hence prevents cleavage of the MT1-MMP biosensor. As such, cells treated with or without GM6001 contain mainly intact or cleaved biosensors, respectively.
Initially, all of the HT1080 cells were imaged through the ECFP channel at a modulation frequency of 40 MHz. When the phase delays and modulation ratios from the two sets of HT1080 cells were collected and projected on a polar plot, there was no substantial difference between cells treated with inhibitor and those not treated (Fig. 5a).
However, analysis of the phase delays and modulation ratios collected through the YPet channel with a modulation frequency of 40 MHz did show a separation in the polar coordinates on polar plots based on the inhibitor treatment (Fig. 5b). The polar coordinate describing the cells incubated with the MT1-MMP inhibitor moved toward the outside of the semi-circle close to position predicted in the in-vitro measurements. Likewise, when cells were not given the inhibitor treatment, the polar coordinate fell into the semi-circle indicating that intracellular MT1-MMP was readily cleaving the biosensor.
Phase Differential Enhancement Applied to the YPet Channel
Phase differential enhancement was then applied to the HT1080 cell data collected through the YPet channel. In this case, the phase difference between the two sets of HT1080 cells containing the two conformations of the biosensor (intact and cleaved) was approximately 3.7°. Figure 6a shows the average of eight intensity images collected at each detector phase for the HT1080 cells treated with GM6001 inhibitor (n=5) and the HT1080 cells not treated with inhibitor (n=5), normalized to their corresponding DC offset on a pixel-wise basis. Although not shown in Fig. 6a, the standard deviation of each of the plotted intensities was less than 4% of the associated mean value. Separate measurements of the fluorescein standard were acquired during the acquisition of each set of images (before and after each FLIM measurement on the cells). The variance in these standard measurements was always well within the difference of the phases between the two states of the biosensor.
Fig. 6.
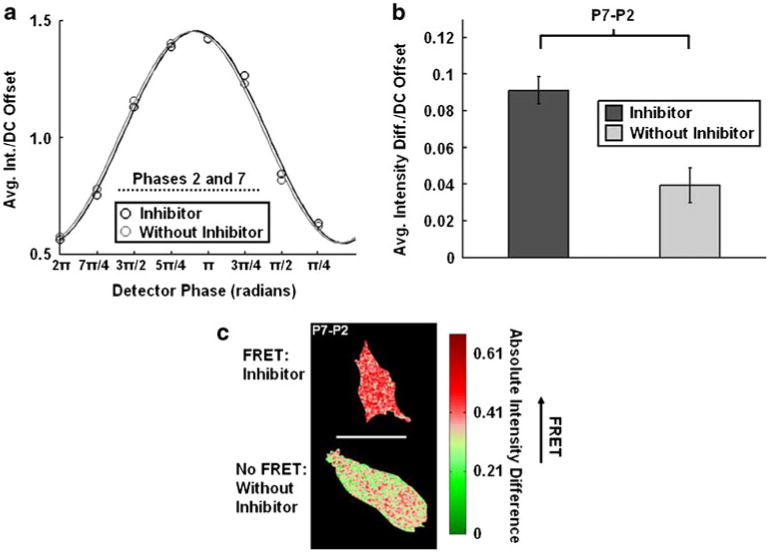
Examination of the MT1-MMP biosensor transfected into HT1080 cells using phase differential enhancement: a The average intensity of each phase image taken of the MT1-MMP biosensor in HT1080 cells treated with (intact biosensor with FRET, black, n=5) or without (cleaved biosensor without FRET, light gray, n=5) GM6001 for 12–18 h are shown as measured through the YPet channel. The corresponding fits used to extract the phase delay and modulation ratio for homodyne detection are also plotted. b The bar graphs show the average intensity differences/DC offset of the MT1-MMP biosensor HT1080 cells treated with (intact biosensor with FRET, black, n=5) or without (cleaved biosensor without FRET, light gray, n=5) GM6001 for 12–18 h after the phase image #7 was subtracted from that of #2 (P7-P2). The error bars represent standard errors. c This representative cell image resulted from the phase differential enhancement in the P7-P2 group
Subtraction of the phase-7 image from the phase-2 image acquired with the YPet channel, substantially reduced the magnitude of the resulting intensity difference from HT1080 cells not treated with inhibitor (Fig. 6b). Although there was a larger degree of cell-to-cell heterogeneity for HT1080 cells as compared to HeLa cells, the resulting intensity differences representing the two sets of HT1080 cells varied in magnitude by approximately a factor of 2. The images resulting from phase differential enhancement revealed a fairly homogenous distribution of the biosensor in the cases of HT1080 cells treated with and without GM6001 inhibitor. Representative HT1080 cell images containing intensity differences resulting from phase differential enhancement are shown in Fig. 6c.
Discussion
We have shown how phase differential enhancement aids in distinguishing changes between the two conformations of our FRET-based MT1-MMP biosensor using frequency-domain FLIM. The increased discrimination provided by phase differential enhancement allowed us to visualize subtle, but physiologically significant, signal changes from the MT1-MMP sensor in live cells, even when a direct polar plot analysis yielded only slight changes in the polar coordinates. With phase differential enhancement, we were able to visualize better the two conformations of the biosensor and highlight differences between its conformations directly in the image. By combining FLIM and FRET, this precise visualization of MT1-MMP activity in live cells should advance our insight into the complex function of MT1-MMP and its role in regulating cancer invasion.
When the FRET-based MT1-MMP biosensor was introduced into the HT1080 and HeLa cells, the measured changes in the coordinates of our biosensor on the polar plot were diminished when examined globally or using various regions of interest within the cells, as compared to the corresponding measurements taken in vitro. The differences between the measured data taken from the live cells and the in-vitro data likely stem from environmental differences (specific to each cell type) [59–62], auto-fluorescence from the cells, indiscriminate proteolytic activity and imperfect expression of the biosensor. For example, when the conformations of the MT1-MMP biosensor were measured in the HT1080 cells as a function of the inhibitor (GM6001) treatment, almost no change in polar coordinate was detected in the ECFP channel. However, there was a definite change in the polar coordinate observed during the same experiment in the YPet channel. Therefore, the data collected in the ECFP channel indicated a high background of cell auto-fluorescence perhaps combined with a large population of unpaired ECFP, particularly when the cells were treated with GM6001 inhibitor preventing cleavage of the biosensor. Despite being small, the variations in polar coordinates measured in both cell types (HT1080 and HeLa) paralleled the trends in the in-vitro measurements which indicated a conformational change in the biosensor.
Of course, the quality of a phase differential enhancement analysis relies on the accuracy of the phase shift, which is directly measured in our frequency-domain FLIM experiment. As with all optical signals, visible contrast between pixels with different phase delays obtainable by phase differential enhancement is only possible within limits of signal to noise and instrument stability. If these conditions are satisfied, we have shown that phase differential enhancement is a useful analysis to highlight small changes in two conformations of our biosensor, which may be more difficult to visualize using more standard methods, such as a direct polar plot analysis or iterative lifetime fitting. In addition, the data acquisition time is significantly reduced in simple phase differential enhancement, which allows faster measurements when observing changes in live cells.
When the biosensor was transfected into HT1080 cells, the results presented here show that the biosensor can monitor changes in the activity of MT1-MMP in response to the established MMP inhibitor GM6001. This set of data can potentially lead to assays where the MT1-MMP enzyme could be activated by the removal of the inhibitor. As a result, the kinetics of the biosensor may be able to be monitored following the activation of MT1-MMP and spatiotemporal information about the enzyme could be derived in settings approximating those found in tissues. However, the incorporation of new FP variants with improved characteristics for FLIM-based FRET studies, such as Cerulean [63] or the teal FP (mTFP1) [64], will improve the ability of FLIM-FRET for resolving changes in the conformation of intracellular biosensors in single cells. In addition, the recent developments of long wavelength fluorescent proteins that absorb and emit light at wavelengths greater than 500 nm can further enhance the contrast in FLIM-FRET imaging by reducing auto-fluorescence and providing greater changes in FRET efficiencies [65, 66].
Supplementary Material
Acknowledgments
This work is supported in part by grants from NIH HL098472, CA139272, NS063405, NSF CBET0846429, CMMI0800870, the Wallace H. Coulter Foundation and Beckman Laser Institute, Inc. (Y.W.). We thank ISS for the use of the cuvette-based lifetime equipment.
Although the derivations of frequency-domain methods [26, 28, 29], the polar plot [44, 49, 50] and homodyne detection [26, 27, 30, 31, 33, 34] have been presented elsewhere, the following appendices are provided here to assist the reader.
Appendix A: Frequency-Domain Lifetime Measurements for Systems with Sets of Discrete Lifetimes
The fluorescence response of a sample with more than one lifetime component that is illuminated with a very short excitation pulse is described by Fδ(t),
| (A.1) |
The fluorescence emission of each component with species fraction (ai) decays with a lifetime (τi).
The fundamental fluorescence response from a multi-component sample excited by excitation light E(t) repetitively modulated at radial frequency ωE, arises from the convolution of the Fδ(t) with E(t) to give the resulting function S(t),
| (A.2) |
The measured modulation depth (the AC amplitude) of S(t) and its phase delay relative to E(t) are dependent on all the separate lifetimes of the sample and the frequency of light modulation. The phase delay and modulation ratio of the multi-component fluorescence response are defined and calculated as in Eqs. 3 and 4 of the text; however, there is no simple relation between phase and modulation and the fluorescence lifetimes.
In the analysis of frequency-domain data, S(t) is normalized by the DC fluorescence, E0 Σ iaiτi, which normalizes the measurement for the sample concentration and brightness.
Appendix B: The Polar Plot
The analysis of S(t) on the polar plot begins by normalizing S(t) by its DC offset yielding the function S(t)/SS (SS = steady-state intensity averaged over a complete period of oscillation, which can be referred to as the DC offset),
| (B.1) |
In this equation,(αi) is the fractional contribution that each lifetime component (i) has to the measured steady-state intensity,
| (B.2) |
This function S(t)/SS can also be described by Eq. (B.3), where (M) and (φ) are the measured modulation ratio and phase delay of the fluorescence signal due to all the contributing fluorescence components.
| (B.3) |
A simple re-arrangement of terms will then lead to following two equations,
| (B.4) |
| (B.5) |
The coordinate transforms of the polar plot stem from the equivalence of Eq. (B.4) and Eq. (B.5). By applying a trigonometric identity, Eq. (B.4) can be written as,
| (B.6) |
Taking Eq. (B.6), the M cos(ωEt)cos(φ) term is in phase with the excitation light's modulation, and the M sin(ωEt)sin(φ) term is out of phase with (orthogonal to) the excitation light's modulation. Thus, M cos(ωEt−φ) can be represented on a Cartesian [x,y] coordinate plot: this is called the polar plot. We only need to consider the amplitudes of the in-phase and out-of-phase components, which can be represented as a vector,
| (B.7) |
| (B.8) |
Likewise, any term (j) in the sum in Eq. (B.5) can also be re-written as shown below,
| (B.9) |
By applying a similar argument, each constituent lifetime of a measured multi-component lifetime can be represented as a vector on the polar plot. In this case, the resulting vector is weighted by the fractional contribution of lifetime (j) to the steady-state intensity (αj).
Therefore, since , one can see that the vector on the polar plot describing M cos(ωEt−φ) is the sum of the vectors of each lifetime component (i) weighted by its corresponding fractional contribution to the steady-state intensity (αi).
Appendix C: Homodyne Detection for Measuring Lifetimes in the Frequency Domain
The acquisition of the phase delay and modulation ratio reflected by the lifetimes in the frequency domain is often accomplished by mixing the intensity-modulated fluorescence response of the sample S(t) with a harmonic signal injected in the detector G(t),
| (C.1) |
The signal that acquired by the detector is product of S(t) and G(t). This multiplication results in a DC offset term, several high frequency terms (with frequencies ωE, ωG, and ωG+ ωE) and a term oscillating at a frequency equal to difference between ωG and ωE, as shown below,
| (C.2) |
The homodyne detection method included in this paper applies a signal to our detector that has the same frequency as the waveform which modulates the intensity of the excitation light (ωG = ωE). When averaged for times in the millisecond range, the high frequency terms in [G(t)S(t)], go to zero and consequently, the terms that remain have no time dependence,
| (C.3) |
To define various points on this sinusoid (Eq. (C.3)) so that the modulation ratio and phase delay indicative of the sample can be found, the phase of the detector (φG), is changed through a set of angles over a full period of cosine. All other parameters are kept constant throughout the acquisition. Hence, the varying phase of G(t) applied in this process is often written relative to the constant phase of E(t) as (φE − φG). The intensity of the sample ([G(t)S(t)]LF, Homp) is then collected at each phase sampled in the detector. A fitting, normalization to the DC offset and comparison to a known reference standard are then performed on the data collected by homodyne methods to determine the phase delay and the modulation ratio of the sample.
In this paper, [G(t)S(t)]LF, Homp refers to a discrete intensity collected for a single pixel in a lifetime image or something similar to a cuvette-based sample in a fluorom-eter. The entire lifetime image containing these types of curves at each pixel is denoted by D(φE − φG) as shown in Supplemental Figure 1.
Footnotes
Personal Communication
Electronic supplementary material The online version of this article (doi:10.1007/s10895-011-0871-x) contains supplementary material, which is available to authorized users.
Contributor Information
John Paul Eichorst, Center of Biophysics and Computational Biology, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA.
He Huang, Department of Bioengineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA.
Robert M. Clegg, Email: rclegg@uiuc.edu, Center of Biophysics and Computational Biology, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA, Department of Physics and Department of Biochemistry, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA.
Yingxiao Wang, Email: yingxiao@uiuc.edu, Center of Biophysics and Computational Biology, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA, Department of Bioengineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA, Beckman Institute for Advanced Science and Technology, Department of Molecular and Integrative Physiology, Neuroscience Program, and Institute of Genomic Biology, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA.
References
- 1.Lynch CC, Matrisian LM. Matrix metalloproteinases in tumor-host cell communication. Differentiation. 2002;70(9–10):561–573. doi: 10.1046/j.1432-0436.2002.700909.x. [DOI] [PubMed] [Google Scholar]
- 2.Poincloux R, Lizarraga F, Chavrier P. Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J Cell Sci. 2009;122(Pt 17):3015–3024. doi: 10.1242/jcs.034561. [DOI] [PubMed] [Google Scholar]
- 3.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nyalendo C, et al. Src-dependent phosphorylation of membrane type I matrix metalloproteinase on cytoplasmic tyrosine 573: role in endothelial and tumor cell migration. J Biol Chem. 2007;282(21):15690–15699. doi: 10.1074/jbc.M608045200. [DOI] [PubMed] [Google Scholar]
- 5.Itoh Y, Seiki M. MT1-MMP: a potent modifier of pericellular microenvironment. J Cell Physiol. 2006;206(1):1–8. doi: 10.1002/jcp.20431. [DOI] [PubMed] [Google Scholar]
- 6.Uekita T, et al. Cytoplasmic tail-dependent internalization of membrane-type 1 matrix metalloproteinase is important for its invasion-promoting activity. J Cell Biol. 2001;155(7):1345–1356. doi: 10.1083/jcb.200108112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mori H, et al. CD44 directs membrane-type 1 matrix metalloproteinase to lamellipodia by associating with its hemopexin-like domain. EMBO J. 2002;21(15):3949–3959. doi: 10.1093/emboj/cdf411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seiki M. Membrane-type 1 matrix metalloproteinase: a key enzyme for tumor invasion. Cancer Lett. 2003;194(1):1–11. doi: 10.1016/s0304-3835(02)00699-7. [DOI] [PubMed] [Google Scholar]
- 9.Sabeh F, et al. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J Cell Biol. 2004;167(4):769–781. doi: 10.1083/jcb.200408028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu X, et al. FAK-mediated src phosphorylation of endophilin A2 inhibits endocytosis of MT1-MMP and promotes ECM degradation. Dev Cell. 2005;9(2):185–196. doi: 10.1016/j.devcel.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Kajita M, et al. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J Cell Biol. 2001;153(5):893–904. doi: 10.1083/jcb.153.5.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Endo K, et al. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J Biol Chem. 2003;278(42):40764–40770. doi: 10.1074/jbc.M306736200. [DOI] [PubMed] [Google Scholar]
- 13.Udayakumar TS, et al. Membrane type-1-matrix metal-loproteinase expressed by prostate carcinoma cells cleaves human laminin-5 beta3 chain and induces cell migration. Cancer Res. 2003;63(9):2292–2299. [PubMed] [Google Scholar]
- 14.Miyawaki A, et al. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388(6645):882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- 15.Förster T. Fluoreszenz Organischer Verbindungen. Van-denhoek & Ruprecht; Göttingen: 1951. [Google Scholar]
- 16.Förster T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann Phys. 1948;437(2):55–75. [Google Scholar]
- 17.Förster T. Intermolecular energy migration and fluroes-cence. Ann Phys. 1948;2:55–75. [Google Scholar]
- 18.Clegg RM. Fluorescence resonance energy transfer and nucleic acids. Meth Enzymol. 1992;211:353–388. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
- 19.Ouyang M, et al. Simultaneous visualization of protumorigenic Src and MT1-MMP activities with fluorescence resonance energy transfer. Cancer Res. 70(6):2204–2212. doi: 10.1158/0008-5472.CAN-09-3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen AW, Daugherty PS. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat Biotechnol. 2005;23(3):355–360. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]
- 21.Ouyang M, et al. Visualization of polarized membrane type 1 matrix metalloproteinase activity in live cells by fluorescence resonance energy transfer imaging. J Biol Chem. 2008;283(25):17740–17748. doi: 10.1074/jbc.M709872200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyawaki A, Tsien RY. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Meth Enzymol. 2000;327:472–500. doi: 10.1016/s0076-6879(00)27297-2. [DOI] [PubMed] [Google Scholar]
- 23.Jovin TM, Arndt-Jovin D. FRET microscopy: digital imaging of fluorescence resonance energy transfer. In: Kohen E, Hirschberg J, editors. Application in cell biology, in cell structure and function by microspectrofluorometry. Academic; San Diego: 1989. pp. 99–115. [Google Scholar]
- 24.Young RM, et al. Quantitation of fluorescence energy transfer between cell surface proteins via fluorescence donor photobleaching kinetics. Biophys J. 1994;67(2):881–888. doi: 10.1016/S0006-3495(94)80549-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mattheyses AL, Hoppe AD, Axelrod D. Polarized fluorescence resonance energy transfer microscopy. Biophys J. 2004;87(4):2787–2797. doi: 10.1529/biophysj.103.036194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider P, Clegg RM. Rapid acquisition, analysis and display of fluorescence lifetime-resolved images for real-time applications. Rev Sci Instrum. 1997;68(11):4107–4119. [Google Scholar]
- 27.Spring BQ, Clegg RM. FLIM microscopy in biology and medicine. Chapman & Hall/CRC; New York: 2010. Frequency-domain FLIM; pp. 115–142. [Google Scholar]
- 28.Lakowicz J. Principles of fluorescence spectroscopy. Springer; New York: 2006. p. 954. [Google Scholar]
- 29.Valeur B. Molecular fluorescence: principles and applications. WILEY-VCH; Weinheim: 2002. [Google Scholar]
- 30.Clegg RM. Fluorescence lifetime-resolved imaging microscopy (FLIM) In: Scheggi AM Verga., editor. Biomedical optical instrumentation and laser-assisted biotechnology: proceedings of the NATO advanced study institute, Etice, Italy November 10-22, 1995. Kluwer Academic Publishers; Dordrecht: 1996. pp. 143–156. [Google Scholar]
- 31.Gadella TWJ, Jr, Jovin TM, Clegg RM. Fluorescence lifetime imaging micorscopy (FLIM): spatial resolution of micro-structures on the nanosecond time scale. Biophys Chemist. 1993;48:221–239. [Google Scholar]
- 32.Lakowicz JR, et al. Fluorescence lifetime imaging of free and protein-bound NADH. Proc Natl Acad Sci USA. 1992;89(4):1271–1275. doi: 10.1073/pnas.89.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clegg RM, Gadella TWJ, Jr, Jovin TM. Lifetime-resolved fluorescence imaging. SPIE. 1994;2137:105–118. [Google Scholar]
- 34.Clegg RM, et al. Time resolved imaging fluorescence microscopy. SPIE Time Resolved Laser Spectrosc Biochem III. 1992;1640:448–460. [Google Scholar]
- 35.Holub O, et al. Fluorescence lifetime imaging (FLI) in realtime—a new technique in photosynthesis research. Photosynthetica. 2000;38(4):581–599. [Google Scholar]
- 36.Veselova TV, Cherkasov AS, Shirokov VI. Fluorometric method for individual recording of spectra in systems containing two types of luminiscent centers. Opt Spectrosc. 1970;29:617–618. [Google Scholar]
- 37.Veselova TV, Shirokov VI. A spectral fluorometric study of the luminescence of exiplexes of 3-amino-AT-methylphthalimide with acetone, pyridine, and dimethylformamide. Iz Akad Nauk SSSR Ser Fiz. 1972;36:1024. [Google Scholar]
- 38.Jameson DM, Gratton E, Hall RD. The measurement and analysis of hetergeneous emissions by multifrrequency phase and modulation fluorometry. Appl Spectrosc Rev. 1984;20(1):55–106. [Google Scholar]
- 39.McGown LB, Bright FV, Demas JN. Phase-resolved fluorescence in chemical analysis. Crit Rev Anal Chem. 1987;18(3):245–298. [Google Scholar]
- 40.Lakowicz JR, et al. Background suppression in frequency-domain fluorometry. Anal Biochem. 2000;277(1):74–85. doi: 10.1006/abio.1999.4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lakowicz JR, Cherek H. Phase-sensitive fluorescence spectroscopy: a new method to resolve fluorescence lifetimes or emission spectra of components in a mixture of fluorophores. J Biochem Biophys Meth. 1981;5(1):19–35. doi: 10.1016/0165-022x(81)90030-0. [DOI] [PubMed] [Google Scholar]
- 42.Lakowicz JR, Cherek H. Resolution of heterogeneous fluorescence by phase-sensitive fluorescence spectroscopy. Biophys J. 1982;37(1):148–150. doi: 10.1016/S0006-3495(82)84645-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spencer R, Weber G. Measurements of subnanosecond fluorescence lifetimes with a cross-correlation phase fluorometer. Ann NY Acad Sci. 1969;158:361–376. [Google Scholar]
- 44.Redford GI, Clegg RM. Polar plot representation for frequency-domain analysis of fluorescence lifetimes. J Fluoresc. 2005;15(5):805–815. doi: 10.1007/s10895-005-2990-8. [DOI] [PubMed] [Google Scholar]
- 45.Gratton E, Jameson DM, Hall RD. Multifrequency phase and modulation fluorometry. Annu Rev Biophys Bioeng. 1984;13:105–124. doi: 10.1146/annurev.bb.13.060184.000541. [DOI] [PubMed] [Google Scholar]
- 46.Lakowicz JR, et al. Analysis of fluorescence decay kinetics from variable-frequency phase shift and modulation data. Biophys J. 1984;46(4):463–477. doi: 10.1016/S0006-3495(84)84043-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weber G. Resolution of fluorescence lifetimes in a heterogeneous system by phase and modulation measurements. J Phys Chem. 1981;85:949–953. [Google Scholar]
- 48.Kremers GJ, et al. Quantitative lifetime unmixing of multiexponentially decaying fluorophores using single-frequency fluorescence lifetime imaging microscopy. Biophys J. 2008;95(1):378–389. doi: 10.1529/biophysj.107.125229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Digman MA, et al. The phasor approach to fluorescence lifetime imaging analysis. Biophys J. 2008;94(2):L14–L16. doi: 10.1529/biophysj.107.120154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clayton AH, Hanley QS, Verveer PJ. Graphical representation and multicomponent analysis of single-frequency fluorescence lifetime imaging microscopy data. J Microsc. 2004;213(Pt 1):1–5. doi: 10.1111/j.1365-2818.2004.01265.x. [DOI] [PubMed] [Google Scholar]
- 51.Chen YC, et al. General concerns of FLIM data representation and analysis: frequency-domain model-free analysis. In: Periasamy A, Clegg RM, editors. FLIM microscopy in biology and medicine. Chapman & Hall/CRC; New York: 2010. pp. 291–335. [Google Scholar]
- 52.Gratton E, Limkeman M. A continuously variable frequency cross-correlation phase fluorometer with picosecond resolution. Biophys J. 1983;44(3):315–324. doi: 10.1016/S0006-3495(83)84305-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schiller PW. The measurement of intramolecular distances by energy transfer. In: Chen RF, Edelhoch H, editors. Biochemical fluorescence: concepts. Marcel Dekker Inc; New York: 1975. pp. 285–303. [Google Scholar]
- 54.Lakowicz JR, Balter A. Analysis of excited-state processes by phase-modulation fluorescence spectroscopy. Biophys Chem. 1982;16(2):117–132. doi: 10.1016/0301-4622(82)85013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lakowicz JR, Balter A. Theory of phase-modulation fluorescence spectroscopy for excited-state processes. Biophys Chem. 1982;16(2):99–115. doi: 10.1016/0301-4622(82)85012-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, et al. Visualizing the mechanical activation of Src. Nature. 2005;434(7036):1040–1045. doi: 10.1038/nature03469. [DOI] [PubMed] [Google Scholar]
- 57.Spring BQ, Clegg RM. Image analysis for denoising full-field frequency-domain fluorescence lifetime images. J Microsc. 2009;235(2):221–237. doi: 10.1111/j.1365-2818.2009.03212.x. [DOI] [PubMed] [Google Scholar]
- 58.Otsu N. A threshold selection method from gray-level histograms. IEEE Trans Syst Man Cybern. 1979;9:62–66. [Google Scholar]
- 59.Borst JW, et al. Effects of refractive index and viscosity on fluorescence and anisotropy decays of enhanced cyan and yellow fluorescent proteins. J Fluoresc. 2005;15(2):153–160. doi: 10.1007/s10895-005-2523-5. [DOI] [PubMed] [Google Scholar]
- 60.Jose M, et al. Photophysics of Clomeleon by FLIM: discriminating excited state reactions along neuronal development. Biophys J. 2007;92(6):2237–2254. doi: 10.1529/biophysj.106.092841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Millington M, et al. High-precision FLIM-FRET in fixed and living cells reveals heterogeneity in a simple CFP-YFP fusion protein. Biophys Chem. 2007;127(3):155–164. doi: 10.1016/j.bpc.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 62.Villoing A, et al. Complex fluorescence of the cyan fluorescent protein: comparisons with the H148D variant and consequences for quantitative cell imaging. Biochemistry. 2008;47(47):12483–12492. doi: 10.1021/bi801400d. [DOI] [PubMed] [Google Scholar]
- 63.Rizzo MA, et al. An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol. 2004;22(4):445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- 64.Day RN, Booker CF, Periasamy A. Characterization of an improved donor fluorescent protein for Forster resonance energy transfer microscopy. J Biomed Opt. 2008;13(3):031203. doi: 10.1117/1.2939094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shaner NC, et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22(12):1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 66.Shaner NC, et al. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat Meth. 2008;5(6):545–551. doi: 10.1038/nmeth.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.