

Abstract
Objectives
The steroids estradiol (E2), estrone (E1), and estriol (E3) are the major estrogens. E1/E2 and their metabolite 16-hydroxyestrone (16-OHE1, known to be carcinogenic) could be involved in the development of many cancers including human breast cancer. The aim of the current study was to develop a rapid and simple high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) assay to simultaneously measure E1, E2, E3 and 16-OHE1 in human serum without the need for solid phase extraction or derivatization.
Methods
An API-5000 triple-quadrupole mass spectrometer coupled with electrospray ionization (ESI) source and Shimadzu HPLC system was used employing isotope dilution with deuterium-labeled internal standard (IS) for each analyte. Quantitation by multiple reaction monitoring (MRM) analysis was performed in negative ion mode.
Results
The limits of detection were 1.0 pg/mL for E1 and 16-OHE1 and 2.0 pg/mL for E2 and E3. Within-day CVs were <6.5% for all analytes tested and between-day CVs ranged from 4.5% to 9.5%. Recovery ranged from 88% to 108%.
Conclusion
This method allows for the simultaneous measurement of four estrogens in human serum within 8 min. It can be routinely employed in a clinical environment and is attractive because of its simplicity in sample processing, micro sample requirement, and high throughput.
Keywords: Estradiol, Estriol, Estrone, 16-Hydroxyestrone, HPLC-tandem mass spectrometry, Estrogen profiling
Introduction
Estrone (E1), estradiol (E2), and estriol (E3) are the major estrogens in human serum. E1/E2 and their metabolite 16-hydroxyestrone (16-OHE1, a known carcinogen) are thought to be involved in the development of human breast cancer [1–3]. Immunoassays (IAs) are the most widely used technique for measuring estrogens and afford good sensitivity, but lack specificity due to the cross-reaction of the antibodies used in the assays with other steroids and interfering substances in the matrix. The total overestimation of estradiol by interfering substances such as estrone, estrone sulphate and other estrogens can be more than 60% [4] and is concentration dependent. Estriol was also found to interfere with the measurement of estradiol by commonly used immunoassays [5], and there is a significant lack of agreement between the results of different immunoassays for the measurement of estradiol [5,6]. Several liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) assays are reported to measure E1, E2, and E3 [7–12]. In the past, the low concentrations of estrogens in human blood necessitated the employment of either liquid–liquid extraction (LLE) or solid phase extraction (SPE) prior to derivatization in all of the reported HPLC-MS/MS methods. These methods are highly sensitive and specific, but are very labor-intensive and often derivatization can compromise the specificity of the analysis [13]. In this paper we report a rapid, simple, specific and highly sensitive HPLC-MS/MS assay to simultaneously measure E1, E2, E3 and 16-OHE1 in human serum without the need for LLE/SPE or derivatization.
Materials and methods
Materials
E2, E3, E1, 16-OHE1, and bovine albumin were obtained from Sigma Chemical Co. (St. Louis, MO, USA). Two deuterated internal standards, estriol-d2, estrone-d4 were purchased from C/D/N Isotopes, Inc. (Pointe-Claire, Quebec, Canada), estradiol-d4 was supplied by Cambridge Isotope Laboratories, Inc. (Andover, MA, USA) and aldosterone-d6 (used as internal standard for 16-OHE1) was custom synthesized by IsoSciences, LLC (King of Prussia, PA, USA). All steroid estrogens and their deuterated analogues have reported chemical and isotopic purity greater than 95% and were used without further purification. HPLC-grade water, acetonitrile, and methanol were obtained from Fisher Scientific (Fair Lawn, NJ, USA). Deionized water was prepared from Barnstead B-PURE Water System (Dubuque, IA, USA).
Preparation of internal standards, calibration standards, and in-house controls
A stock internal standard solution (50 μg/mL) was prepared by dissolving 1 mg of individual deuterated internal standard in 20 mL of methanol. Stock solutions of each analyte were prepared separately in methanol to produce a concentration of 1.0 mg/mL. Working profile calibrator solution was prepared by mixing appropriate amounts of individual analyte stock solution to obtain a mixture containing 10 μg/mL of each estrogen. The stock solutions of estrogens, working profile solution, and their internal standards were stored at −80 °C until use. Calibration standards at 5, 10, 50, 500, and 1000 pg/mL were prepared by diluting the working profile solution with 4% albumin solution by factors of 10,000, 20,000, 200,000, 1,000,000, and 2,000,000. In-house quality controls for 16-OHE1 were prepared at 12.5, 100, and 1250 pg/mL in 4% albumin solution. External quality controls for E1, E2 and E3 were provided by the New York State Department of Health-Wadsworth Center Endocrinology Proficiency Testing Program (E2, E3) [Albany, NY, USA] and the College of American Pathologist Proficiency Testing Program (E1) [Northfield, IL, USA]. Samples for comparison studies for E1 and E2 were obtained from Mayo Clinic (Rochester, MN, USA) and from the College of American Pathologist Proficiency Testing Program for E1.
HPLC-tandem mass spectrometry instrumentation and conditions
An API 5000 triple-quadrupole mass spectrometer (Applied Biosystems/MDS SCIEX, Foster City, CA, USA/Concord, Ontario, Canada) coupled with electrospray ionization (ESI) source was operated in the negative ion mode. The liquid chromatography system consisted of three Shimadzu LC-20 AD pumps, a Shimadzu SIL-HTA autosampler, and a Shimadzu DGU-20A5 degasser (Shimadzu Scientific Instruments, Columbia, MD, USA). Isotope dilution with deuterium-labeled internal standard (IS) for each analyte was employed in the analysis. Following a 3 min wash with mobile phase A (methanol: water 2:98, v/v) at a flow rate of 1 mL/min, the switching valve was activated to initiate the binary gradient program which eluted the steroids at a flow rate of 600 μL/min as follows: 80% A and 20% B (methanol) for 3 min, 52% A and 48% B going to 42%A and 58% B from 3.0–7.1 min, and finally 10% A and 90% B from 7.1–8.0 min. There is a 1 min re-equilibration with mobile phase A at 1 mL/min between each run. The ESI source was operated with ionspray voltage at −4500 V and heater temperature at 600 °C. Nitrogen and zero grade air were produced by a high purity nitrogen generator (Peak Scientific Instrument Ltd., model NM20z, Renfrewshire, Scotland). Gas settings were as follows: curtain gas 25 arbitrary units, collision gas 6 arbitrary units, nebulizer gas 40 arbitrary units, and heater gas 35 arbitrary units. Dwell time per transition was set at 200 ms. Nitrogen was used as curtain gas and collision gas in the Q2 collision cell. Unit mass resolution was set in both mass-resolving quadrupole Q1 and Q3. Data were acquired on a Dell Precision 370 workstation and were processed by Analyst 1.4.1 software package (MDS SCIEX, Concord, Ontario, Canada). Chromatographic separation was carried out on a reverse-phase C-8 analytical column (Supelco LC-8-DB, 3.3 cm×3.0 mm, 3 μm particle size) protected by a Supelco Discover C-8 (3.0 mm) guard column of the same packing material (Supelco, St. Louis, MO, USA) at room temperature.
Quantitation by multiple reaction mode (MRM) analysis was performed in the negative ion mode. The MRM conditions for the deprotonated molecules [M−H]− of the estrogens and their deuterium-labeled internal standards and retention times are shown in Table 1. In order to ensure maximum sensitivity and minimum interference from neighboring peaks, the total ion chromatogram was divided into two sections (Table 1 and Fig. 1).
Table 1.
MRM conditions for the estrogen steroids and deuterium-labeled internal standard in negative ion mode
| Period (min) | Estrogen steroid | MRM transition | Retention time (min) | DP | EP | CE | CXP |
|---|---|---|---|---|---|---|---|
| Period I (5.5) | Estriol | 287/171 | 4.46 | −120 | −10 | −55 | −15 |
| Estriol (2nd) | 287/145 | 4.46 | −120 | −10 | −55 | −15 | |
| Estriol-d2 | 289/147 | 4.45 | −120 | −10 | −55 | −15 | |
| 16-Hydroxyestrone | 285/145 | 4.77 | −120 | −10 | −55 | −15 | |
| 16-Hydroxyestrone (2nd) | 285/143 | 4.77 | −120 | −10 | −78 | −17 | |
| Aldosterone-d6 | 365/337 | 4.63 | −60 | −10 | −23 | −15 | |
| Period II (2.5) | Estrone | 269/145 | 6.88 | −120 | −10 | −55 | −15 |
| Estrone (2nd) | 269/143 | 6.88 | −120 | −10 | −55 | −15 | |
| Estrone-d4 | 273/147 | 6.86 | −120 | −10 | −55 | −15 | |
| Estradiol | 271/145 | 6.93 | −120 | −10 | −55 | −15 | |
| Estradiol (2nd) | 271/183 | 6.93 | −120 | −10 | −55 | −15 | |
| Estradiol-d4 | 275/147 | 6.89 | −120 | −10 | −55 | −15 |
Note: for estriol, 16-hydroxyestrone, estrone, and estradiol, the first MRM transitions were listed for quantitation, and the second transitions were added for confirmation.
Fig. 1.

High performance liquid chromatography-electrospray ionization-tandem mass spectrometry multiple reaction monitoring (negative ion mode) chromatographic profiles of estrogen steroids corresponding to 100 pg/mL in standard solution (4%(w/w) bovine albumin). Chromatograms were obtained using two periods. Period I: 0→5.50 min; Period II: 5.50→8.00 min.
Calibration, using internal standardization, was done by linear regression. Calibration curves for each estrogen analyte were constructed by plotting peak area ratio between target analyte and its internal standard versus the amount of target analyte.
Sample preparation
200 μL of serum sample, calibration standard, or quality control was deproteinized by adding 300 μL of acetonitrile containing 1 ng/mL of deuterated internal standards (IS) in 1.7 mL conical plastic centrifuge tubes. The tubes were capped, vortexed vigorously for 30 s, and centrifuged at 13,000 rpm for 10 min by Eppendorf Centrifuge 5415D (Hamburg, Germany). After centrifugation, 350 μL of supernatant was transferred into autosampler vials and diluted with 1400 μL of deionized water. 600 μL aliquot was injected into the LC-ESI-MS/MS system. Sample preparation was performed at room temperature.
Assay validation
To assess the accuracy and precision of this method, results were compared with those from the Mayo Clinic, College of American Pathologists (CAP) Proficiency Testing Program, and New York State Department of Health-Wadsworth Center Endocrinology Proficiency Testing Program, which were all assayed for E1, E2, and E3. The inter-day and between-day (10 batches) precisions (coefficients of variation CV) were also determined using New York State Department of Health-Wads-worth Center Endocrinology Proficiency Testing Program and in-house controls.
Results and discussion
Lin et al. reported that the signal intensity of dansyl derivatives in positive ESI mode was significantly higher (38–101 times) than that produced by underivatized estrogens in negative ESI mode [10]. To achieve the significant increase in analytical sensitivity, all previously reported studies involved the employment of liquid/liquid extraction or solid phase extraction before the derivatization step [7–12]. These sample pretreatments use lengthy steps as well as careful pH and temperature control, and thus are quite laborious and time consuming. The large pH changes (from pH 3 to pH 10) during the pretreatment steps may also potentially contribute to the hydrolysis of conjugated estrogens in serum samples, leading to erroneous and falsely high results in the measurement of estrogens. A simple and rapid estrogen measurement by LC-MS/MS is important in clinical and research laboratories, and we developed such a method by taking advantage of the superior analytical sensitivity offered by API 5000 triple-quadrupole tandem mass spectrometer.
A LC-MS/MS chromatogram of a standard calibrator containing 100 pg/mL of each estrogen analyte is shown in Fig. 1. E3, 16-OHE1 and E2 are baseline separated chromatographically, while E1 and E2 have very close retention times under current elution conditions. Considering the unique specificity provided by tandem mass spectrometry, the interference and cross-talk between E1 and E2 are negligible.
The limits of detection were 1.0 pg/mL for E1 and 16-OHE1 and 2.0 pg/mL for E2 and E3. Dividing the chromatogram into 2 segments allowed fewer ions to be monitored in each segment thereby enhancing the dwell times and increasing the sensitivity of all the analytes measured. Sensitivity can be further enhanced and lowered by a factor of 2 by injection of a 1200 μL (instead of the routine 600 μL) aliquot after precipitation of the serum proteins. Within-day CVs were <6.5% for all analytes tested and between-day CVs ranged from 4.5% to 9.5%. Recovery ranged from 88% to 108% in 4% (w/w) albumin solution. Accuracy for E1 and E2 was assessed by correlation with the Mayo Clinic’s results (estrogens), and correlation coefficients were r=0.995 for E2 and r=0.996 for E1, respectively (Fig. 2). Accuracy for E3 was evaluated by comparison of our results with those from the College of American Pathologists (CAP) Proficiency Testing Program, 2006 (Y-01 to Y-04), and New York State Department of Health Endocrinology Proficiency Testing, 2007 (E-01 to E-05) and 2006 (E-76 to E95). Both CAP and New York State Department of Health Proficiency Testing Programs document the results by various immunoassays from a large number of clinical laboratories. The IA All Method Mean was compared to our results (Fig. 3). Our results for estriol are very close to the immunoassay All Method Mean and the correlation coefficient was r=0.992 (Fig. 3). 16-OHE1 could not be assessed for accuracy in this way due to the inability to locate another laboratory to measure this analyte and thereby allowing such a comparison.
Fig. 2.

Correlation between Georgetown University Bioanalytical Core Laboratory (GU) and Mayo Clinic. The Mayo Clinic’s results were obtained using tandem mass spectrometry [7].
Fig. 3.
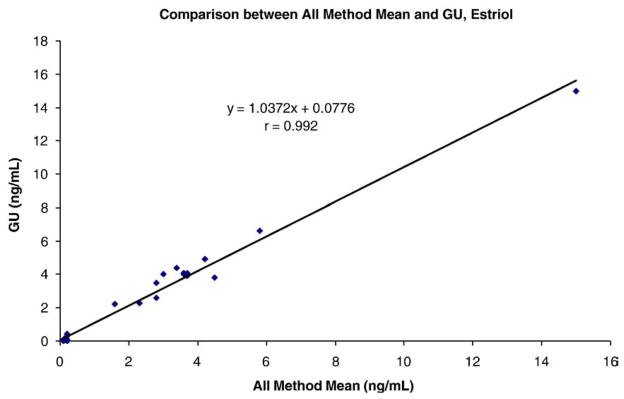
Correlation between Georgetown University Bioanalytical Core Laboratory (GU) and All Methods and Instruments Mean for estriol.
There is no commercially available deuterium-labeled internal standard (IS) for 16-OHE1, and we used aldosterone-d6 as its IS instead, which has a retention time of 4.63 min and m/z 365/337.
In our previous studies [14–17], estradiol was reported to be determined in the positive ion mode with transition pair of m/z 255/159 using atmospheric pressure photoionization (APPI) source. The species of [M+H−H2O]+ is less abundant in the positive ion mode compared to that of [M−H]− in the negative ion mode, especially in human serum matrix, and is subject to interference from neighboring peaks co-eluting with E2 in the chromatogram. We found that E2 has at least a 10 fold increase in sensitivity in the ESI negative ion mode over the APPI positive ion mode and does not suffer from interferences.
The manuscript of Xu et al. [12] describes a tandem mass spectrometric method for the measurement of 15 estrogens. The method they describe is very time consuming, requires extraction and derivatization, needs a 500 μL aliquot of serum and has a chromatography time of 70 min. Method comparison studies were also not performed by these authors and the paper studied only the estrogens in some 6 sera. Finally no comment was made by them regarding the lack of stability of the 2 phenols, 2-OHE1 and 4-OHE1. We were unable to stabilize 2-OHE1 in sera even though partial stabilization was achieved by addition of dithiothreitol to the samples. For all the above reasons, the method of Xu et al. [12] is clearly unsuitable for the measurement of the 3 most frequently requested estrogens, estradiol, estrone and estriol in the routine clinical chemistry laboratory.
Conclusion
We have developed an estrogen profile assay employing an isotope dilution LC-ESI-MS/MS-based method without derivatization. The method requires 200 μL serum and is run in the negative ion mode. The analysis was performed in the MRM mode, which provides high specificity and enough sensitivity for the measurement of four estrogens in human serum. This method correlates very well for E1 and E2 with other MS/MS-based methods developed at the Mayo Clinic. For E3, the method was validated by utilizing proficiency testing materials from CAP and New York State Department of Health-Wadsworth Medical Center. This method can simultaneously measure four estrogens in human serum within 8 min. It is reliable, can be routinely employed in a clinical environment and is attractive because of its simplicity in sample work-up (no derivatization), great sensitivity, micro sample requirement and high throughput.
Acknowledgments
This study was partially supported by NIH GCRC grant # 5-MO1-RR-13297-S1, by grant 1 U10HD45993-02 of the National Institute of Child Health and Development, Bethesda, MD and by Applied Biosystems, Foster City, CA. (SJS); and NIH/NICHD grant 5U10HD047890-03 and the Office of Research on Women’s Health grant 5U10HD047890-03 Obstetrics and Pharmacology Research Unit (OPS).
References
- 1.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med. 2006;354:270–82. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 2.Travis RC, Key TJ. Oestrogen exposure and breast cancer risk. Breast Cancer Res. 2003;5:239–47. doi: 10.1186/bcr628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clemons M, Goss P. Mechanisms of disease: estrogen and the risk of breast cancer. N Engl J Med. 2001;344:276–85. doi: 10.1056/NEJM200101253440407. [DOI] [PubMed] [Google Scholar]
- 4.Farre M, Kuster M, Brix R, Rubio F, Lopez de Alda MJ, Barcelo D. Comparative study of an estradiol enzyme-linked immunosorbent assay kit, liquid chromatography-tandem mass spectrometry, and ultra performance liquid chromatography-quadrupole time of flight mass spectrometry for part-per-trillion analysis of estrogens in water samples. J Chromatogr, A. 2007;1160(1–2):166–75. doi: 10.1016/j.chroma.2007.05.032. [DOI] [PubMed] [Google Scholar]
- 5.Cao Z, Swift TA, West CA, Rosano TG, Rej R. Immunoassay of estradiol: unanticipated suppression by unconjugated estriol. Clin Chem. 2004;50:160–5. doi: 10.1373/clinchem.2003.023325. [DOI] [PubMed] [Google Scholar]
- 6.College of American Pathologists Proficiency Testing Program (CAP PT) Surveys 2002 Y-A ligands (Special) Northfield, Ill: College of American Pathologists; 2002. [Google Scholar]
- 7.Nelson RE, Grebe SK, O’Kane DJ, Singh RJ. Liquid chromatography-tandem mass spectrometry assay for simultaneous measurement of estradiol and estrone in human plasma. Clin Chem. 2004;50:373–84. doi: 10.1373/clinchem.2003.025478. [DOI] [PubMed] [Google Scholar]
- 8.Tai SC, Welch MJ. Development and evaluation of a reference measurement procedure for the determination of estradiol-17β in human serum using isotope-dilution liquid chromatography-tandem mass spectrometry. Anal Chem. 2005;77:6359–63. doi: 10.1021/ac050837i. [DOI] [PubMed] [Google Scholar]
- 9.Xu X, Keefer LK, Ziegler RG, Veenstra TD. A liquid chromatography-mass spectrometry method for the quantitative analysis of urinary endogenous estrogen metabolites. Nat Protoc. 2007;2:1350–5. doi: 10.1038/nprot.2007.176. [DOI] [PubMed] [Google Scholar]
- 10.Lin YH, Chen CY, Wang GS. Analysis of steroid estrogens in Water by LC/MS/MS. Rapid Commun Mass Spectrom. 2007;21:1973–83. doi: 10.1002/rcm.3050. [DOI] [PubMed] [Google Scholar]
- 11.Yamashita K, Okuyama M, Watanabe Y, Honma S, Kobayashi S, Numazawa M. Highly sensitive determination of estrone and estradiol in human serum by liquid chromatography-electrospray ionization tandem mass spectrometry. Steroids. 2007;72(11–12):819–27. doi: 10.1016/j.steroids.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Xu X, Roman JM, Issaq HJ, Keefer LK, Veenstra TD, Ziegler RG. Quantitative measurement of endogenous estrogens and estrogen metabolites in human serum by liquid chromatography-tandem mass spectrometry. Anal Chem. 2007;79:7813–21. doi: 10.1021/ac070494j. [DOI] [PubMed] [Google Scholar]
- 13.Malekinejad H, Scherpenisse P, Bergwerff AA. Naturally occurring estrogens in processed milk and in raw milk (from gestated cows) J Agric Food Chem. 2006;54(26):9785–91. doi: 10.1021/jf061972e. [DOI] [PubMed] [Google Scholar]
- 14.Holst JP, Soldin OP, Guo T, Soldin SJ. Steroid hormones: relevance and measurement in the clinical laboratory. Clin Lab Med. 2004;24(1):105–18. doi: 10.1016/j.cll.2004.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo T, Chan M, Soldin SJ. Steroid profiles using liquid chromatography tandem mass spectrometry with atmospheric pressure photoionization source. Arch Pathol Lab Med. 2004;128:469–75. doi: 10.5858/2004-128-469-SPULCM. [DOI] [PubMed] [Google Scholar]
- 16.Soldin OP, Guo T, Weiderpass E, Tractenberg RE, Hilakivi-Clarke L, Soldin SJ. Steroid hormone levels in pregnancy and 1 year postpartum using isotope dilution tandem mass spectrometry. Fertil Steril. 2005;84(3):701–10. doi: 10.1016/j.fertnstert.2005.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo T, Taylor RT, Singh RJ, Soldin SJ. Simultaneous determination of 12 steroids by isotope dilution liquid chromatography-photospray ionization tandem mass spectrometry. Clin Chim Acta. 2006;372:76–82. doi: 10.1016/j.cca.2006.03.034. [DOI] [PubMed] [Google Scholar]