

Abstract
The sirtuins (SIRTs) have gained preeminence for their roles in the response to caloric restriction and the regulation of aging and lifespan. A new study now identifies gene promoters that bind the transcription factor AP1 as targets for silencing by SIRT6, providing possible links between SIRT6 deficiency and dysregulation of insulin-like growth factor signaling, hypertrophic cardiomyopathy and heart failure (pages 1643–1650).
The SIRTs are a family of class III histone deacetylases (HDACs), distinguished from other HDAC classes by their requirement for nicotinamide adenine dinucleotide (NAD) in the deacetylation reaction. SIRTs can catalyze deacetylation of histone and nonhistone lysines. The requirement for NAD constitutes an ancient and evolutionarily conserved mechanism that may link the expression of SIRT-regulated genes with metabolism and nutritional state1.
SIRTs were first discovered in yeast, in which an extra copy of SIR2 was shown to extend lifespan by 50%, whereas its deletion shortened lifespan1. In worms (Caenorhabditis elegans) and flies (Drosophila melanogaster) the respective SIR2 homologs seemed to similarly regulate lifespan, and in worms this regulation was found to be mediated by DAF-16, a homolog of human Forkhead (FOXO) transcription factors that regulate stress-related genes, apoptotic factors, antioxidants and metabolism2, 3. Regulation of glucose homeostasis is a recurring theme linking SIRTs with caloric restriction, aging and lifespan throughout evolution. Daf2, an insulin-like receptor that regulates PI3K, also regulates DAF-16 and is downregulated by Sir2 or caloric restriction4.
Compelling evidence now implicates each of the mammalian nuclear SIRTs 1, 6 and 7 in the regulation of aging, with links to metabolism, caloric restriction and, especially in the case of SIRT6, glucose homeostasis and the insulin and insulin-like growth factor (IGF) signaling pathways5, 6. In this issue of Nature Medicine, Sundaresan et al.7 now directly link SIRT6 with heart disease, which is currently the main determinant of human lifespan in Western populations. Their work shows that SIRT6 selectively downregulates multiple components of the IGF pathway in mouse hearts by binding the transcription factor c-Jun and directing its deacetylase activity selectively to gene promoters with binding sites for the transcription factor Fos/Jun (AP1). The authors show that SIRT6 expression is decreased in failing human hearts, and, in parallel transgenic mouse models, they present plausible evidence that SIRT6 deficiency–induced chronic activation of the IGF signaling pathway and its effectors promotes hypertrophic cardiomyopathy, adverse cardiac remodeling and heart failure (Fig. 1).
Figure 1. Derepression of IGF signaling–related genes and hypertrophic cardiomyopathy by SIRT6 deficiency.
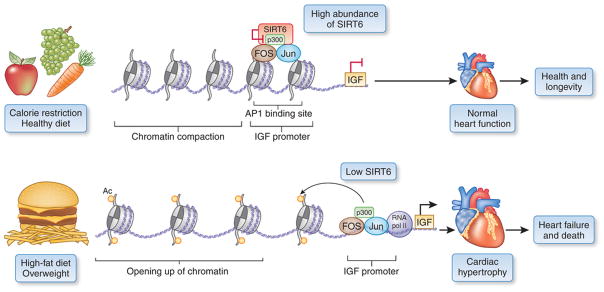
Sundaresan et al.7 provide evidence to suggest that in healthy individuals SIRT6 suppresses IGF signaling–related genes by binding and silencing expression from AP1-dependent promoters, and this contributes to metabolic homeostasis and the maintenance of a healthy heart. Low expression of SIRT6 possibly caused by a high-fat diet, sedentary lifestyle and/or genetics results in reduced SIRT6 silencing, and overexpression of IGF and possibly also of other AP1-dependent genes, thus promoting cardiomyopathy, heart failure and early death. Ac, acetylated H3K9.
SIRT6-deficient mice show the most severe phenotype of all SIRT gene knockouts, with premature aging that includes features of osteoporosis, spinal curvature, absence of subcutaneous fat and severe metabolic imbalance, lymphopenia and acute onset hypoglycemia that results in death of mice within 1 month of age2. In contrast, transgenic overexpression of Sirt6 protects mice against the adverse consequences of a high-fat diet, including the ‘metabolic syndrome’ (characterized by obesity, insulin resistance, elevated serum triglycerides and cholesterol), which is commonly considered a forerunner of cardiovascular disease6.
SIRT6 selectively targets and deacetylates histone H3 lysine 9 (H3K9), and before the study by Sundaresan et al.7 three SIRT6-specific targets had been identified that contribute to metabolic dysfunction and aging. The first targets to be discovered were H3K9 sites on human telomeres, at which deacetylation by SIRT6 was required to prevent telomere-dependent genome instability and premature senescence8. Subsequently, it was shown that the RELA subunit of the transcription factor NF-κB selectively bound SIRT6, thus directing its deacetylase function to the chromatin of NF-κB–dependent genes and resulting in their silencing9. More recently, SIRT6 was shown to bind and exert similar control over HIF1-α-regulated genes, and increased expression of genes encoding products such as glucose transporters and glycolytic enzymes in Sirt6-knockout mice substantially accounted for their hypoglycemia phenotype and premature death10.
The studies by Sundaresan et al.7 identify a new cardiomyopathy phenotype of SIRT6-deficient mice and shed additional light on the targets and mechanism of action of SIRT6 that may be relevant to heart disease and, consequently to life span, if the results extend to humans. Using Sirt6-knockout mice that survive up to 1 year of age (due to backcrossing the original 129sv strain with Black Swiss/FVB mice), the authors showed that these mice age more rapidly compared with wild-type mice on the same background and develop classical symptoms of hypertrophic cardiomyopathy (HCM), with contractile dysfunction, chamber dilation and, ultimately, heart failure. To confirm that SIRT6 has a heart-selective role, the authors showed that cardiac-specific deletion of Sirt6 conferred the same cardiomyopathy as whole-body knockout of Sirt6. The Sirt6-knockout phenotype includes defects in mitochondrial structure and organization, increased apoptosis and overexpression of fetal cardiac genes, typical of classical HCM. Sundaresan et al.7 then showed that SIRT6-overexpressing transgenic mice were protected from HCM that was either driven pharmacologically or caused by pressure overload.
The authors attribute the cardiomyopathy in Sirt6-knockout mice to overactivity of the IGF–Akt pathway caused by SIRT6 deficiency. Whereas acute activation of this pathway is known to be protective in the setting of acute myocardial infarction, there is also evidence that chronic IGF–Akt overactivity mediates hypertrophic growth11. Sundaresan et al.7 found that the hearts of Sirt6-knockout mice had elevated levels of the pathway members IGF-1R, InsR, Akt, GSK3 and IGF-2 and increased activities of multiple signaling components and downstream effectors, including eIF4E, S6P and p70S6K. They found no hypoglycemia and no changes in the expression of NF-κB– or HIF-1α–dependent genes in these mice. Nicotinamide, a sirtuin inhibitor, and PQ401, a partially selective inhibitor of IGF signaling, both rescued the cardiomyopathy in Sirt6-knockout mice, supporting direct roles for SIRT6 and the IGF pathway in heart disease.
To define the underlying mechanism, the authors used chromatin immunoprecipitation assays to show that SIRT6 binds the promoters of IGF-related genes, and they found that H3K9 at these sites was hypoacetylated in Sirt6-knockout mice. Using in silica analyses, they revealed that AP1-binding sites are common to multiple IGF-responsive gene promoters, implicating these as candidate targets for SIRT6. Indeed, the results confirm that the AP1 component c-Jun is a binding partner for SIRT6, allowing its deacetylase activity to be selectively delivered to AP1 sites of IGF-related genes in the heart and theoretically allowing the complex to bind and fine-tune all promoters with active AP1-binding sites. Knockdown of c-Jun decreased the association of SIRT6 with IGF-associated genes, and inhibition of AP1 downregulated the expression of the same genes in the Sirt6-knockout mice.
If the results of Sundaresan et al.7 extrapolate to the human heart, SIRT6 may be a new susceptibility marker for HCM and, possibly, a therapeutic target. The regulation of SIRT6 and dysregulation during HCM becomes an important issue. Clearly, energy state and diet could have roles because SIRT6 expression is responsive to caloric restriction. Interestingly, SIRT1 is also regulated by caloric restriction and can bind and activate the SIRT6 promoter, possibly amplifying the response to caloric restriction, diet or both12. AP1 is now the third SIRT6-regulated transcription factor to be identified, and, along with NF-κB and HIF-1α, it also regulates major stress response pathways. It may also be pertinent that the opposing histone acetyltransferase activity for SIRT6 at genomic sites that bind these transcription factors is the adaptor protein p300/CREB-binding protein (CBP), which is known to have a major role in HCM. This may be a coincidence or there may be a selective regulatory relationship between SIRT6 and p300/CBP, a possibility that deserves further investigation.
Footnotes
Competing financial interests
The author declares no competing financial interests.
References
- 1.Ralser M, et al. Front Pharmacol. 2012;3:32. doi: 10.3389/fphar.2012.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haigis MC, Guarente LP. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 3.Oellerich MF, Potente M. Circ Res. 2012;110:1238–1251. doi: 10.1161/CIRCRESAHA.111.246488. [DOI] [PubMed] [Google Scholar]
- 4.Herndon LA, et al. Nature. 2002;419:808–814. doi: 10.1038/nature01135. [DOI] [PubMed] [Google Scholar]
- 5.Xiao C, et al. J Biol Chem. 2010;285:36776–36784. doi: 10.1074/jbc.M110.168039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanfi Y, et al. Aging Cell. 2010;9:162–173. doi: 10.1111/j.1474-9726.2009.00544.x. [DOI] [PubMed] [Google Scholar]
- 7.Sundaresan NR, et al. Nat Med. 2012;18:1643–1650. doi: 10.1038/nm.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michishita E, et al. Nature. 2008;452:492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawahara TL, et al. Cell. 2009;136:62–74. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong L, et al. Cell. 2010;140:280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu CH, et al. Endocrinology. 2009;150:2723–31. doi: 10.1210/en.2008-0975. [DOI] [PubMed] [Google Scholar]
- 12.Kim HS, et al. Cell Metab. 2010;12:224–236. doi: 10.1016/j.cmet.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]