

Abstract
Background
Our previous studies indicated that NMDA receptor (NMDAR) deletion from a subset of corticolimbic interneurons in the mouse brain during early postnatal development is sufficient to trigger several behavioral and pathophysiological features resembling the symptoms of human schizophrenia. Interestingly, many of these behavioral phenotypes are exacerbated by social isolation stress. However, the mechanisms underlying the exacerbating effects of social isolation are unclear.
Methods
Using GABAergic interneuron-specific NMDAR hypofunction mouse model (Ppp1r2-cre/fGluN1 KO mice), we investigated whether oxidative stress is implicated in the social isolation-induced exacerbation of schizophrenia-like phenotypes and further explored the underlying mechanism of elevated oxidative stress in KO mice.
Results
The reactive oxygen species (ROS) level in the cortex of group-housed KO mice was normal at eight weeks although increased at 16 weeks old. Post-weaning social isolation (PWSI) augmented the ROS levels in KO mice at both ages, which was accompanied by the onset of behavioral phenotype. Chronic treatment with apocynin, an ROS scavenger, abolished markers of oxidative stress and partially alleviated schizophrenia-like behavioral phenotypes in KO mice. Markers of oxidative stress following PWSI were especially prominent in cortical parvalbumin (PV)-positive interneurons. The vulnerability of PV interneurons to oxidative stress was associated with down-regulation of peroxisome proliferator-activated receptor α coactivator-13 (PGC-1α), a master regulator of mitochondrial energy metabolism and antioxidation.
Conclusions
These results suggest that a PWSI-mediated impairment in antioxidant defense mechanisms, presumably mediated by PGC-1α downregulation in the NMDAR-deleted PV-positive interneurons, results in oxidative stress, which, in turn, may contribute to exacerbation of schizophrenia-like behavioral phenotypes.
Keywords: interneuron, NMDA receptor hypofunction, oxidative stress, parvalbumin, PGC-1α, schizophrenia, social isolation, transgenic mice
While the etiology of schizophrenia is not fully elucidated, genetic and environmental factors appear to interact in its onset and/or course (1). For example, the 41–65% monozygotic twin concordance rate (2) suggests that environmental triggers play a significant role. Epidemiological studies show that these triggers include social disadvantage, such as compromised family structures and restricted social networks (3). However, the exact molecular mechanisms linking environmental or social factors to the precipitation of schizophrenia psychosis still remain unclear.
In rodents, the social isolation paradigm models the impact of environmental insults on brain function at the cellular and molecular levels, which is thought to be mediated by cellular oxidative stress (4,5). Oxidative stress occurs when antioxidant defense mechanisms fail to counterbalance and control endogenous reactive oxygen species (ROS) or reactive nitrogen species (RNS), which are generated from normal oxidative metabolism or from pro-oxidant environmental exposure. Overproduction of ROS and RNS can cause oxidative damage of lipids, proteins and DNA.
Schizophrenia patients exhibit increased lipid peroxidation and protein modification and reduced antioxidant enzyme levels, suggesting that oxidative stress plays a role in the pathogenesis of schizophrenia (6–10). Oxidative stress has also been implicated in animal models of psychiatric disorders (11). Chronic perinatal administration of the NMDA receptor (NMDAR) antagonist phencyclidine reduced glutathione levels and produced long-term alteration of antioxidant defense in the corticolimbic areas of the rat brain (12). Similarly, repetitive exposure to ketamine, another NMDAR antagonist, activated the superoxide-producing enzyme NADPH oxidase Nox2, accompanied by schizophrenia-like pathophysiology including decreased expression of glutamic acid decarboxylase-67 (GAD67) and parvalbumin (PV) and reduced inhibitory tone in the medial prefrontal cortex (mPFC) (13,14).
Previously we generated Ppp1r2-cre/floxed-GluN1 (NR1) knockout mice with early postnatal deletion of NMDARs from 40–50% of corticolimbic interneurons, >70% of which are PV-positive, and these mutants displayed schizophrenia-like behavioral and pathophysiological features (15). Social isolation exacerbated many of these symptoms, such as deficits in nest building, mating, anhedonia, and anxiety-like behaviors. In the present study, we used Ppp1r2-cre/fGluN1 KO mice to investigate whether and through what mechanisms social isolation-induced oxidative stress contributes to schizophrenia-like phenotypes in these mice.
Materials and Methods
All experimental procedures were carried out in accordance with guidelines published in the National Research Council Guide for the Care and Use of Laboratory Animals, and were approved by the National Institute of Mental Health Animal Care and Use Committee. For detailed experimental procedures, see Methods and Materials in Supplement 1.
Animals
Ppp1r2-cre/fGluN1 knockout mice or simply KO mice was generated as previously described (15). Briefly, the protein phosphatase 1, regulatory subunit 2 (Ppp1r2)-cre line was bred to a loxP-flanked GluN1 line (16) to elicit the GluN1 deletion from the postnatal second week in a subset of cortical and hippocampal Ppp1r2-positive interneurons, a majority of which are parvalbumin-containing. Double in situ hybridization immunocytochemistry showed that, at postnatal day 14, no GluN1 mRNA was detected in ~30% of Cre-targeted interneurons in the S1 cortex of Ppp1r2-Cre/homozygously-floxed-GluN1 knockout mice (Zhang S., BS, unpublished, 2012). GluN1 deletion appeared to be completed by postnatal day 21. In contrast, there was no GluN1 deletion in the homozygously-floxed GluN1 (fGluN1 or flox) or Ppp1r2-cre littermate control mice. All mice were maintained after backcrossing to C57BL/6NTac (B6) strain six times. Female KO mice were crossed with homozygously-floxed GluN1 male mice. Wild-type B6 mice were also used for PGC-1α in situ hybridization.
Post weaning social isolation (PWSI)
Both male and female mice were used for immuno-histological and biochemical studies, and only male mice for behavioral studies. At weaning (postnatal day 21), pups were separated from their mothers and reared either in social isolation (two mice were separated by a median barrier in one mouse cage) or in social groups (2 to 5 mice per cage). Mice were given access to food and water ad libitum. This experimental protocol lasted until animals were used for behavioral, biochemical, or histological analysis. No obvious differences were detected between sex in histological and biochemical studies.
In vivo detection of reactive oxygen species level
In vivo detection of reactive oxygen species (ROS) in mouse brains was performed as previously described (13). Briefly, two serial i.p. injections of freshly prepared dihydroethidium (DHE, 27 mg/kg, Invitrogen) were given at 30 min intervals. Eighteen hr later, mice were perfused with 4% paraformaldehyde in PBS. Brains were coronally sectioned on a vibratome with 35-μm thickness and counterstained with DAPI (4′,6-diamidino-2-phenylindole). Six coronal sections from each animal were observed under a confocal microscope (mPFC - bregma −2.10 mm and −1.54 mm; S1 cortex −1.10 mm, 0.02 mm, 0.82 mm, and 1.82 mm). Red fluorescence intensity of oxidized DHE by ROS was quantified with NIH Image-J after converting confocal images to gray scale. Relative fluorescence intensity from mPFC or S1 area (covering through layer I–VI) was normalized by the average value from age-matched group-housed fGluN1 controls.
Chronic treatment with apocynin and behavioral analysis
From the age of 2 weeks, male KO mice and their littermate fGluN1 control mice received apocynin (APO, 1-(4-Hydroxy-3-methoxyphenyl) ethanone, Sigma-Aldrich, St. Louis, MO) in drinking water (7.5μg/ml) until behavioral evaluation (Figure 2A). Given that mice with body weight of 20 g drink 13 ml of water per day (13), APO was orally administered at 5 mg/kg per day. Since studies showed no detectable decay of apocynin at room temperature in 7 days (17), freshly prepared APO water bottles were replaced weekly. Apocynin is an antioxidant and scavenger of ROS (18) although it may act as an NADPH oxidase inhibitor in the presence of myeloperoxidase activity (19). Group-housed or PWSI animals underwent the following behavioral tests, the protocol of which were previously described (15; see Supplement 1): 1) the elevated plus maze and open field tasks at 8 weeks of age; 2) the spontaneous alternation Y-maze test and pre-pulse inhibition at 8 and 12 weeks of age; 3) acoustic startle reflex at 12 weeks of age; 4) the saccharine preference test at 14 weeks of age; 5) the nest building behavioral test at 8, 12, and 16 weeks of age.
Non-radioisotopic double in situ hybridization
DNA fragments corresponding to murine PV cDNA (GenBank accession number: NM_013645) and PGC-1α cDNA (GenBank accession number: NM_008904.2) were amplified by reverse transcription polymerase chain reaction (RT-PCR) using the following primers: 5′ GGGCCTGAAGAAAAAGAACC 3′ and 5′AGTACCAAGCAGGCAGGAGA 3′ for PV (20), and 5′GACAGTGTGTGTGTGTGTGTCC 3′ and 5′ TATCAGAGGCCATGCTAGTGAA 3′ for PGC-1α (exon 13). To detect mouse PV and PGC-1α mRNAs, the complementary RNA probe for PV was labeled with 2, 4-dinitrophenyl (DNP), and the complementary RNA (cRNA) probe for PGC-1α was labeled with digoxygenin (DIG). Double non-radioisotopic in situ hybridization was performed as previously described (15).
Measurement of superoxide dismutase activity
Medial PFC tissue was dissected and homogenized in cold PBS (10% w/v) with a tissue sonicator. The supernatant was assayed for superoxide dismutase (SOD) using a determination kit (Sigma-Aldrich) after centrifugation at 21,000 g for 10 min at 4°C as per the manufacturer’s instructions.
Statistics
Differences between groups were assessed for normally distributed data using a Student’s t-test (two groups), one- or two-way ANOVA, or repeated measures ANOVA followed by post-hoc tests for group comparisons. Data were presented as mean ± s.e.m. Statistical analyses were conducted using Statistica 7.0 (StatSoft Inc., Tulsa, OK).
Results
Post-weaning social isolation (PWSI) augmented cortical ROS levels in KO mice
To investigate the involvement of oxidative stress in Ppp1r2-cre/fGluN1 KO mice, cortical ROS levels in brain sections from KO animals (8- and 16-week-old) were examined after i.p. injection of dihydroethidium (DHE). DHE has been used to monitor cortical ROS production in vivo (13), because DHE is oxidized by ROS, forming ethidium bromide, which emits red fluorescence once it intercalates with DNA (21). A prominent increase of cortical ROS level was observed in group-housed KO animals compared with fGluN1 or Ppp1r2-cre (Figure S1A, B) controls at 16 but not at 8 weeks old (Figure 1). However, PWSI sharply augmented ROS levels in mPFC and S1 cortex of KO mice both at 8 and 16 weeks of age. Overall, the oxidized DHE signals were neither excitatory nor inhibitory neuron-specific, as assessed by excitatory neuronal marker neurogranin and various interneuron markers, such as parvalbumin, calretinin, and calbindin (Figure S1C in Supplement 1).
Figure 1.

Elevated cortical reactive oxygen species (ROS) production in Ppp1r2-cre/fGluN1 KO mice following post-weaning social isolation (PWSI). (A & B) PWSI exacerbated cortical ROS production in KO animals and chronic treatment with apocynin (+APO), an ROS scavenger, diminished cortical ROS production. Floxed-GluN1 (flox) or KO animals that underwent group-housing (group) or PWSI until 8 or 16 weeks old were injected with dihydroethidium (DHE) (4~5 animals per group). ROS levels in mPFC (A) and S1 cortex (B) were visualized by oxidized DHE (in red) in sections counterstained with DAPI (blue). For drug treatment, animals were subjected to apocynin treatment (+APO) from postnatal 2 weeks (4~5 animals per group) until injected with DHE at the age of 8 weeks or 16 weeks. (C & D) Relative ROS levels in mPFC (C) and S1 cortex (D) were quantified and normalized to the levels of 8 week-old group-housed fGluN1 controls. Without APO pretreatment, regardless of brain areas, PWSI ROS levels were elevated only in 8-week-old KO mice. PWSI also exacerbated the ROS elevation in the KO mice at 16 weeks old. Two-way ANOVA, F(1,12)=6.36, p= .027 for genotype x housing condition interaction at 8 week-old in mPFC. F(1,14)=14.01, p= .002 for genotype at 16 week-old in mPFC. F(1,12)=7.75, p= .017 for genotype x housing condition interaction at 8 week-old in S1 cortex. F(1,13)=5.39, p= .037 for genotype at 16 week-old in S1 cortex. Chronic APO treatment reversed the PWSI-induced ROS elevation at both ages in both brain regions. Post-hoc Fisher least significant difference test, *p < .05, #p< .01. Scale bar in (B): 50 μm.
Next we tested whether apocynin (APO), an antioxidant and ROS scavenger, alleviated the elevation of cortical ROS levels in KO mice after PWSI. Animals were treated with APO from P14 and subjected to PWSI from P21 (Figure 2A). Cortical ROS levels in PWSI KO mice were largely abolished by chronic APO treatment (Figures 1C & D, decrease to 35.4 ± 11.1% in mPFC and decrease to 28.7 ± 5.0% in S1 cortex).
Figure 2.

Oxidative stress exacerbated schizophrenia-like behavioral phenotypes in KO mice. (A) Diagram of the ages of the animals used for apocynin treatment, social isolation and behavioral tests. (B) Chronic treatment with APO rescued the anxiety-like behavior of KO animals at 8 weeks of age following PWSI. Group-housed KO mice did not show anxiety-like behavioral; however, PWSI reduced a time spent in open arms by KO mice, but not by fGluN1 controls. F(1, 40) = 6.25, p= .0166. Post-hoc Fisher’s LSD test, flox vs KO, *p < .02 for PWSI). APO treatment increased KO’s stay time in open arms to the control level after PWSI (F(1, 51) = 6.20, p = .016 for genotype x drug treatment interaction, post-hoc Fisher LSD test, *p < .01.
(C) Novelty-induced hyperlocomotion of KO animals was unaffected by APO. Repeated measures ANOVA showed significant genotype effect, F(10, 33) = 2.36, p = .031; but no drug effect, F(10, 33) = .70, p = .715, post-hoc Fisher’s LSD test, *p < .05, KO vs flox.
(D) Chronic APO treatment rescued Y-maze spontaneous alternation deficits of KO mice at 12 weeks old. Before drug treatment, KO animals had significant reductions in alternation compared to flox controls regardless of housing conditions, and PWSI exacerbated the deficit (F(1,54) = 16.67, p = .00015 for genotype effect, F(1,54) = 8.47, p = .0052 for housing condition effect. This deficit in KO mice was reversed by APO treatment (F(1, 51)=4.49, p= .039 for drug effect, F(1, 51)=6.02, p= .018 for genotype effect. Post-hoc Fisher LSD, *p< .05, #p< .01.
(E) Impaired PPI in KO animals at 12 weeks of age was partially rescued by APO treatment. Without APO treatment, KO mice showed PPI deficits, in particular at 72 and 74 dB prepulse intensities, irrespective of PWSI. Two-way repeated measure of ANOVA, F(4, 62)=4.88, p= .0017 for genotype effect, F(4.62)=0.32, p= .86 for genotype x housing condition interaction. Interestingly, PWSI impaired PPI of fGluN1 (flox) controls at 72 dB. APO treatment reversed the impairment of PPI at 74 dB~78 dB for PWSI KO mice, but not at 72 dB. PPI deficits at 72dB for PWSI flox controls were also alleviated by APO treatment. Post-hoc Fisher LSD test, *p< .05, #p< .01.
(F) APO did not affect auditory startle reflex of the mice at the age of 12 weeks, regardless of genotype, housing condition, or drug treatment.
(G) PWSI impaired hedonic-like/reward-seeking behavior of KO mice, but not fGluN1 controls, in saccharine preference test and APO treatment alleviated this deficit. Before drug treatment, F(1, 42)=4.30, p= .044 for genotype x housing condition interaction in the 0.03% saccharine solution test. After chronic treatment with APO, F(1, 43)=7.44, p= .0092 for drug effect in 0.03% saccharine solution test. Post-hoc Fisher LSD test, *p < .05, #p< .01 vs other groups.
(H) Nest building deficits precipitated by PWSI were partially reversed by APO treatment. Before drug treatment, F(1, 198)=38.54, p< .00001 for genotype effect and F(1, 198)=9.35, p= .0025 for genotype x housing condition interaction. Post-hoc Fisher’s LSD test, *p< .05, compared with flox controls; ¶ p< .001, PWSI KO vs group-housed KO. APO partially alleviated nesting deficits at 12 or 16 week-old (F(1, 191)=47.25, p< .00001 for genotype effect, and F(1, 191)=5.31, p= .022 for APO effect. #p < .05, KO (+APO) vs KO (−APO); *p< .05, KO (+APO) vs flox (+APO)). Number of animals is indicated in parentheses or inside plot bars. ns, no significant difference; other abbreviations as in Figure 1.
Oxidative stress exacerbated schizophrenia-like behavioral phenotypes
Behavioral tasks were conducted after chronic treatment with APO (Figure 2A) to investigate whether a reduction in PWSI-induced oxidative stress was associated with behavioral improvement. APO treatment significantly improved performance on tests of anxiety (Figure 2B), spatial memory (Figure 2D), prepulse inhibition (Figure 2E), saccharine preference (Figure 2G), and nest building (Figure 2F), while having no effect on overall activity (Figure 2C) or startle responses (Figure 2F). Considering the actions of APO, these findings suggest that PWSI mediates its effects on behavior by enhancing oxidative stress.
Concordant with the emergence of oxidative stress by PWSI, the time spent in unprotected open arms of the PWSI KO mice in the elevated plus maze was significantly reduced compared to that of fGluN1 controls, an impairment absent in group-housed KO mice of the same age (Figure 2B). Chronic APO treatment alleviated this PWSI-induced anxiety-like behavior in the KO mice (Figure 2B).
Spatial working memory deficits were evaluated using the spontaneous Y-maze alternation task, which relies on the natural tendency of mice to alternate the choice of maze arms. At 8 weeks of age this natural tendency was not as pronounced or working memory ability was not fully matured, because the group-housed fGluN1 controls alternated less at 8 weeks old than at 12 weeks old (Figure S2A in Supplement 1). Thus we tested KO animals at 12 weeks old. At this age, group-housed KO mice displayed a reduction in alternation which was exacerbated by PWSI (Figure 2D), suggesting a spatial working memory deficit and echoing the results of our previous study (15). Notably, chronic oral APO administration for 10 weeks increased the alternation index of PWSI KO mice to a similar level as the PWSI control (Figure 2D).
Prepulse inhibition (PPI) of the acoustic startle reflex is a measure of sensorimotor gating. In this task, as in the spatial Y-maze, the control mice performance was not fully mature at 8 weeks old, performance significantly increased at 12 weeks of age (Figure S2B in Supplement 1). Therefore, we tested at 12 weeks old, where KO mice were significantly impaired in PPI (Figure 2E) albeit with normal startle reflex amplitudes (Figure 2F). APO treatment only partially rescued PPI deficits; KO mice showed normal PPI at 74 dB and 78 dB (Figure 2E). This was presumably because the genotype effect on PPI deficits is more robust than the social isolation environmental effect. Indeed, we found no effect of PWSI or adolescent social isolation on the extent of PPI deficits of KO mice (Figure S2C in Supplement 1).
The KO mice regardless of housing conditions showed novelty-induced hyperlocomotion at 8 weeks of age in the first three minutes of spontaneous exploration in the open field test (Figure 2C). Like PPI, this effect was not rescued by APO treatment.
A two-bottle saccharine preference tested the hedonic-like/reward-seeking behavior. While KO mice at 14 weeks old show no deficit under group-housed condition, they displayed a reduced preference for sweet solutions following PWSI (Figure 2G) with no change in total fluid intake (Figure S2D in Supplement 1). Chronic treatment of APO increased their preference for saccharine to the control level.
Nest building behavior assessed social behavior. Without drug treatment, fGluN1 control mice consistently formed an identifiable nest in a new cage from all of the provided cotton nestlet. In contrast, KO mice showed a robust deficit at both 12 and 16 weeks, and PWSI exacerbated their impairment (Figure 2F). Interestingly, APO reduced the amount of unused nestlet to a similar level as the group-housed KO mice.
PWSI exacerbated oxidative stress in cortical PV interneurons in KO mice
The consequences of oxidative stress were examined by DNA oxidation and lipid peroxidation. Immunostaining with anti-8-hydroxy-2′-deoxyguanosine (8-OH-dG) (Figure 3A) and anti-4- Hydroxy-2-nonenal (4-HNE) (Figure 3B), which are markers for DNA oxidative damage and lipid peroxidation, respectively, revealed significantly increased immunoreactivity of 8-OH-dG and 4-HNE in the cortex of 8-week-old KO mice after PWSI compared with age-matched fGluN1 controls. Interestingly, cortical PV interneurons in PWSI-exposed KO animals showed prominent staining with anti-8-OH-dG (Figure 3A) and anti-4-HNE (Figure 3B), suggesting that PV interneurons in KO animals are highly vulnerable to oxidative stress. Moreover, consistent with non-specific staining of DHE (Figure S1C in Supplement 1), weak 8-OH-dG and 4-HNE immunostaining was present in non-PV neurons in the KO animals after PWSI, presumably in the excitatory neurons.
Figure 3.

Post-weaning social isolation (PWSI) increased DNA and lipid oxidative stress in KO mice, particularly in cortical PV interneurons. Brain sections containing S1 cortex from group-housed or PWSI-treated flox control and KO animals at 8 weeks old were double-stained with PV antibody (red) and DNA oxidative stress marker 8-OH-dG (green in A), or lipid peroxidation marker 4-HNE (green in B). Scale bar: 50 μm (A) and 200 μm (B).
To further evaluate the impact of oxidative stress on PV interneurons, we quantified parvalbumin-IR (PV-IR). Overall PV-IR intensity was significantly reduced in mPFC and S1 cortex of KO mice after 5 weeks of PWSI, but not in fGluN1 controls (Figure 4). Unlike PV, fluorescence immunoreactivity intensity for calretinin (Figure S3A, C in Supplement 1) and calbindin (Figure S3B, C in Supplement 1), another calcium binding proteins, did not differ between experimental groups. Interestingly, reduction of PV-IR upon PWSI in KO mice was prevented by chronic treatment of apocynin (Figure 4), suggesting that reduced PV expression in PWSI KO mice was associated with elevated oxidative stress in PV neurons.
Figure 4.
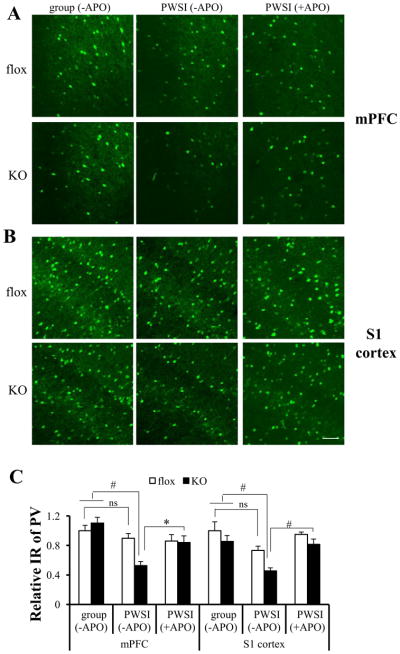
Cortical PV expression in KO mice was down-regulated by post-weaning social isolation (PWSI) and was reversed by chronic treatment of apocynin (+APO). (A, B) Brain sections from group-housed or PWSI animals or PWSI animals under APO treatment (4~6 animals for each group, 8 week-old) were immunostained with parvalbumin (PV)antibody and visualized by Alexa488 (green fluorescence). (C) PV expression was significantly decreased in the mPFC and S1 cortex of KO animals after PWSI, but not in fGluN1 (flox) controls. However, PV IR reduction in KO animals can be alleviated by chronic treatment of apocynin. Post-hoc Tukey’s HSD test following two-way ANOVA, #p < .01, *p < .05. Scale bar: 100 μm. ns: no significant difference; other abbreviations as in Figure 1 and 3.
PWSI downregulated cortical PGC-1α in KO mice
To investigate possible causes of increases in oxidative stress and concomitant PV-IR decrease in the PV-positive interneurons, we evaluated the expression levels of the transcriptional coactivator PGC-1α (peroxisome proliferator-activated receptor γ coactivator α). PGC-1α, a master regulator of mitochondrial energy metabolism and anti-oxidation (22), is activated by ROS overproduction and stimulates the transcription of ROS-detoxifying enzyme genes (23). Previous study has also suggested that PGC-1α appears to be necessary and sufficient for neuronal PV expression (24). First, to verify the PV interneuron-predominant expression of PGC-1α, we performed double in situ hybridization using DIG-labeled PGC-1α mRNA antisense probe and DNP-labeled PV mRNA antisense probes (Figure 5A, B). In wild-type control brains, almost all the PV-containing cells were PGC-1α-positive in S1 cortex (97.5% of PV cells in layer II/III and 100% in layer IV-VI), while of the total population of PGC-1α positive cells the majority were PV positive (88.5%, 22.5%, and 64.4% in layer II/III, layer IV/V, and layer VI, respectively in Figure 5B). Interestingly, PGC-1α mRNA signals were dramatically reduced following PWSI in the cortex of KO mice (Figure 5C). Quantitative RT-PCR using mPFC total RNA confirmed that PWSI elicits down-regulation of PGC-1α expression only in KO mice at 8 weeks old (Figure 5D). Reduced PGC-1α in KO mice following PWSI was also detectable at the protein level, as assessed by Western blots (Figure 5E). Interestingly, while PGC-1α mRNA levels were reduced at 16 weeks old regardless of housing conditions, APO treatment encompassing the entire PWSI period in KO mice alleviated the reduction (Figure 5F). As over 80% of cortical PV-positive cells in the KO mice are cre-targeted (15) and most of PV cells are PGC-1α positive, this suggested that the down regulation of PGC-1α in the KO mice are mostly in the GluN1-deleted neurons. While 24% of cre-targeted cells are Reelin positive, there was no PGC-1α expression in these Reelin-positive cre cells (Figure S4 in Supplement 1).
Figure 5.

PGC-1α in KO mice was down-regulated by post-weaning social isolation (PWSI). (A, B) DIG-labeled PGC-1α mRNA antisense probe was used to detect PGC-1α mRNA (visualized by NBT-BCIP, blue precipitate) (A) or together with DNP-labeled PV mRNA antisense probe (visualized by AEC, red precipitate) to detect PGC-1α expression in PV-positive interneurons (B). Purple arrows in B-1 and B-2 indicate neurons with both PGC-1α and PV expression. Blue arrows indicate neurons with PGC-1α expression only. No red cells were detected, suggesting that PGC-1α is expressed in all the PV neurons.
(C–E) Cortical expression of PGC-1α was significantly reduced in KO animals at 8 weeks old after PWSI. PGC-1α mRNA levels were examined by in situ hybridization using DIG-labeled PGC-1α mRNA antisense probe (C) and quantitative RT-PCR (5~6 animals for each group) with RNA from mPFC (D). Post-hoc Tukey’s HSD test following two-way ANOVA revealed no difference between group-housed GluN1 (flox) controls and KO animals, p = .958; however, PWSI exacerbated the reduction of PGC-1α mRNA in KO mice, but not in fGluN1 (flox) controls, *p < .02). PGC-1α protein levels in mPFC (5~8 animals for each group) were examined by Western blot (E). Post-hoc Tukey’s HSD test following two-way ANOVA revealed no difference between group-housed fGluN1 (flox) and KO animals at 8 week-old (p = .254); however, PWSI exacerbated the reduction of PGC-1α protein in KO mice, but not in fGluN1 (flox) controls, #p < .003. (F) A reduction in PGC-1α protein levels was observed in both group-housed and PWSI KO mice at the age of 16 weeks, F(1, 17)=34.90, p< .0001 for genotype effect, #p< .01, post-hoc Tukey’s HSD test. Chronic treatment with APO reversed the PGC-1α protein reduction in PWSI KO mice. Post-hoc Fisher LSD test, *p< .05. Scale bar: 200 μm. ns: no significant difference; other abbreviations as Figure 1.
KO mice were impaired in the cortical antioxidant defense system
Several key ROS-detoxifying enzymes, such as superoxide dismutase 1 (SOD1), SOD2, catalase 1 (CAT1), and glutathione peroxidase 1 (GPX1), function as downstream gene targets of PGC-1α (23). We therefore examined the basal transcription level of these antioxidant defense system genes in brain using quantitative RT-PCR. The mRNA expression levels of SOD1, SOD2, CAT1, and GPX1 were considerably reduced in mPFC of KO mice after PWSI compared with group-housed fGluN1 controls (Figure 6A). In contrast, no significant reduction in the mRNAs of these ROS-detoxifying enzymes was observed in either group-housed KO or PWSI-treated fGluN1 controls. Down-regulation of the antioxidant defense system was also confirmed by the evaluation of SOD enzyme activity (Figure 6B). These results suggested that the antioxidant defense system in the cortex is defective in KO mice following PWSI.
Figure 6.

Cortical ROS defense system was down-regulated by post-weaning social isolation (PWSI) in KO mice. (A) ROS-detoxifying enzymes GPX1, CAT1, SOD1, and SOD2 were down-regulated in the cortex of KO mice at 8 week-old after PWSI, as examined by quantitative RT-PCR using RNA from the medial prefrontal cortex (mPFC) (5~8 animals for each group). Post-hoc Fisher’s LSD test after two-way ANOVA indicated that expression of these enzymes in fGluN1 (flox) controls were not significant decreased after PWSI (p = .987 for GPX1, p =.750 for CAT1, p = .251 for SOD1, p = .146 for SOD2). However, the reduction was observed in KO mice. Post-hoc Fisher’s LSD test after two-way ANOVA, *p < .05, #p < .01. (B) PWSI exacerbated SOD activity (U/mg protein) in the mPFC of KO mice, not fGluN1 (flox) controls (animal number is indicated inside bars). Post-hoc Fisher’s LSD test after two-way ANOVA, *p < .05. ns: no significant difference; other abbreviations as Figure 1 and 5.
Discussion
The present study had several salient findings suggesting that a failure of the antioxidant mechanism may be critical to the environmental effect on schizophrenia onset. Specifically, (i) PWSI augmented cortical ROS production in Ppp1r2-cre/fGluN1 KO mice; (ii) scavenging ROS from the early postnatal period via chronic administration of APO alleviated some of behavioral deficits observed in the KO mice; (iii) oxidative stress was prominent in cortical PV-positive interneurons, a majority of which have genetically eliminated NMDARs; (iv) down-regulation of PGC-1α was associated with a downregulation of PV and increased ROS production. Taken together, these results suggest that failure of antioxidant defense mechanisms in fast-spiking PV interneurons led to excessive ROS production in the cortex, which exacerbates schizophrenia-like behavior in this animal model.
Vulnerability of PV interneurons to oxidative stress
Cortical PV interneurons have unique fast-spiking properties involved in driving synchronous oscillatory activity (25–28). To maintain high frequency firing, PV neurons contain high concentrations of cytochrome oxidase c and a greater number of mitochondria than pyramidal cells (29). Mitochondria in these fast-spiking neurons produce much more ROS and ATP compared to other cell types. Thus, fast-spiking neurons are equipped with potent anti-oxidation mechanisms to counterbalance increased ROS production. In the present study, cortical PV interneurons in Ppp1r2-cre/fGluN1 KO mice that underwent PWSI showed prominent staining in response to 8-OH-dG and 4-HNE, markers of DNA oxidation and lipid peroxidation, respectively. Cortical PV immunoreactivity, but not calretinin or calbindin immunoreactivity, was also reduced following PWSI in KO mice, which is consistent with recent findings indicating that PV neurons are vulnerable to chronic stress (4,30,31). These results suggest that PV interneurons in KO animals are highly vulnerable to oxidative stress.
PGC-1α is a master regulator of mitochondrial energy metabolism and anti-oxidation (22) and is known to work in concert with other proteins to drive the expression of ROS-detoxifying enzymes in response to ROS elevations (23). PGC-1α-mediated anti-oxidation is thus one possible anti-oxidation mechanism in PV neurons because it’s most highly concentrated in PV-positive neurons (32; Figure 5). Indeed, in KO mice after PWSI, we demonstrated down-regulation of cortical PGC-1α and several of its downstream targets important for antioxidant function. PGC-1α is also required for the normal expression of the calcium buffer PV (24); thus, as expected, decreases in PV expression accompanied decreases in PGC-1α in PWSI-exposed KO mice. Taken together, the data suggest that a decrease in PGC-1α abundance compromises multiple transcriptional pathways, augmenting oxidative stress in PV-containing fast-spiking neurons.
One important question concerns how PGC-1α expression is down-regulated in the cortex of KO mice following social isolation. Accumulating evidence suggests that neural expression of PGC-1α is activity-dependent and requires intracellular calcium influx. For example, depolarization of neurons by NMDAR activation or high-potassium solution increases PGC-1α expression (33), and NMDA antagonists or calcium channel blockade prevent such increases (34,35). Conversely, blockade of neuronal activity by tetrodotoxin or sensory deprivation decreases PGC-1α expression in the visual cortex (36). In the present study, it is possible that protracted maturation of the NMDAR-deleted PV neurons (Zsiros V., PhD, unpublished, 2012) might impair the activity-dependent expression of PGC-1α after social isolation. Additional studies are necessary to delineate the mechanisms involved.
Comparison to other animal models of psychiatric disorders
It was proposed by maternal immune activation (MIA) theory of schizophrenia that MIA stimulates microglia and enhances oxidative stress, which in turn diminishes neural functioning (6,37,38). Indeed, both post-mortem studies (39–42) and positron emission tomography (PET) study on recent-onset schizophrenia patients (43) has implicated the involvement of microglia in the pathophysiology of schizophrenia. Interestingly, involvement of microglial cells seems to be negligible in our NMDAR hypofunction mouse model. Neither proliferation of microglial cells (by anti-Iba1 staining, Figure S5C, D in Supplement 1), or microglial activation (by anti-CD68 staining, Figure S5E in Supplement 1), was observed in the cortex of Ppp1r2-cre/fGluN1 KO animals. Lack of microglial activation/proliferation in our mouse model may be due to the reason that the manipulation is targeted downstream of MIA in the developmental process of schizophrenia pathogenesis.
Behrens and colleagues elegantly demonstrated that Nox2 contributes to oxidative stress and dysfunction of GABAergic interneurons after subchronic ketamine exposure in mice (13,44). Furthermore, Sorce and colleagues (45) suggested that Nox2 is a major source of ROS production in mPFC following acute ketamine exposure. Neuronal production of interleukin-6 (IL-6) appears to be necessary and sufficient for ketamine-mediated activation of NADPH oxidase in mouse brains (44). However, Nox2 seems not to play a major role in ROS production in our KO mice, because PWSI-induced exacerbation of oxidative stress was still robustly observed in mPFC and S1 cortex of the Nox2-deleted Ppp1r2-cre/fGluN1 KO animals (Figure S5A, B in Supplement 1). Our preliminary result using quantitative real-time RT-PCR also showed no detectable increase in IL-6 or Nox2 mRNA level in cortical tissues of KO mice compared to the fGluN1 controls (Figure S5F in Supplement 1), suggesting that NADPH oxidase Nox2 is not the main source of the elevated ROS in our animal model. We postulate that the impaired antioxidant capacity of PV interneurons is due to PGC-1α down-regulation enhanced ROS levels in response to social isolation. Alternatively, albeit not mutually exclusive, we do not exclude the possible contribution of other Nox family proteins to the ROS increase in PWSI KO mice. For example, NADPH oxidase Nox4 mRNA level appeared to be elevated in 16-week-old KO mice; otherwise at the level near the detection limit in the fGluN1 controls (Figure S5F in Supplement 1). Nox4 is known to produce hydrogen peroxide (46), cell membrane-permeable ROS (47), which might cause ROS elevation in many cell-types (Figure 1). Further study is necessary to clarify the cellular mechanisms of the ROS level elevation in the PWSI KO mice.
The interaction of genes and environment in schizophrenia-like NMDAR hypofunction
An interaction between genes and environment that impacts both early and late brain development may be necessary to trigger the onset of schizophrenia, which typically emerges in early adulthood (48). The present study demonstrated that PWSI-induced stress augmented cortical ROS levels in Ppp1r2-cre/GluN1 KO mice, and that ROS elevation is associated with the onset of schizophrenia-like symptoms. We further found that impaired antioxidant capacity due to PGC-1α down-regulation correlates with oxidative stress in PV interneurons in these KO mice. It is notable that no significant change in PGC-1α expression in the KO animals was observed until post-adolescent period (i.e., 8-week-old). Moreover, PWSI exacerbated the reduction of PGC-1α only in KO mice, which is consistent with a notion of gene x environment interaction. Interestingly, genetic association studies on chromosome 4p (49–51) implicated the PPARGC1A (gene for PGC-1α) locus in schizophrenia and bipolar disorder. Based on these findings, we postulate that PGC-1α is a possible candidate for the underlying mechanism of interplay between genetic predisposition and environmental insults in schizophrenia. In future experiments, it will be interesting to investigate whether overexpressing PGC-1α in Cre-targeted interneurons can alleviate oxidative stress and thereby rescue social isolation-exacerbated behavioral deficits.
It is notable that PWSI had little impact on the behavior of the fGluN1 control mice, except partial impairment of PPI (Figure 2E). PWSI was also unable to induce a robust oxidative stress response, measured by DHE (Figure 1), anti-8-OH-dG or 4-HNE staining (Figure 3). These signs of murine resilience to PWSI stress, as recently reported in wild-type mice (52), appear to be in contrast to the recent reports in rats (4,53); however, underlying mechanisms explaining species’ differences are unclear since few such studies have been published with mice (54).
Taken together, the results of the present study suggest that high vulnerability of corticolimbic fast-spiking interneurons to oxidative stress may provide a potential explanation for the key mechanism in the diathesis-stress (55) or “two hit” model (56) for the etiology of schizophrenia.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Mental Health (NIMH), National Institutes of Health (NIH), and MH077955-05 (RMC). We thank Ioline Henter and Stefan Kolata for their outstanding editorial assistance.
Footnotes
Conflict of Interest: The authors have no biomedical financial interests or potential conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fulker DW. A biometrical genetic approach to intelligence and schizophrenia. Soc Biol. 1973;20:266–275. doi: 10.1080/19485565.1973.9988053. [DOI] [PubMed] [Google Scholar]
- 2.Cardno AG, Gottesman II. Twin studies of schizophrenia: from bow-and-arrow concordances to star wars Mx and functional genomics. Am J Med Genet. 2000;97:12–17. [PubMed] [Google Scholar]
- 3.Veling W, Susser E. Migration and psychotic disorders. Expert Rev Neurother. 2011;11:65–76. doi: 10.1586/ern.10.91. [DOI] [PubMed] [Google Scholar]
- 4.Schiavone S, Sorce S, Dubois-Dauphin M, Jaquet V, Colaianna M, Zotti M, et al. Involvement of NOX2 in the development of behavioral and pathologic alterations in isolated rats. Biol Psychiatry. 2009;66:384–392. doi: 10.1016/j.biopsych.2009.04.033. [DOI] [PubMed] [Google Scholar]
- 5.Möller M, Du Preez JL, Emsley R, Harvey BH. Isolation rearing-induced deficits in sensorimotor gating and social interaction in rats are related to cortico-striatal oxidative stress, and reversed by sub-chronic clozapine administration. Eur Neuropsychopharmacol. 2011;21:471–483. doi: 10.1016/j.euroneuro.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 6.Do KQ, Cabungcal J-H, Frank A, Steullet P, Cuenod M. Redox dysregulation, neurodevelopment, and schizophrenia. Curr Opin Neurobiol. 2009;19:220–230. doi: 10.1016/j.conb.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Yao JK, Keshavan MS. Antioxidants, redox signaling, and pathophysiology in schizophrenia: an integrative view. Antioxid Redox Signal. 2011;15:2011–2035. doi: 10.1089/ars.2010.3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reddy R, Sahebarao MP, Mukherjee S, Murthy JN. Enzymes of the antioxidant defense system in chronic schizophrenic patients. Biol Psychiatry. 1991;30:409–412. doi: 10.1016/0006-3223(91)90298-z. [DOI] [PubMed] [Google Scholar]
- 9.Mukerjee S, Mahadik SP, Scheffer R, Correnti EE, Kelkar H. Impaired antioxidant defense at the onset of psychosis. Schizophr Res. 1996;19:19–26. doi: 10.1016/0920-9964(95)00048-8. [DOI] [PubMed] [Google Scholar]
- 10.Coughlin JM, Ishizuka K, Kano SI, Edwards JA, Seifuddin FT, Shimano MA, et al. Marked reduction of soluble superoxide dismutase-1 (SOD1) in cerebrospinal fluid of patients with recent-onset schizophrenia. Mol Psychiatry. 2013;18:10–11. doi: 10.1038/mp.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powell SB, Sejnowski TJ, Behrens MM. Behavioral and neurochemical consequences of cortical oxidative stress on parvalbumin-interneuron maturation in rodent models of schizophrenia. Neuropharmacology. 2012;62:1322–1331. doi: 10.1016/j.neuropharm.2011.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Radonjić NV, Knezević ID, Vilimanovich U, Kravić-Stevović T, Marina LV, Nikolić T, et al. Decreased glutathione levels and altered antioxidant defense in an animal model of schizophrenia: long-term effects of perinatal phencyclidine administration. Neuropharmacology. 2010;58:739–745. doi: 10.1016/j.neuropharm.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 13.Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, et al. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318:1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Behrens MM, Lisman JE. Prolonged exposure to NMDAR antagonist suppresses inhibitory synaptic transmission in prefrontal cortex. J Neurophysiol. 2008;100:959–965. doi: 10.1152/jn.00079.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dang MT, Yokoi F, Yin HH, Lovinger DM, Wang Y, Li Y. Disrupted motor learning and long-term synaptic plasticity in mice lacking NMDAR1 in the striatum. Proc Natl Acad Sci U S A. 2006;103:15254–15259. doi: 10.1073/pnas.0601758103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clinical Investigation. 2008;118:659–670. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heumüller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schröder K, et al. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 19.Simons JM, Hart BA, Ip Vai Ching TR, Van DH, Labadie RP. Metabolic activation of natural phenols into selective oxidative burst agonists by activated human neutrophils. Free Radic Biol Med. 1990;8:251–258. doi: 10.1016/0891-5849(90)90070-y. [DOI] [PubMed] [Google Scholar]
- 20.Tanahira C, Higo S, Watanabe K, Tomioka R, Ebihara S, Kaneko T, et al. Parvalbumin neurons in the forebrain as revealed by parvalbumin-Cre transgenic mice. Neurosci Res. 2009;63:213–223. doi: 10.1016/j.neures.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 21.Benov L, Sztejnberg L, Fridovich I. Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med. 1998;25:826–831. doi: 10.1016/s0891-5849(98)00163-4. [DOI] [PubMed] [Google Scholar]
- 22.Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 23.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 24.Lucas EK, Markwardt SJ, Gupta S, Meador-Woodruff JH, Lin JD, Overstreet-Wadiche L, et al. Parvalbumin deficiency and GABAergic dysfunction in mice lacking PGC-1α. J Neurosci. 2010;30:7227–7235. doi: 10.1523/JNEUROSCI.0698-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soltesz I, Deschênes M. Low- and high-frequency membrane potential oscillations during theta activity in CA1 and CA3 pyramidal neurons of the rat hippocampus under ketamine-xylazine anesthesia. J Neurophysiol. 1993;70:97–116. doi: 10.1152/jn.1993.70.1.97. [DOI] [PubMed] [Google Scholar]
- 26.Klausberger T, Magill PJ, Márton LF, Roberts JD, Cobden PM, Buzsáki G, et al. Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature. 2003;421:844–848. doi: 10.1038/nature01374. [DOI] [PubMed] [Google Scholar]
- 27.Hajos N, Palhalmi J, Mann EO, Nemeth B, Paulsen O, Freund TF. Spike timing of distinct types of GABAergic interneuron during hippocampal gamma oscillations in vitro. J Neurosci. 2004;24:9127–9137. doi: 10.1523/JNEUROSCI.2113-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cunningham MO, Hunt J, Middleton S, LeBeau FE, Gillies MJ, Davies CH, et al. Region-specific reduction in entorhinal gamma oscillations and parvalbumin-immunoreactive neurons in animal models of psychiatric illness. J Neurosci. 2006;26:2767–2776. doi: 10.1523/JNEUROSCI.5054-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gulyás AI, Buzsáki G, Freund TF, Hirase H. Populations of hippocampal inhibitory neurons express different levels of cytochrome c. Eur J Neurosci. 2006;23:2581–2594. doi: 10.1111/j.1460-9568.2006.04814.x. [DOI] [PubMed] [Google Scholar]
- 30.Czeh B, Simon M, van der Hart MG, Schmelting B, Hesselink MB, Fuchs E. Chronic stress decreases the number of parvalbumin-immunoreactive interneurons in the hippocampus: prevention by treatment with a substance P receptor (NK1) antagonist. Neuropsychopharmacology. 2005;30:67–79. doi: 10.1038/sj.npp.1300581. [DOI] [PubMed] [Google Scholar]
- 31.Hu W, Zhang M, Czéh B, Flügge G, Zhang W. Stress impairs GABAergic network function in the hippocampus by activating nongenomic glucocorticoid receptors and affecting the integrity of the parvalbumin-expressing neuronal network. Neuropsychopharmacology. 2010;35:1693–1707. doi: 10.1038/npp.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cowell RM, Blake KR, Russell JW. Localization of the transcriptional coactivator PGC-1α to GABAergic neurons during maturation of the rat brain. J Comp Neurol. 2007;502:1–18. doi: 10.1002/cne.21211. [DOI] [PubMed] [Google Scholar]
- 33.Meng H, Liang HL, Wong-Riley M. Quantitative immuno-electron microscopic analysis of depolarization-induced expression of PGC-1α in cultured rat visual cortical neurons. Brain Res. 2007;1175:10–16. doi: 10.1016/j.brainres.2007.07.063. [DOI] [PubMed] [Google Scholar]
- 34.Luo Y, Zhu W, Jia J, Zhang C, Xu Y. NMDA receptor dependent PGC-1α up-regulation protects the cortical neuron against oxygen-glucose deprivation/reperfusion injury. J Mol Neurosci. 2009;39:262–268. doi: 10.1007/s12031-009-9196-5. [DOI] [PubMed] [Google Scholar]
- 35.Liang HL, Dhar SS, Wong-Riley MT. p38 mitogen-activated protein kinase and calcium channels mediate signaling in depolarization-induced activation of peroxisome proliferator-activated receptor gamma coactivator-1α in neurons. J Neurosci Res. 2010;88:640–649. doi: 10.1002/jnr.22222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang HL, Wong-Riley MT. Activity-dependent regulation of nuclear respiratory factor-1, nuclear respiratory factor-2, and peroxisome proliferatoractivated receptor gamma coactivator-1 in neurons. Neuroreport. 2006;17:401–405. doi: 10.1097/01.wnr.0000204980.98876.11. [DOI] [PubMed] [Google Scholar]
- 37.Patterson PH. Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav Brain Res. 2009;204:313–321. doi: 10.1016/j.bbr.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 38.Meyer U. Developmental neuroinflammation and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2011 doi: 10.1016/j.pnpbp.2011.11.003. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 39.Bayer TA, Buslei R, Havas L, Falkai P. Evidence for activation of microglia in patients with psychiatric illnesses. Neurosci Lett. 1999;271:126–128. doi: 10.1016/s0304-3940(99)00545-5. [DOI] [PubMed] [Google Scholar]
- 40.Radewicz K, Garey LJ, Gentleman SM, Reynolds R. Increase in HLA-DR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenics. J Neuropathol Exp Neurol. 2000;59:137–150. doi: 10.1093/jnen/59.2.137. [DOI] [PubMed] [Google Scholar]
- 41.Steiner J, Mawrin C, Ziegeler A, Bielau H, Ullrich O, Bernstein HG, et al. Distribution of HLA-DR-positive microglia in schizophrenia reflects impaired cerebral lateralization. Acta Neuropathol. 2006;112:305–316. doi: 10.1007/s00401-006-0090-8. [DOI] [PubMed] [Google Scholar]
- 42.Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C, et al. Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res. 2008;42:151–157. doi: 10.1016/j.jpsychires.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 43.van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers E, et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry. 2008;64:820–822. doi: 10.1016/j.biopsych.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 44.Behrens MM, Ali SS, Dugan LL. Interleukin-6 mediates the increase in NADPH-oxidase in the ketamine model of schizophrenia. J Neurosci. 2008;28:13957–13966. doi: 10.1523/JNEUROSCI.4457-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sorce S, Schiavone S, Tucci P, Colaianna M, Jaquet V, Cuomo V, et al. The NADPH oxidase NOX2 controls glutamate release: a novel mechanism involved in psychosis-like ketamine responses. J Neurosci. 2010;30:11317–11325. doi: 10.1523/JNEUROSCI.1491-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takac I, Schröder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, et al. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J Biol Chem. 2011;286:13304–13313. doi: 10.1074/jbc.M110.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 48.Brown AS. The environment and susceptibility to schizophrenia. Prog Neurobiol. 2011;93:23–58. doi: 10.1016/j.pneurobio.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blackwood DH, He L, Morris SW, McLean A, Whitton C, Thomson M, et al. A locus for bipolar affective disorder on chromosome 4p. Nat Genet. 1996;12:427–430. doi: 10.1038/ng0496-427. [DOI] [PubMed] [Google Scholar]
- 50.Christoforou A, Hellard SL, Thomson PA, Morris SW, Tenesa A, Pickard BS, et al. Association analysis of the chromosome 4p15-p16 candidate region for bipolar and schizophrenia. Mol Psychiatry. 2007;12:1011–1025. doi: 10.1038/sj.mp.4002003. [DOI] [PubMed] [Google Scholar]
- 51.Christoforou A, McGhee KA, Morris SW, Thomson PA, Anderson S, McLean A, et al. Convergence of linkage, association and GWAS findings for a candidate region for bipolar disorder and schizophrenia on chromosome 4p. Mol Psychiatry. 2010;16:240–242. doi: 10.1038/mp.2010.25. [DOI] [PubMed] [Google Scholar]
- 52.Hsiao YH, Chen PS, Chen SH, Gean PW. The involvement of Cdk5 activator p35 in social isolation-triggered onset of early Alzheimer’s disease-related cognitive deficit in the transgenic mice. Neuropsychopharmacology. 2011;36:1848–1858. doi: 10.1038/npp.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hermes G, Li N, Duman C, Duman R. Post-weaning chronic social isolation produces profound behavioral dysregulation with decreases in prefrontal cortex synaptic-associated protein expression in female rats. Physiol Behav. 2011;104:354–359. doi: 10.1016/j.physbeh.2010.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fone KCF, Porkess MV. Behavioural and neurochemical effects of post-weaning social isolation in rodents-relevance to developmental neuropsychiatric disorders. Neurosci Biobehav Rev. 2008;32:1087–1102. doi: 10.1016/j.neubiorev.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 55.Nuechterlein KH, Dawson ME. A heuristic vulnerability/stress model of schizophrenic episodes. Schizophr Bull. 1984;10:300–312. doi: 10.1093/schbul/10.2.300. [DOI] [PubMed] [Google Scholar]
- 56.Bayer TA, Falkai P, Maier W. Genetic and non-genetic vulnerability factors in schizophrenia: the basis of the “two hit hypothesis”. J Psychiatr Res. 1999;33:543–548. doi: 10.1016/s0022-3956(99)00039-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.