

Abstract
Cohesin, a multiunit complex of SMC1A, SMC3 and Rad21, associates with chromatin after mitosis and holds sister chromatids together following DNA replication. It has been reported that SMC1A is mutated in some cancer types, leading to genomic instability and abnormal cell growth. In this study, we investigated the role of SMC1A in human glioma. We found that SMC1A was expressed at abnormally high levels in human glioma tissue and in cultured U251 glioma cells. Knocking down SMC1A expression in U251 cells with SMC1A-targeted interfering RNAs inhibited cell growth and induced G2/M cell cycle arrest. Furthermore, expression of the cell cycle associated gene CCNB1IP1 was dramatically increased, whereas expression of Cyclin B1 was decreased in SMC1A-deficienct U251 cells. These results suggest that SMC1A upregulation is involved in the pathogenesis of glioma.
Keywords: SMC1A, knocking down, human glioma
Introduction
Cohesin is a multi-subunit complex that associates with chromatin after mitosis and is important for holding sister chromatids together following DNA replication [1]. Cohesin is composed of four subunits: a pair of SMC (Structural Maintenance of Chromosome) proteins, SMC1A and SMC3, and two non-SMC proteins, RAD21/Scc1 and STAG/Scc3/Sa. SMC proteins are characterized by a globular hinge domain flanked by two a-helical domains, which fold back on themselves at the hinge, forming a long antiparallel a-helical coiled coil arm that brings the N- and C-termini together. The N-terminal contains the Walker A box (or P-loop), which binds ATP. The C-terminal holds the DNA binding domain Walker B. SMC1A and SMC3 dimerize at the hinge domains, forming a V-shaped structure through hydrophobic interactions.
The cohesin complex acts as a tripartite ring in which SMC1A and SMC3 are connected by their hinge domains on one side, and RAD21 closes the ring by connecting the SMC1A and SMC3 head domains on the other side [2,3]. Based on this proposed ring shape, it is thought that the interaction between cohesin and DNA is topological, which is known as the “ring” or “embrace” model. According to this model, cohesin enwraps sister chromatids by opening up at the head domain allowing the sister chromatid to enter, which is then locked in when the RAD21 subunit closes overtop [3-5]. Transient dissociation of the SMC hinge domains also seems to contribute to the entry of DNA into the cohesion ring [6]. The cohesin binding to chromatin is dynamic and occurs during the G1/S phase in the budding yeast or in the telophase of the cell cycle in vertebrates [7].
Recent studies have concluded that SMC1A is involved in the pathogenesis of cancer [8,9]. SMC1A mutations have been identified in colorectal cancers, and RNAi-mediated down-regulation of SMC1A results in chromosomal instability and chromatid cohesion defects in human cells [8]. It is believed that genetic instability plays a critical role in the development of cancer [10,11]. Therefore, it is suggested that SMC1A mutations induced defective sister chromatid cohesion, caused chromosome instability in human cancer. In addition, one study demonstrated an upregulation of SMC1A mRNA in cervix cancer cells compared to normal cervix [12]. Other studies have found an association between SMC1A expression levels and age in patients with acute myeloid leukemia. Increasing age was associated with significantly decreased mRNA levels of SMC1A. Importantly, patients with low protein expression levels of SMC1A experienced significantly shortened event free (2.6 months versus 10.3 months, p=0.003) and overall survival (10.4 months versus 22.6 months, p=0.015). The SMC1A protein expression level remained a significant prognostic factor for event free survival (p=0.014) with a borderline significance for overall survival (p=0.066) in a multivariate analysis. It is implicated that SMC1A protein in leukemia prognosis and suggesting that treatments that increase SMC1A may offer a clinical benefit [9]. These findings suggest that cohesin function may be compromised in a wide variety of as yet unknown cancer types.
Gliomas are primary brain tumors derived from glial cells. The World Health Organization (WHO) classifies glioma tumors into four grades (I–IV) based on the presence of histopathological features that correlate with prognosis [13]. Current therapies for the treatment of glioma have poor response rates and survival rates for patients with high grade glioma is extremely short (approximately 12–14 months [14]. In spite of the current troubling clinical outlook for gliomas, our knowledge of the molecular underpinnings of the pathogenesis of primary brain tumors is rapidly advancing, and numerous genetic and epigenetic factors have been identified in different tumor types.
In this study we investigated the expression and activity of SMC1A in gliomas. We found that SMC1A is highly expressed in human glioma tissues compared with normal brain tissues. Downregulating SMC1A in cultured U251 glioma cells with SMC1A-targeted shRNA constructs inhibited cell growth, induced G2/M phase cell cycle arrest, and led to increased CCNB1IP1 expression and decreased Cyclin B1 expression. These results suggest that SMC1A is abnormally expressed in gliomas and reducing its levels suppresses glioma growth in vitro.
Materials and methods
Reagents and antibodies
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), TRIzol Reagent were purchased from Invitrogen (Carlsbad, CA, USA); 3-(4,5-dimethylthylthiazol-2yl-)-2,5-diphenyl tetrazolium bromide (MTT) was from Sigma (St. Louis, USA); dimethylsulfoxide (DMSO) and propidium iodide (PI) were from Sibas (Shanghai, China); bicinchoninic acid (BCA) protein assays were from HyClone-Pierce (South Logan, UT, USA); M-MLV reverse transcription kits and SYBR green Master Mixture were from Takara (Takara Biochemical, Kyoto, Japan); oligo-dT was from Sangon Biotech (Shanghai, China); Goat anti-SMC1α, HRP-conjugated mouse anti-GAPDH and Donkey anti-Goat IgG were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit anti-Cyclin B1 was from Booster(Wuhan, China). Mouse anti-Survivin was from Cell signaling Technology (Cell signaling Technology, MA, USA). All other chemicals used were laboratory grade.
Human glioma tissues and cell culture
Human glioma tissues graded were obtained from Shanghai Huashan Hospital (Chinese Hospital). Rat glial cells were obtained from rat brain. U251 glioma cells and C6 glioma cells were purchased from the Shanghai Institutes for Biological Sciences (Chinese Academy of Sciences). Cells were cultured in DMEM supplemented with 10% FBS, penicillin and streptomycin at 37°C in an atmosphere of 5% CO2 and 95% air (Gibco BRL, Life Technologies, NY) and harvested as indicated.
SMC1A-targeted shRNAs and lentivirus infection
Interfering short hairpin RNAs (shRNA) targeting SMC1A were synthesized at Neuron Biotech Inc. (Shanghai, China). Target sequences and primers used were as follows: SMC1A#1 (5’-CGGCGTATTGATGAAATCAAT-3’) was amplified with 5’-CCGGCGGCGTATTGATGAAATCAATCTCGAGATTGATTTCATCAATACGCCGTTTTTTG-3’ (forward) and 5’-AATTCAAAAAACGGCGTATTGATGAAATCAATCTCGAGATTGATTTCATCAATACGCCG-3’ (reverse); SMC1A#2 (5’-CCAACATTGATGAGATCTATA-3’) was amplified with 5’-CCGGCCAACATTGATGAGATCTATACTCGAGTATAGATCTCATCAATGTTGGTTTTTTG-3’ (forward) and 5’-AATTCAAAAAACCAACATTGATGAGATCTATACTCGAGTATAGATCTCATCAATGTTGG-3’ (reverse); and SMC1A#3 (5’-TAGGAGGTTCTTCTGAGTACA-3’) was amplified with 5’-CCGGTAGGAGGTTCTTCTGAGTACATTCAAGAGATGTACTCAGAAGAACCTCCTATTTTTTG-3’ (forward) and 5’-AATTCAAAAAATAGGAGGTTCTTCTGAGTACATCTCTTGAATGTACTCAGAAGAACCTCCTA-3’ (reverse); and SMCA-Scr (5’-TTCTCCGAACGTGTCACGT-3’) was amplified with 5’-CCGGTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTTTG-3’ (forward) and 5’-AATTCAAAAAATTCTCCGAACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGAA-3’ (reverse). shRNA constructs were cloned into the Age I and EcoR I sites of pGCSIL-GFP vector to generate pGCSIL-GFP-SMC1A. For lentivirus infection, 3x105 U251 cells were seeded in 10 cm dishes and cultured for 24 hrs. Cells were then infected with control or SMC1A-shRNA lentivirus at a MOI (multiplicity of infection) of 10. To determine the infection efficiency, GFP expression was assessed under a fluorescence microscope (Nikon Ti-s, Nikon) 4 days after infection. Then the cells were passaged and subcultured 4 days for western blot and Cell cycle analysis.
Quantitative real-time RT-PCR
mRNA was isolated from uninfected and infected U251 cells using TRIzol reagent according to the manufacturer’s instructions (Invitrogen). Reverse transcription was performed using MMLV Reverse Transcriptase and oligo (dT) primers. Real time PCR was conducted with a TAKARA Thermal Cycler Dice Real-Time System using SYBR green Master Mixture. The reaction mixture (20 μl) contained 10 μl SYBR green Master Mixture, 125 nM gene-specific primers, and 1 μl of template was amplified under the following conditions: 95°C for 15 s, followed by 45 cycles of 95°C for 5 s and 60°C for 30 s. TAKARA Thermal Dice Real Time System software Ver3.0 was used to establish the baseline and threshold for each reaction. Data were analyzed using the comparative Ct method and normalized to β-actin. The generation of specific PCR products was confirmed by melting curve analysis. Primers used were SMC1A: 5’-TCGGACCATTTCAGAGGTTCA-3’ (forward) with 5’-TCCTCAGAGTAGACCATGCTG-3’ (reverse); β-actin: 5’-GGCGGCACCACCATGTACCCT-3’ (forward) with 5’-AGGGGCCGGACTCGTCATACT-3’ (reverse); and CCNB1IP1 5’-TCGAAAGTGTCGCATCAAACT-3’ (forward) with 5’-CACAGAAGATGTGAGAGCAGG-3’ (reverse).
Western blot analysis
Glioma tissues were homogenized on ice with a tissue homogenizer (Handheld Homogenizer Model PT 1300D, Equl, China) in homogenization buffer. U251 cells and tissue homogenate were lysed with RIPA lysis buffer containing protease inhibitors (20 mM Tris-HCl, 1 mM EDTA, 1 mM EGTA, 1 mM sodium vanadate, 0.2 mM phenylmethylsulfonyl fluoride, 0.5% NP-40). About 25 μg of total protein from each sample was loaded on 12% SDS-PAGE gel. Western blots were developed using standard methods and the following primary antibodies: polyclonal goat anti-human SMC1A (1:500), polyclonal rabbit anti-Cyclin B1 antibody (1:400), monoclonal mouse anti-Survivin antibody (1:1000) and horseradish peroxidase-conjugated mouse anti-GAPDH (1:10,000). Signals were visualized on ECL plus Western blotting detection system (DNR Bio-imaging system, Israel). GAPDH protein levels were used as loading controls.
MTT assay
Cell proliferation was measured using MTT assays. Briefly, uninfected and infected U251 cells were plated on 96 well plates containing 1,500 cells/well in 150 μl medium. After 4 hrs (day 0, baseline) or 1, 2, 3, 4, days of incubation, 20 μl of MTT stock solution (5 mg/ml) was added into each well, and cells were further incubated at 37°C for 4 hrs. The reaction was stopped by replacing the MTT-containing medium with 100 μl DMSO and formazan salts dissolved by gentle shaking for about 10 min at room temperature. For colorimetric analysis, absorbance at 490 nm was recorded using a Microplate Reader (IMark 10303, Bio-Rad, USA). Each assay was repeated at least in triplicate and the ratio of the absorbance relative to baseline was calculated.
Colony formation assay
Uninfected and infected cells (1000 cells/well) were diluted in complete medium and seeded in 6-well plates. After incubation for 14 days at 37°C in 5% humidified CO2 (culture medium changed at regular intervals), adherent cells were washed twice with PBS, stained with 0.5% Crystal Violet in methanol for 15 min at room temperature, washed again and counted. The experiment was performed in triplicate.
Cell cycle analysis
The distribution of cells in the cell cycle was determined using FACS analysis. Briefly, uninfected and infected cells were collected and washed, centrifuged, then fixed in 70% ethanol at 4°C for half an hour. Prior to analysis, cells were resuspended in PBS containing 20 μg/ml RNase A and 50 μg/ml propidium iodide, and then incubated at 4°C for 30 min in the dark. Cells were analyzed by flow cytometry using a FACSCalibur flow cytometer (Becton–Dickinson, San Jose, CA, USA). The data was analyzed with FlowJo 7.6.1 software (Tree Star, Inc., Ashland, OR).
Statistical analysis
All experiments were performed at least in triplicate. Statistical differences were determined by the Student’s t-test and P<0.05 was considered statistically significant. Data are expressed as the means ± S.D.
Results
SMC1A expression in rat C6 glioma cell and human glioma tissues
We began our study by examining levels of SMC1A protein in rat C6 glioma cells and glial cells by western blot. We found that SMC1A was expressed at higher levels in rat C6 glioma cells than normal rat glial cells (Figure 1A). Then human glioma tissues and healthy brain tissue were also examined. Similarly SMC1A was expressed at higher levels in glioma tissue than healthy brain tissue, but there was no significant correlation between SMC1A expression levels and the grade of malignancy (Figure 1B).
Figure 1.
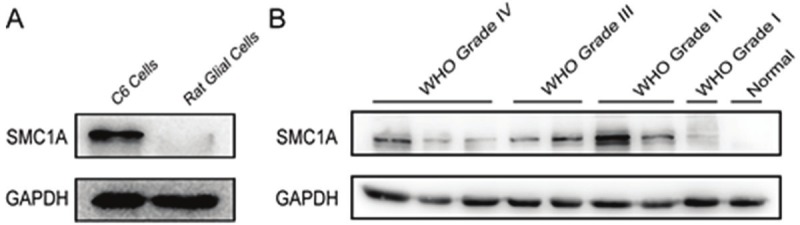
SMC1A is up-regulated in rat C6 glioma cells human glioma tissues by western blot. A. SMC1A protein levels in rat C6 glioma cells and normal rat glial cells. B. SMC1A protein levels in human glioma tissues compared with healthy brain tissues.
SMC1A silencing in U251 cells
To examine the role of SMC1A in human glioma, we generated three recombinant lentivirus constructs each harboring a unique SMC1A targeting shRNA, and transduced these into U251 cells. The infection efficiency was more than 95%, with almost all cells expressing the GFP reporter protein 4 days after infection (Figure 2A). Western blot and real-time RT-PCR revealed that SMC1A-shRNAs significantly reduced SMC1A expression in U251 cells compared with Control and SMC1A-Scr U251 cells (Figure 2B and 2C), demonstrating that lentivirus-mediated delivery of SMC1A-targeting shRNA constructs is an effective method for knocking down SMC1A mRNA and protein in U251 cells. Although all three sequences were effective, the following experiments were conducted using shRNAs SMC1A#2 and SMC1A#3.
Figure 2.

Lentivirus-mediated shRNA stably decreases SMC1A expression in U251 cells. A. GFP expression in 4 day old cultures of control U251 cells or cells infected with SMC1A-Scr lentivirus or lentivirus harboring SMC1A-shRNAs (SMC1A#1, #2 or #3). Top, bright field; bottom, GFP signal. B. Representative western blot showing SMC1A levels in control U251 cells and U251 cells expressing SMC1A-shRNA. GAPDH included as a loading control. C. Quantification of real-time RT-PCR measurements of SMC1A expression normalized to β-actin in U251 cells infected as indicated. The protein and RNA were extracted at 4 days after subculture.
Effect of SMC1A down-regulation on U251 cell growth inhibition
To investigate the effects of SMC1A silencing on cell growth and proliferation in U251 cells, we performed MTT and colony formation assays. After 5 days in culture, our MTT assays revealed U251 cells infected with SMC1A#3 had virtually not grown, whereas growth was reduced by about 30% in SMC1A#2-infected cells compared with control cells (Figure 3A). Consistently, the number of SMCA#2 and #3 infected cell colonies were near zero 14 days after plating while numerous colonies had developed in control plates (Figure 3B and 3C). Thus, both assays show that knocking-down SMC1A severely hinders U251 cell growth.
Figure 3.
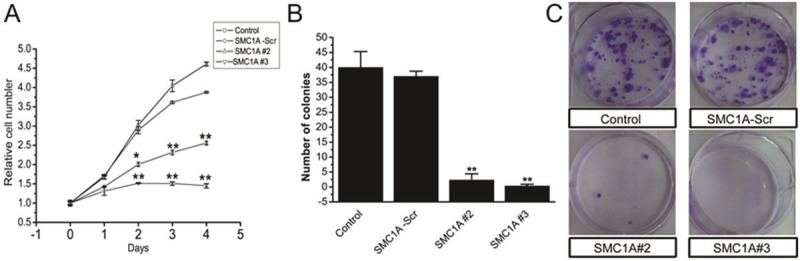
SMC1A silencing inhibits U251 cell growth. A. MTT assays showing the growth of cultured U251 cells infected as indicated over 4 days. B. Representative image of colonies formed in dishes seeded with 1000 U251 cells and cultured for 14 days. C. Quantification of colony formation assays.
Effect of SMC1A down-regulation on U251 cell cycle dynamics
To further evaluate the effects of SMC1A silencing on cell growth, we analyzed the cell cycle phase distribution of SMC1A-deficient U251 cells by flow cytometry (FACS) analysis after propidium iodide staining of cellular DNA. As shown in Figure 4A, down-regulation of SMC1A induced an accumulation of U251 cells in the G2/M phase compared with control cells. The G2/M phase fraction increased from about 25% in control-transduced cells to over 40% in SMC1A shRNA-infected U251 cells (Figure 4B).
Figure 4.
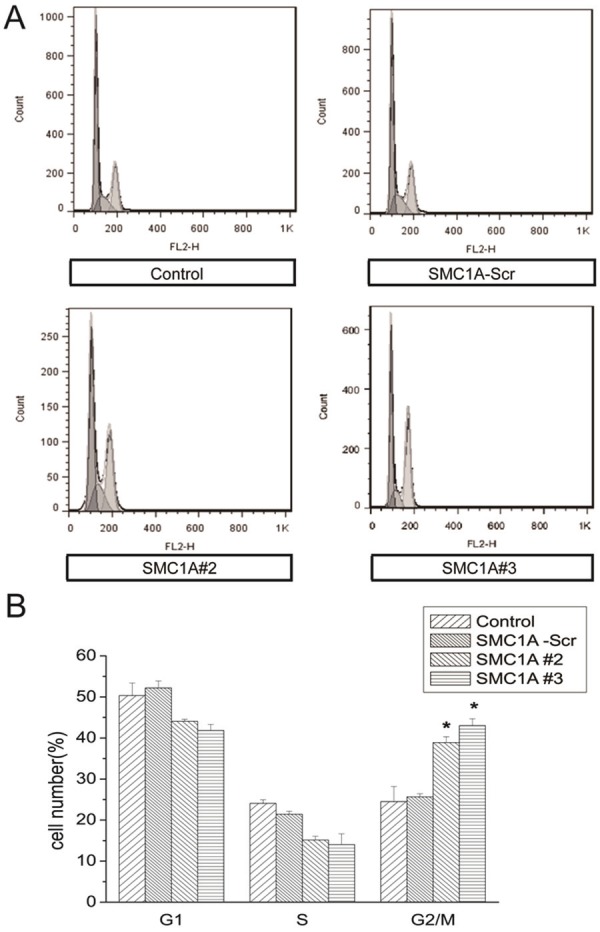
SMC1A silencing arrests the U251 cell cycle in G2/M. A. Representative FACS results showing cell cycle phase distributions of U251 cells at 4 days after subculture. B. Quantification of FACS analyses.
Effect of SMC1A silencing on CCNB1IP1 mRNA and Cyclin B1 expression
To begin to investigate the mechanism underlying cell growth inhibition and cell cycle arrest in SMC1A-deficient U251 cells, we examined the expression of the cell cycle associated gene CCNB1IP1, a modulator of cyclin B1 levels that regulates progression through the G2 phase [15-17]. We found that CCNB1IP1 was dramatically increased and Cyclin B1 was decreased in SMC-1A#2 and #3-infected U251 cells (Figure 5A and 5B). However, Survivin, inhibitor of cell death, did not change (Figure 5B). The results demonstrated that SMC1A gene silencing inhibit cell growth possibly by increasing CCNB1IP1 expression and reducing Cyclin B1 expression in U251 cells.
Figure 5.
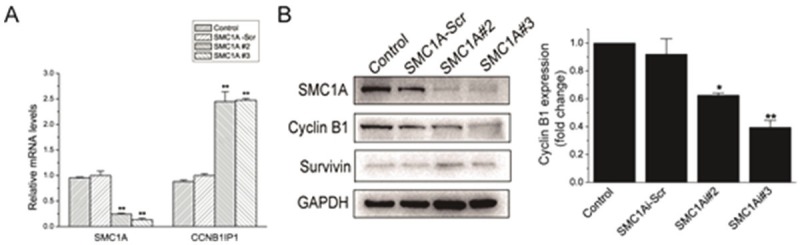
SMC1A silencing increases mRNA expression of CCNB1IP1 and reduces Cyclin B1 expression. A. Quantitative RT-PCR data showing CCNB1IP1 mRNA expression normalized to β-actin in U251 cells subcultured for 4 days. B. Left, Western blot showing Cyclin B1 and Survivin expression. Right, gray scale analysis on Cyclin B1 expression, compared with Control group. Data represents the mean ± SD of three-independent experiments. *P<0.05, **P<0.01, compared with Control group.
Discussion
Glioma is the most common form of primary brain tumor and includes morphologically distinct cancers such as astrocytoma, ependymoma and oligodendroglia [13]. In the ongoing effort to characterize these diseases, numerous studies have shown correlations between the pathogenesis or pathophysiology of glioma and specific genetic alterations; together these studies have revealed considerable variability in the genetic factors and phenotypes associated with gliomas. This diversity underscores the need for more accurate biomarkers, as well as putative targets for the development of new therapeutic paradigms [18-20].
In the study, we present evidence to show that SMC1A is highly expressed in advanced human glioma tissue compared with healthy brain tissue and Grade I glioma tissue. It is consistent with the results that an upregulation of SMC1A mRNA in cervix cancer cells compared to normal cervix [12].
To begin to explore the possibility of using SMC1A-based therapies for the treatment of glioma, we employed lentivirus-mediated RNAi silencing of endogenous SMC1A expression to elucidate the effect of suppressing SMC1A function in U251 cells. Silencing SMC1A inhibited cell growth and caused the U251 cell cycle to arrest in the G2/M phase (Figures 3 and 4). This observation is consistent with a previous report showing that down-regulation of SMC1A leads to G2/M phase cell cycle arrest in HCT116 in human colorectal cancer cells [8]. Proliferating eukaryotic cells possess checkpoint mechanisms to prevent cell division in the event of unreplicated or damaged DNA. Cyclin B1 is essential for controlling cell cycle progression at the G2/M transition [21]. And CCNB1IP1 could interact with Cyclin B1 levels to regulate progression through the G2 phase [15]. In our experiments, silencing SMC1A was associated with a significant increase in CCNB1IP1 mRNA expression (Figure 5A). CCNB1IP1 belongs to a divergent class of E3 ubiquitin ligases and may directly interact with Cyclin B1 in the control of protein degradation that occurs as cells enter into mitosis [15]. Consistently, it was found that Cyclin B1 decreased in U251 cells of SMC1A silencing (Figure 5B). However, survivin was a member of inhibitor apoptosis gene family, which prevent apoptotic cell death [22]. Results show that survivin expression remains unchanged (Figure 5B), which indicated that SMC1A silencing may not induce cell death. In addition, CCNB1IP1 has been identified as a novel merlin-binding partner [17]. As merlin is a tumor suppressor and participates in cell cycle regulation, it is possible that SMC1A upregulation in glioma cells contributes to increased tumor cell growth via the repression of CCNB1IP1 and merlin.
In conclusion, this study is the first report, to our knowledge, to show aberrant SMC1A expressed in human glioma tissues. Furthermore, shRNA-mediated knock down of SMC1A prevents the growth of U251 glioma cells in vitro and arrests the cell cycle in G2/M. The growth suppressing effect of SMC1A shRNA may be due to the upregulation of CCNB1IP1 and down-regulation of Cyclin B1 that occurs in U251 cells in the absence of SMC1A expression.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No.30600577).
References
- 1.Losada A, Hirano T. Dynamic molecular linkers of the genome: the first decade of SMC proteins. Genes Dev. 2005;19:1269–1287. doi: 10.1101/gad.1320505. [DOI] [PubMed] [Google Scholar]
- 2.Anderson DE, Losada A, Erickson HP, Hirano T. Condensin and cohesin display different arm conformations with characteristic hinge angles. J Cell Biol. 2002;156:419–424. doi: 10.1083/jcb.200111002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haering CH, Löwe J, Hochwagen A, Nasmyth K. Molecular Architecture of SMC Proteins and the Yeast Cohesin Complex. Mol Cell. 2002;9:773–788. doi: 10.1016/s1097-2765(02)00515-4. [DOI] [PubMed] [Google Scholar]
- 4.Gruber S, Haering CH, Nasmyth K. Chromosomal Cohesin Forms a Ring. Cell. 2003;112:765–777. doi: 10.1016/s0092-8674(03)00162-4. [DOI] [PubMed] [Google Scholar]
- 5.Ivanov D, Nasmyth K. A Physical Assay for Sister Chromatid Cohesion In Vitro. Mol Cell. 2007;27:300–310. doi: 10.1016/j.molcel.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Gruber S, Arumugam P, Katou Y, Kuglitsch D, Helmhart W, Shirahige K, Nasmyth K. Evidence that Loading of Cohesin Onto Chromosomes Involves Opening of Its SMC Hinge. Cell. 2006;127:523–537. doi: 10.1016/j.cell.2006.08.048. [DOI] [PubMed] [Google Scholar]
- 7.Haering CH, Schoffnegger D, Nishino T, Helmhart W, Nasmyth K, Löwe J. Structure and Stability of Cohesin’s Smc1-Kleisin Interaction. Mol Cell. 2004;15:951–964. doi: 10.1016/j.molcel.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 8.Barber TD, McManus K, Yuen KW, Reis M, Parmigiani G, Shen D, Barrett I, Nouhi Y, Spencer F, Markowitz S, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C, Hieter P. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc Natl Acad Sci U S A. 2008;105:3443–3448. doi: 10.1073/pnas.0712384105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Homme C, Krug U, Tidow N, Schulte B, Kohler G, Serve H, Burger H, Berdel WE, Dugas M, Heinecke A, Buchner T, Koschmieder S, Muller-Tidow C. Low SMC1A protein expression predicts poor survival in acute myeloid leukemia. Oncol Rep. 2010;24:47–56. doi: 10.3892/or_00000827. [DOI] [PubMed] [Google Scholar]
- 10.Duesberg P, Rasnick D, Li R, Winters L, Rausch C, Hehlmann R. How aneuploidy may cause cancer and genetic instability. Anticancer Res. 1999;19:4887–4906. [PubMed] [Google Scholar]
- 11.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 12.Narayan G, Bourdon V, Chaganti S, Arias-Pulido H, Nandula SV, Rao PH, Gissmann L, Dürst M, Schneider A, Pothuri B, Mansukhani M, Basso K, Chaganti RS, Murty VV. Gene dosage alterations revealed by cDNA microarray analysis in cervical cancer: Identification of candidate amplified and overexpressed genes. Genes Chromosomes Cancer. 2007;46:373–384. doi: 10.1002/gcc.20418. [DOI] [PubMed] [Google Scholar]
- 13.Louis D, Ohgaki H, Wiestler O, Cavenee W, Burger P, Jouvet A, Scheithauer B, Kleihues P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathologica. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 15.Toby GG, Gherraby W, Coleman TR, Golemis EA. A Novel RING Finger Protein, Human Enhancer of Invasion 10, Alters Mitotic Progression through Regulation of Cyclin B Levels. Mol Cell Biol. 2003;23:2109–2122. doi: 10.1128/MCB.23.6.2109-2122.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh MK, Nicolas E, Gherraby W, Dadke D, Lessin S, Golemis EA. HEI10 negatively regulates cell migration by inhibiting cyclin B/Cdk1 and other promotility proteins. Oncogene. 2007;26:4825–4832. doi: 10.1038/sj.onc.1210282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gronholm M, Muranen T, Toby GG, Utermark T, Hanemann CO, Golemis EA, Carpen O. A functional association between merlin and HEI10, a cell cycle regulator. Oncogene. 2006;25:4389–4398. doi: 10.1038/sj.onc.1209475. [DOI] [PubMed] [Google Scholar]
- 18.Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1:97–117. doi: 10.1146/annurev.pathol.1.110304.100043. [DOI] [PubMed] [Google Scholar]
- 19.Mason WP, Cairncross JG. Invited Article: The expanding impact of molecular biology on the diagnosis and treatment of gliomas. Neurology. 2008;71:365–373. doi: 10.1212/01.wnl.0000319721.98502.1b. [DOI] [PubMed] [Google Scholar]
- 20.Sathornsumetee S, Rich JN. Designer therapies for glioblastoma multiforme. Ann N Y Acad Sci. 2008;1142:108–32. doi: 10.1196/annals.1444.009. [DOI] [PubMed] [Google Scholar]
- 21.Gavet O, Pines J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell. 2010;18:533–43. doi: 10.1016/j.devcel.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–21. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]