

Abstract
Peutz-Jeghers syndrome (PJS) is a rare inherited autosomal dominant disease characterized by mucocutaneous pigmentation and multiple polyps in the gastrointestinal tract. We report on an 18-year-old Chinese male who complained with pigmentation on face and extremities for over 10 years. Colonoscopy revealed more than ten polyps from transverse colon to rectum. The family survey included 15 family members from three generations, and 6 PJSs (4 males and 2 females) were found. This case is reported because of its rarity of Peutz-Jeghers syndrome and the survey of family history.
Keywords: Peutz-Jeghers syndrome, family survey
Introduction
Peutz-Jeghers syndrome (PJS) is a rare inherited autosomal dominant disease, characterized by mucocutaneous pigmentation and multiple hamartomatous polyps in the gastrointestinal tract. Researchers had confirmed the relativity of PJS and STK11 gene. To improve the understanding, diagnosis and treatment of PJS, we now report a case of PJS and present the survey of the family of interest.
Case present
An 18-year-old Chinese male presented to us with pigmentation on face and extremities for over 10 years. The patient had scattered needle-size dark blue macules on the mucosa of lower lip from age three, and similar macules on distant parts of his fingers from age seven. Since then, he occasionally suffered abdomen pain, and the pain recessed without particular medication. In last two years, the patient complained the number of dark macules increased, especially on mucosa of lower lip. Family history confirmed that some other family members had similar symptoms.
On physical examination, we found out that there are scattered hyperpigmented macules around his lower lip, nostril and nasolabial fold, round or irregular shape, size from needle to rice. These pigmentations had definite boundaries, did not fade under pressure, and did not protrude from skin. Especially, there were more hyperpigmented macules around his lower lip, and these macules were darker and presented irregular shapes (Figure 1A). Besides, hyperpigmented macules, size from rice to bean, were also detected on palms and soles, while no abnormality was found on nails or hairs (Figure 1B and 1C).
Figure 1.
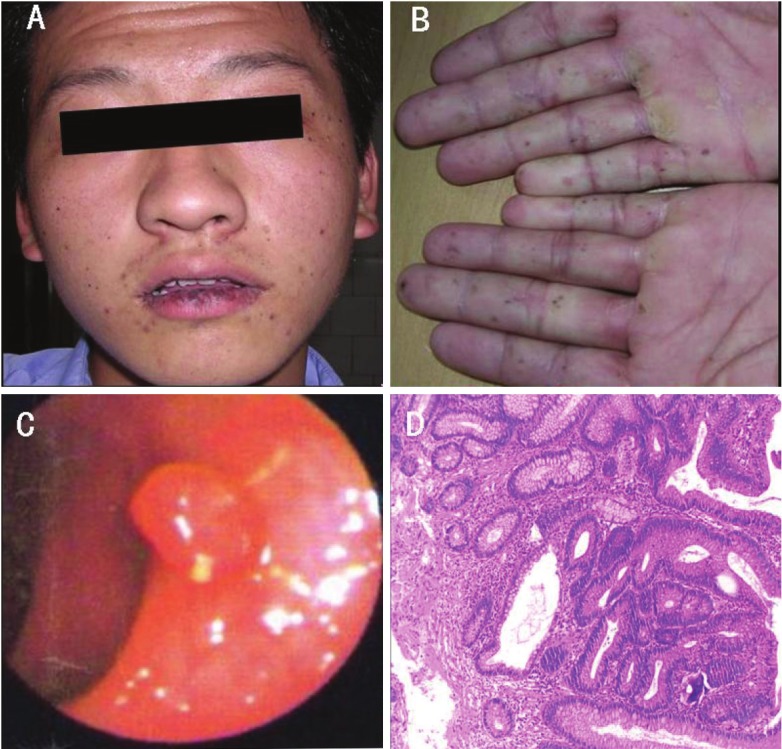
Images showing hyperpigmented macules on the patient’s lips (A) and fingers (B); Endoscopic view of pedunculated colonic polyp (C); Histology image, showing a typical Peutz-Jeghers polyp (HE staining), demonstrating glandular disorganisation, hamartomatous appearance and ramifying branching bundles of smooth muscle (D).
Full Gastrointestinal Contrast revealed no abnormal protruding or recessing; however, jejunum and ileum were found gut cavity dilatation, mucosal morphology bulky, intestinal canal peristalsis hyperfunction and functional disorder. Colonoscopy revealed more than ten polyps (Figure 1D) from transverse colon to rectum, size from 0.5 cm to 1.5 cm. We diagnosed him with Peutz-Jeghers syndrome.
Family survey
This survey included 15 family members from three generations, and 6 PJSs (4 males and 2 females) were found. We found PJS in each generation without a sexual difference. No consanguineous marriage was found in this family, so it goes well with PJS’s trait, a trait of an inherited autosomal dominant disorder (Figure 2). All patients in this family presented peri-oral mucosal pigmentation, especially needle-size brown-black macules on the lower lip, when they were born or at the age around ten. These macules were getting bigger and darker progressively with time, and macules on palms and soles gradually occurred. Except for the patient we just reported, no other family member with PJS complained abdomen pain, bloody stool or other symptoms.
Figure 2.
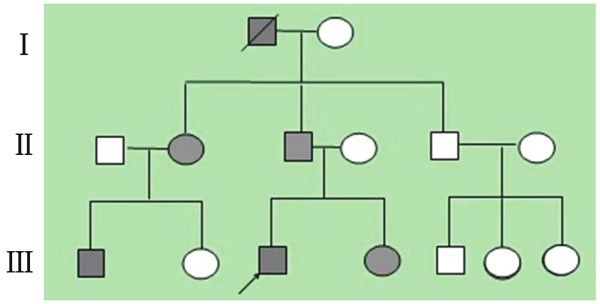
Pedigree of the family with Peutz-Jeghers syndrome. Squares and circles indicate males and females, respectively, and arrow indicates the proband. Black color denotes affected individuals. A slash through the symbol indicates decreased individuals.
Discussion
Peutz-Jeghers syndrome is a more than rare inherited disorder with particular features of mucocutaneous pigmentation macules around mouth and cheek, multiple polyps in the gastrointestinal tract, and recognizable family history. Conditions with either mucocutaneous pigmentation or gastrointestinal polyps are defined as incomplete PJS, which usually presents only typical pigmentation or enterorrhagia or symptoms of intussusception [1,2]. In 1998, Jenne and Hemminki [3,4] colonized PJS-related gene at the proximity of D19S886190kb, and named it STK11. Papp J [5] found, in 21 cases from 13 families in Hungary, that all patients had STK11 mutation. In our family survey, there were patients in all three generations, so gene tests of this family might reveal the mutation point in PJS in this family.
In treating PJS, the treatment for mucocutaneous pigmentations were usually considered as unnecessary; however if it is necessary, laser therapy can be a choice. For polyps’ treatment, opinions diverge. Some consider these polyps as precursors of cancer, which should be paid considerable attention or removed by surgery [6]. However, others found out that canceration of these polyps was rare, and periodic follow-up is enough. In the case we reported, except for the patient, no other family member with PJS complained abdomen pain, or bloody stool or other symptoms. No particular medication was administrated, while periodic follow-up was demanded.
Conclusion
Peutz-Jeghers syndrome should be paid more attention because of the risk of complications related to polyps, and the association with cancers. Peutz-Jeghers syndrome patients should have a periodical inspection of the digestive tract.
References
- 1.Kopacova M, Tacheci I, Rejchrt S, Bures J. Peutz-Jeghers syndrome: diagnostic and therapeutic approach. World J Gastroenterol. 2009;15:5397–408. doi: 10.3748/wjg.15.5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beggs AD, Latchford AR, Vasen HF, Moslein G, Alonso A, Aretz S, Bertario L, Blanco I, Bülow S, Burn J, Capella G, Colas C, Friedl W, Møller P, Hes FJ, Järvinen H, Mecklin JP, Nagengast FM, Parc Y, Phillips RK, Hyer W, Ponz de Leon M, Renkonen-Sinisalo L, Sampson JR, Stormorken A, Tejpar S, Thomas HJ, Wijnen JT, Clark SK, Hodgson SV. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59:975–86. doi: 10.1136/gut.2009.198499. [DOI] [PubMed] [Google Scholar]
- 3.Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Müller O, Back W, Zimmer M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18:38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- 4.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Höglund P, Järvinen H, Kristo P, Pelin K, Ridanpää M, Salovaara R, Toro T, Bodmer W, Olschwang S, Olsen AS, Stratton MR, de la Chapelle A, Aaltonen LA. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–7. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 5.Papp J, Kovacs ME, Solyom S, Kasler M, Børresen-Dale AL, Olah E. High prevalence of germline STK11 mutations in Hungarian Peutz-Jeghers Syndrome patients. BMC Med Genet. 2010;11:169. doi: 10.1186/1471-2350-11-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sinha N, Chatterjee U, Sarkar S. Jejunal carcinoma in a patient with Peutz–Jeghers syndrome. Can J Surg. 2009;52:E299–E300. [PMC free article] [PubMed] [Google Scholar]