

Abstract
The detection and quantification of genetic heterogeneity in populations of cells is fundamentally important to diverse fields, ranging from microbial evolution to human cancer genetics. However, despite the cost and throughput advances associated with massively parallel sequencing, it remains challenging to reliably detect mutations that are present at a low relative abundance in a given DNA sample. Here we describe smMIP, an assay that combines single molecule tagging with multiplex targeted capture to enable practical and highly sensitive detection of low-frequency or subclonal variation. To demonstrate the potential of the method, we simultaneously resequenced 33 clinically informative cancer genes in eight cell line and 45 clinical cancer samples. Single molecule tagging facilitated extremely accurate consensus calling, with an estimated per-base error rate of 8.4 × 10−6 in cell lines and 2.6 × 10−5 in clinical specimens. False-positive mutations in the single molecule consensus base-calls exhibited patterns predominantly consistent with DNA damage, including 8-oxo-guanine and spontaneous deamination of cytosine. Based on mixing experiments with cell line samples, sensitivity for mutations above 1% frequency was 83% with no false positives. At clinically informative sites, we identified seven low-frequency point mutations (0.2%–4.7%), including BRAF p.V600E (melanoma, 0.2% alternate allele frequency), KRAS p.G12V (lung, 0.6%), JAK2 p.V617F (melanoma, colon, two lung, 0.3%–1.4%), and NRAS p.Q61R (colon, 4.7%). We anticipate that smMIP will be broadly adoptable as a practical and effective method for accurately detecting low-frequency mutations in both research and clinical settings.
Genetic heterogeneity underlies phenotypic variation, evolution, and human disease. In multicellular organisms, both germline variation and somatic mutation can lead to phenotypic differences. In humans, somatic mutation in particular can lead to benign phenotypic variation as well as to a variety of clinically important conditions, including all types of cancer. In a given tissue sample, clinically informative mutations may be present at a low frequency because of non-neoplastic cell admixture or tumor heterogeneity (Navin et al. 2011; Carter et al. 2012; Gerlinger et al. 2012; Nik-Zainal et al. 2012). Furthermore, recent pioneering studies have explained the rapid emergence of resistance to targeted therapy in colon cancer by showing that drug-resistance mutations may be present at a very low frequency prior to treatment initiation (Diaz et al. 2012; Misale et al. 2012), although the generality of this phenomenon remains to be established. Post-zygotic mutations also have implications for human disease beyond cancer; for example, some developmental disorders can be caused by somatic mutations, as was recently shown for the overgrowth syndromes MCAP and MPPH (Riviere et al. 2012) and HME (Lee et al. 2012). Subclonal variation also has important consequences in populations of microorganisms, where low-frequency variants can confer drug resistance or facilitate immune evasion.
A wide variety of methods have been developed to identify and characterize genetic variation, including, but not limited to allele-specific PCR, mass spectrometry, microarrays, and DNA sequencing. In general, however, these methods are designed to detect heterozygous and homozygous variation and have poor sensitivity for variation present at lower frequencies. The sensitive and accurate detection of subclonal genetic variation remains challenging, as this necessarily requires a method that is capable of processing a large number of DNA molecules and sensitively identifying a variant at low relative abundance without an excess of false-positive calls. To address these challenges, other innovative methods have been developed, including phenotypic screening, COLD-PCR (Li et al. 2008; Milbury et al. 2012), the random mutation capture assay (Bielas and Loeb 2005), and BEAMing (Dressman et al. 2003). However, these methods are generally limited by some combination of technical complexity, poor sensitivity to variants below 1% frequency, and restriction to one or a small number of mutations interrogated per assay.
The recent advent of massively parallel DNA-sequencing technologies, which have dramatically decreased cost and increased throughput, has transformed many fields, including the study of population genetic variation (Tennessen et al. 2012), gene expression (Mortazavi et al. 2008) and its regulation (Ernst et al. 2011; Patwardhan et al. 2012), and the genetic basis of rare (Ng et al. 2010) and common (O'Roak et al. 2012b) disease. These methods also have attractive properties for the study of subclonal variation, including throughput on the order of 1,000,000,000 molecules per run and a separate readout for each molecule processed. However, the application of massively parallel sequencing to detecting subclonal variation generally faces two technical challenges. First, massively parallel sequencing instruments typically suffer from high per-base substitution error rates, hampering their specificity for detecting low-frequency variants. Second, whereas throughput has improved substantially, whole-genome sequencing is not practical because of the size of mammalian genomes and, consequently, the high sampling depth required for detecting low-frequency variation (typically addressed by targeted sequencing).
Several groups have sought to increase the sensitivity of massively parallel sequencing for low-frequency variation by carefully modeling error processes (Druley et al. 2009; Harismendy et al. 2011; Flaherty et al. 2012; Gerstung et al. 2012); however, such analytical methods do not actively correct errors that occur during the preparation and sequencing processes. An alternative approach, developed by us and others, is to perform “single molecule tagging” to mark sequence reads derived from a common progenitor molecule (that is, the same genomic equivalent in source DNA) (Hiatt et al. 2010; Casbon et al. 2011; Fu et al. 2011; Jabara et al. 2011; Kinde et al. 2011; Kivioja et al. 2012; Shiroguchi et al. 2012), and to subsequently use this information to guide consensus calling on a molecule-by-molecule basis (Hiatt et al. 2010; Jabara et al. 2011; Kinde et al. 2011). However, these methods have generally been either completely untargeted or targeted to only a single locus, and to our knowledge there are no reports integrating single molecule tagging with multiplex targeted sequencing.
Multiplex targeted sequencing has also been an area of extremely active effort (for review, see Mamanova et al. 2010). Emerging methods for multiplex targeted sequencing with particular emphasis on the ability to detect sub-clonal variation have been largely aimed at cancer. These include multiplex hybrid capture (Lipson et al. 2012; Wagle et al. 2012) and highly multiplexed PCR (Tewhey et al. 2009; Harismendy et al. 2011; Forshew et al. 2012). However, each of these methods has important drawbacks that limit practical utility. Hybrid capture typically entails a complex and time-intensive workflow, high per-sample reagent costs, and limited flexibility to reformulate the protocol as the regions of interest change over time. Highly multiplexed PCR often relies on complex instrumentation (Tewhey et al. 2009; Forshew et al. 2012) and may be restricted to a limited number of target genes. Furthermore, to maximize generality, an effective method for sensitive multiplex targeted sequencing must also be robust to relatively small amounts and poor quality of source DNA such as that isolated from formalin-fixed, paraffin-embedded (FFPE) tissue.
We therefore sought to develop a massively parallel sequencing-based method for the detection of low-frequency variation with the following characteristics: (1) minimal error rates via single molecule tagging, (2) targeted to genomic regions of interest, (3) multiplexed across many such regions, (4) simple and scalable experimental protocol, and (5) modular and cost-effective target enrichment reagent. The resulting method, termed smMIP (for single molecule Molecular Inversion Probes), combines the MIP strategy for targeted capture (Turner et al. 2009; Shen et al. 2011; O'Roak et al. 2012a) with single molecule tagging (Hiatt et al. 2010; Casbon et al. 2011; Fu et al. 2011; Jabara et al. 2011; Kinde et al. 2011; Kivioja et al. 2012; Shiroguchi et al. 2012). MIPs represent an attractive platform for targeted capture because of their very low per-sample cost, workflow simplicity, target-set modularity, and low sample input requirements. Single molecule tagging, on the other hand, enables consensus calling for single genomic equivalents present in the input material, thereby facilitating both highly sensitive variant calling and precise quantitation of mutation frequency. The combination of MIPs and single molecule tagging form the basis for an ultra-sensitive, targeted sequencing assay that has additional attractive characteristics from the standpoint of practicality, e.g., speed, ease of use, and compatibility with small quantities of degraded DNA.
To validate our method and establish its utility in a practical context, we designed molecular capture/tagging probes (smMIPs) targeting the coding sequences of 33 cancer genes in which clinically informative mutations may occur. We applied these probes to the targeted capture, sequencing, and mutational analysis of 53 specimens in parallel, comprising 45 clinical cancer specimens and eight HapMap DNA mixtures. We demonstrate that smMIPs enable highly accurate base-calling with substitution error rates below one in 10,000, sensitive and precise detection of subclonal variation, and accurate and comprehensive genotyping of clinically informative variation at clonal and subclonal frequencies. We also demonstrate that the smMIP assay is practical, highly multiplexed and easily scaled, and is compatible with a desktop sequencing instrument for potential rapid return of results in a clinical setting.
Results
Multiplex targeted sequencing using smMIPs
We designed and procured a pool of 1312 smMIP oligonucleotides targeting the coding sequences of 33 cancer-related genes (Supplemental Table S1; MacConaill et al. 2009; O'Roak et al. 2012a). These smMIPs tiled a total of ∼125 kb of genomic sequence, including 80,384 of the 81,190 (99%) coding base pairs (bp) of the 33 targeted genes. Targeted capture with smMIPs involves a standard MIP protocol for “library-free” sequencing (Turner et al. 2009; Shen et al. 2011) with slight modifications (Fig. 1). Following the post-capture PCR amplification, samples are subjected to massively parallel sequencing using the Illumina platform and analyzed using a custom pipeline. Our strategy involves two layers of indexing, with one index sequence (the “sample index”) resolving capture products from distinct source DNAs and another (the “molecular tag”) resolving reads derived from distinct genomic equivalents within individual source DNAs (Supplemental Note S1). Before alignment to the reference genome, overlapping regions of read-pairs are reconciled to produce ∼152-nt forward-reverse reads (“fr-reads”). After alignment, the molecular tag is used to group fr-reads, and groups of fr-reads form the basis for highly accurate single molecule consensus reads (“smc-reads”). All comparisons that we describe are between fr-reads and smc-reads. To our knowledge, this is the first description of molecular tagging integrated with MIPs, and, more generally, with a large-scale multiplex targeted capture strategy.
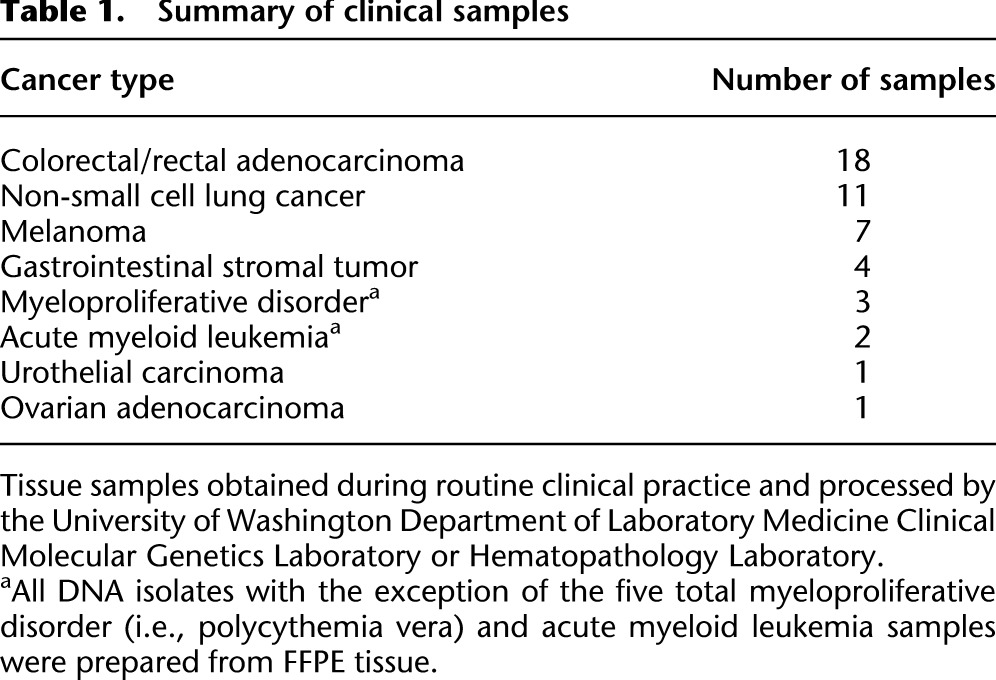
Figure 1.

Schematic of smMIP method. (A) Molecular inversion probes (MIPs) consisting of two 16–24 nt “targeting arms” (dark gray) joined by a constant 28-nt “backbone” sequence (light gray) and a 12-nt degenerate “molecular tag” (red) were designed for the coding exons (light-blue rectangle) of 33 cancer-related genes. Targeting arms were complementary to sequences flanking individual regions of interest, each 112 nt in length. (B) Probes are pooled, hybridized to genomic DNA, and polymerase and ligase were added to “gap-fill” the reverse complement of the genomic DNA to which the probe is hybridized (light-blue) and ligate the probe into a single-stranded circle. (C) After exonuclease treatment and PCR, sequencing library molecules consist of platform compatibility (black), probe backbone (light gray), targeting arm (dark gray), copied target (light blue), molecular tag (red), and sample-specific index introduced during PCR (green). Massively parallel sequencing is used to collect three reads (dark blue). (D) Overlapping read-pairs are reconciled to form “fr-reads” (dark blue), assigned to samples via the sample-specific index sequence (green) and individual capture events via the molecular tag (red). (E) Groups of fr-reads assigned to the same probe via alignment to the reference genome and sharing the same molecular tag and sample index form a “tag-defined read group” (TDRG). Random errors (yellow) that occur during library construction and sequencing may be present in some members of the TDRG at some positions. (F) TDRGs are used to call a single molecule consensus sequence (“smc-read”) for the captured target sequence that is robust to such errors.
To validate the smMIP method, we simultaneously applied it to two sets of samples. First, to assess sensitivity and positive predictive value and the extent to which we could precisely quantify low-frequency variants, we performed smMIP-based targeted sequencing on genomic DNA from two HapMap cell lines (NA12892 and NA19239) and six mixtures of these two gDNAs. Second, to explore the practical utility of the method, we also applied smMIP-based targeted sequencing to a panel of 47 genomic DNA isolates from clinical specimens encompassing a wide range of cancers (Table 1; Supplemental Table S2). All of the nonhematologic DNA isolates were obtained from FFPE-treated tissue (42 of 47 clinical specimens). Importantly, the FFPE specimens were not selected for quality in any way, with most samples isolated from 1 to 3 yr prior to our experiments, and included material that had been isolated as long as 8 yr prior to our experiments and had been processed into genomic DNA as long as 5 yr after FFPE treatment. These specimens thereby represent a stringent and realistic test of “real-world” method performance. We performed 55 capture reactions in parallel, using ∼500 ng of genomic DNA per capture, and carried out sequencing and analysis as outlined above. Two clinical specimens failed to yield sufficient on-target sequence during quality control and were excluded from further analysis, resulting in a success rate of 96% (45 of 47).
Table 1.
Summary of clinical samples
Method performance
We first sought to assess performance of the smMIP assay with respect to sensitivity and positive predictive value for clonal variation and uniformity of target enrichment. Because of the heterogeneous nature of the specimens, the number of sequencing reads obtained per sample varied from 1 to 16 million reads (Supplemental Fig. S1). However, 77% of samples (41 of 53) were within a threefold range, and this distribution could likely be improved by automated pooling (Supplemental Note S2). Fr-reads were then aligned to the reference genome and processed using a custom analysis pipeline to yield smc-reads.
We first explored coverage of targeted regions, finding that mean smc-read coverage of the targeted coding bases was 3538× across the HapMap samples and 1051× across the clinical specimens (Fig. 2; Supplemental Note S3). On average, smc-reads were comprised of 2.03 fr-reads in cell line samples, 1.21 fr-reads in fresh clinical samples, and 2.79 read-pairs in FFPE clinical samples (Supplemental Fig. S2). However, some samples exhibited a much higher rate of fr-reads per smc-read, which was likely due to degraded DNA leading to low-complexity capture. Thus, given a certain sequencing depth, the number of fr-reads per smc-read is directly related to capture complexity and can serve as a useful quality control metric.
Figure 2.

smMIP capture performance and detection of low-frequency variation. (A) Distributions of minimum coverage in a given percentile of total targeted coding positions, rank-ordered by smc-read coverage, for eight HapMap cell line (red) and 45 clinical cancer (blue and green) samples (box plot center line: median; top and bottom edges: quartiles; whiskers: farthest data point within 150% of interquartile range; dots: outliers). Zeroth-percentile indicates maximum coverage. (B) Distributions of fraction of coding positions above a given smc-read coverage cutoff. (C) Observed versus expected variant frequency in smc-read base-calls from mixtures of HapMap genomic DNA samples at known ratios for positions with at least 100× coverage (R = 0.94). Ideal performance is shown as gray line (y = x).
We then used smc-reads to call clonal genotypes using established tools (McKenna et al. 2010) and, for the HapMap samples, compared our calls to 1000 Genomes (“1KG”) pilot project genotypes (The 1000 Genomes Project Consortium 2010). For NA12892, we detected 24 of 25 1KG variant sites; the remaining position was not adequately covered in our data. After discarding three positions that were systematically misgenotyped by our assay (caused by capture of paralogous sequence and subsequent misalignment), we detected two additional variant positions; these calls were supported by manual inspection of more recent 1KG data. For NA19239, we detected 41 of 44 1KG variant sites; the remaining three positions were not adequately covered in our data. Two additional sites had variant genotypes and were again supported by newer 1-KG data. Therefore, based on this limited comparison, our assay is highly accurate at adequately covered positions. Considering all targeted sites, we estimate the sensitivity of our assay for clonal homozygous or heterozygous variation to be 93%–96%, and the positive predictive value to be near 100%.
Subclonal variant detection
To assess whether the smMIP assay was capable of sensitively detecting and accurately quantifying variants present at subclonal frequencies, we applied it to six synthetic mixtures of genomic DNA from the two HapMap cell lines combined in a twofold serial dilution from 1:8 to 1:256 (resulting in low-abundance genome alternate allele frequencies of ∼11% to ∼0.2%). We adopted a custom variant calling strategy to detect subclonal variation and then compared the expected variant frequency to that observed in smc-reads (Fig. 2C). In general, we observed close agreement between the expected and observed frequency for positions with at least 100× smc-read coverage (R = 0.94), with the deviation from expected frequency largely explained by sampling statistics (Supplemental Fig. S3).
We next sought to quantify the absolute error rate of the smMIP assay and to assess potential sources of error (Supplemental Fig. S4). In the samples derived from HapMap cell lines, smc-read base-calls were ∼13-fold more accurate than fr-read base-calls, with a substitution rate of 8.4 × 10−6 per base compared to 1.1 × 10−4 per base for fr-read calls (Table 2). In the hematologic clinical samples, which were not FFPE, the difference was ∼12-fold (9.5 × 10−6 per base compared to 1.1 × 10−4 per base), and for FFPE clinical samples, the difference was approximately fivefold (2.7 × 10−5 per base compared to 1.3 × 10−4 per base). We then explored substitution rates as a function of the expected nucleotide incorporated into the MIP during the gap-fill versus the observed nucleotide in the fr-read or smc-read base-call (Fig. 3A). We observed substantial variation with respect to different pairs of expected/observed nucleotides, as well as an effect of dinucleotide context (Supplemental Fig. S5). In particular, we observed that two single-nucleotide substitutions, cytosine to adenine and guanine to adenine, exhibited elevated rates compared to all other substitutions across all sample types (Fig. 3B).
Table 2.
Substitution error rates
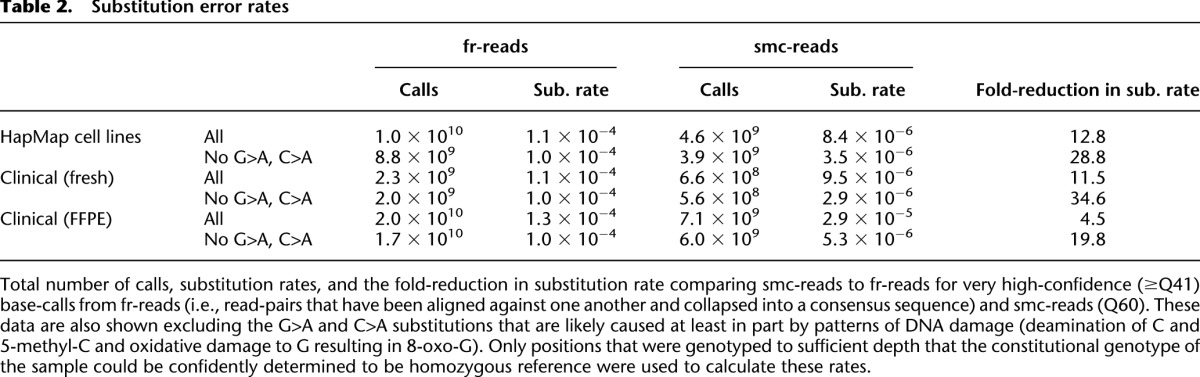
Figure 3.

Substitution error rates as a function of expected and observed nucleotide during gap-fill. (A) Schematic illustrating mononucleotide and dinucleotide substitution dependencies being considered. All rates are shown for a given expected gap-fill mono- or dinucleotide, which is the complementary nucleotide(s) to the nucleotide(s) present in the target genomic DNA, considering only ≥Q41 fr-read base-calls and Q60 smc-read base-calls at putative homozygous positions based on GATK calls. (B) Distributions of substitution error rates for eight HapMap cell line and 45 clinical cancer samples, comparing fr-reads and smc-reads, and all substitutions other than C>A or G>A (W>N + N>B, left) to only C>A (middle), or G>A (right). (C) Distributions of substitution error rates comparing fr-reads and smc-reads, and all G>A substitutions occurring in the non-CG dinucleotide context (DG>DA + GN>AN, left) to G>A substitutions occurring only in the CG dinucleotide context (CG>CA, right).
One potential explanation for these patterns is the occurrence of pro-mutagenic chemical processes in individual progenitor molecules, namely, the oxidatively damaged base 8-oxo-guanine (manifested as a cytosine to adenine substitution) (Shibutani et al. 1991) and spontaneous deamination of cytosine and 5-methyl-cytosine (manifested as a guanine to adenine substitution) (Fig. 3B,C). After smc-read formation, elevated substitution rates potentially attributable to 8-oxo-guanine damage are evident across all sample types, but are most markedly elevated in the FFPE clinical samples, suggesting that FFPE treatment may increase 8-oxo-G formation rates. To further investigate the possibility of cytosine deamination, we asked whether the G-to-A substitutions were more frequent in the CpG context, which could be consistent with deamination of 5-methyl-cytosine (Fig. 3C). Indeed, the G-to-A substitution was more frequent in the CpG context across all sample types compared with all other G-to-A substitutions, but the CpG dinucleotide is rare and is not sufficient to fully explain the elevated G-to-A substitution rate (Supplemental Table S3). Finally, we asked whether the corresponding C-to-T mutation rate was also elevated in the CpG context, which would be expected if 5-methyl-C deamination were occurring spontaneously in a replicating population of cells. We observed modest elevation of C-to-T in the CpG context (Supplemental Fig. S5D). Together, these patterns suggest a combined role of in vivo 5-methyl-cytosine deamination as well as ex vivo deamination of 5-methyl-C-to-T and C-to-U. This is consistent with the properties of the polymerase used during the gap-fill step, which, as a proofreading polymerase, is expected to stall at Uracil residues (Lasken et al. 1996), but also bypasses these lesions at some rate (Greagg et al. 1999).
When we removed the contribution of these potential sources of false-positive substitution calls, smc-read base-calls were even more accurate, with substitution rates of 3.5 × 10−6 per base and 5.1 × 10−6 per base for the HapMap and clinical samples, respectively, while the substitution rates of the fr-read calls did not decrease substantially (Table 2). These patterns are consistent with gap-fill misincorporations due to DNA damage and actual subclonal heterogeneity constituting the major source of substitutions in smc-read base-calls, and with polymerase errors after the initial gap-fill event constituting a major source of substitutions in fr-reads. Smc-reads are, therefore, at least fivefold and as much as 30-fold more accurate than the most confident base-calls in the fr-reads, with substitution rates as low as 3.5 × 10−6 per base when ignoring only two of 12 possible substitutions, or 8.4 × 10−6 per base when considering all possible single nucleotide substitutions.
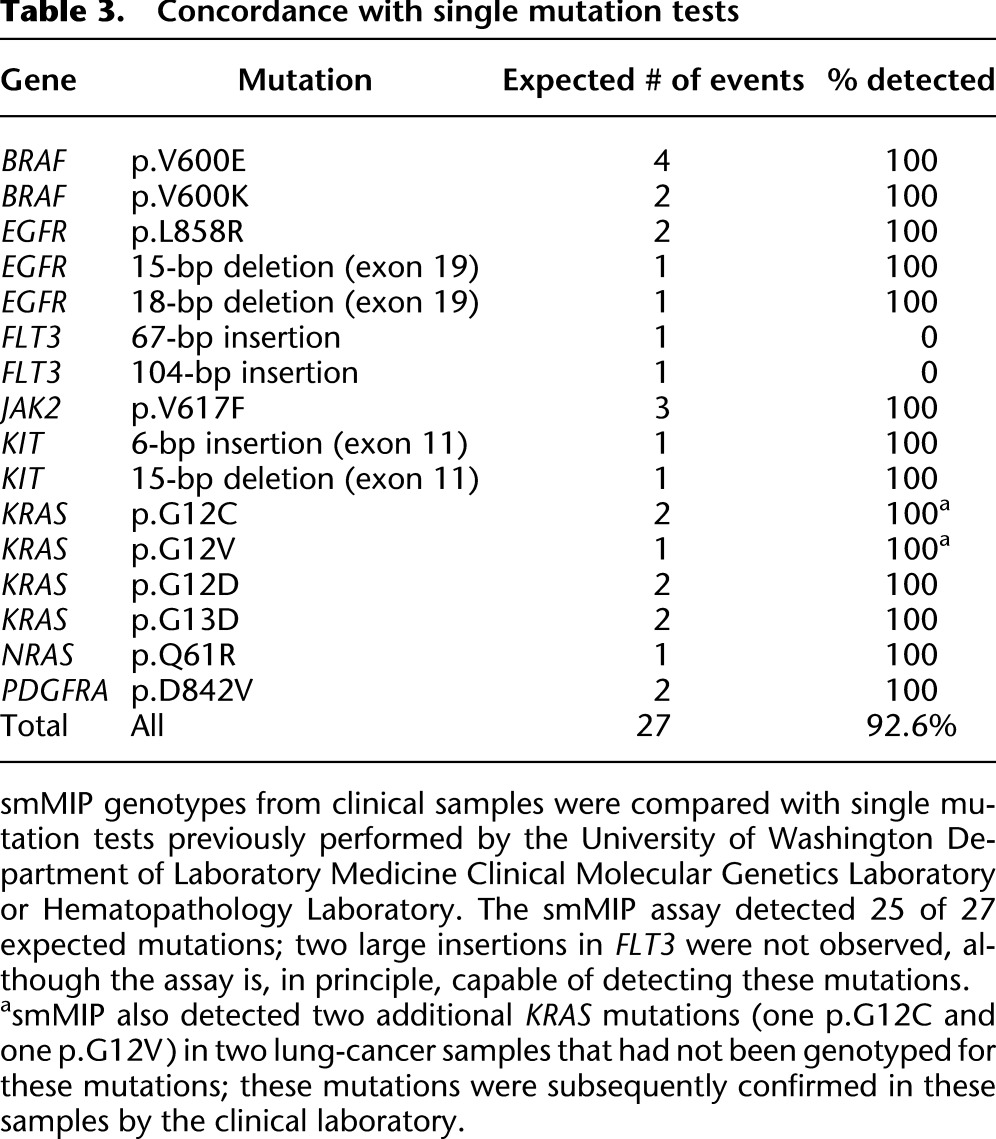
We also sought to determine the sensitivity and false discovery rate (FDR) of smMIP for low-frequency variation more generally. We used the single-nucleotide substitution error rates calculated as described above to compute P-values for each subclonal variant and adjusted these P-values to account for multiple testing. We then explored sensitivity and FDR in the synthetically mixed HapMap samples. This analysis was limited by the small number of sites that were divergent between the two individuals in the targeted coding regions (n = 18). However, we found that smc-reads were generally more sensitive at a given FDR over fr-reads for variation present at frequencies down to 0.2% (Fig. 4). For example, at an FDR of 20%, sensitivity ranged from 94% for variants at 6%–11% frequency to 44% for variants at 0.2%–0.4% frequency, and variant calls from smc-reads were 3%–22% more sensitive than the fr-reads at the same FDR cutoff across the various mixtures.
Figure 4.

Sensitivity and false discovery rates for subclonal variation in synthetic mixtures. Sensitivity versus false discovery rate for low-frequency variants (0.1%–40%) in synthetically mixed HapMap samples for variant calls from fr-reads (red) and smc-reads (blue), for coding positions that were adequately genotyped in both unmixed HapMap samples and for which there was no substantial (binomial adjusted P < 10−10) subclonality in the predominant HapMap sample. Expected subclonal variant frequencies are listed at the top of each panel. Area beneath the curve is shown as an inset in each panel. Candidate subclonal variants occurring in coding sequence and at a frequency of at least 0.1% were prioritized using multiple testing-adjusted binomial P-values that were calculated from substitution error rates.
Detection of somatic variation
Because of the simple experimental workflow, the smMIP assay could also be useful in clinical and high-volume research settings as a replacement for single-gene testing for clinically informative mutations. To assess whether the smMIP method can potentially replace such tests, we performed a blinded comparison of smMIP results to the results of clinical single-gene tests. In particular, a subset of our samples had been previously genotyped for individual actionable substitution and indel mutations in BRAF, EGFR, FLT3, JAK2, KIT, KRAS, NRAS, and PDGFRA. Considering these sites, we detected 25 of 27 (93%) previously identified mutations (Table 3; Supplemental Tables S4, S5). We missed two large (67- and 104-bp) insertions in FLT3, although these could in principle be detected using a more sensitive analysis strategy and/or more densely tiled probes in this region. We further detected two mutations in these sites in two lung cancer samples that had not been previously genotyped at that site (KRAS p.G12C in sample 8 and p.G12V in sample 37); these calls were subsequently confirmed using a melt curve-based assay (data not shown).
Table 3.
Concordance with single mutation tests
To explore other somatic mutations, and because we did not have access to matched normal tissue, we filtered variant sites identified in the clinical cancer specimens against germline variant sites observed in ∼5400 exomes by the Exome Sequencing Project (http://evs.gs.washington.edu/EVS/). We then required at least 30× coverage to remove poorly ascertained sites. Across the 45 clinical samples, this filtering process yielded 134 putative somatic events, of which 74 were found in the Catalogue of Somatic Mutations in Cancer (Supplemental Fig. S6A; Forbes et al. 2011). As expected, several genes were recurrently mutated in specific tumor types (Supplemental Fig. S6B), such as 11 of 16 colon cancer samples harboring at least one APC mutation.
To investigate the possibility of tumor subclones in these specimens, we examined the extent to which putative somatic mutations were observed at similar frequencies within individual samples (Supplemental Fig. S7A). While some clustering of alternate allele frequencies is apparent, we observed substantial variation of alternate allele frequencies within individual samples, which may reflect the presence of multiple, genetically distinct subclones. Copy-number gain is another potential source of alternate allele frequencies substantially different from 0.5 or 1. To explore the possible contribution of copy-number change to allele frequency variation, we also examined alternate allele frequencies for putatively germline variant sites for all clinical samples and the two pure HapMap cell line samples (Supplemental Fig. S7B). Alternate allele frequencies for germline variants appeared substantially more variable in a subset of clinical samples compared with the cell line samples, which is consistent with a contribution of copy-number gain to the observed variation in allele frequencies. We cannot exclude the possibility that other factors, including systematic allelic bias, are also contributing to this phenomenon. However, the general precision observed in the HapMap cell line mixtures decreases the likelihood of widespread systematic allelic bias.
Subclonal somatic variation at clinically informative sites
We next explored the potential for the smMIP assay to detect very low-frequency substitutions at clinically informative sites (BRAF p.V600, EGFR p.L858, JAK2 p.V617, KRAS p.G12/p.G13, NRAS p.Q61, and PDGFRA p.D842) in the clinical samples. These sites were not chosen because of any particular sequence or error rate characteristics, but rather to enable confirmation of subclonal variant calls with established assays and to facilitate interpreting the possible biological significance of putative subclonal variation. Restricting to maximum quality smc-read base-calls but without any filtering for variant frequency or confidence, we detected 17 candidate subclonal variants at these sites across the 45 clinical samples. However, these candidate variants exhibited a highly non-uniform distribution of error probabilities (calculated as described for the HapMap subclonal variants) with seven candidate variants having P < 10−7 and the remaining 10 having P > 10−2. We therefore focused further analysis on the seven high-confidence candidates (Table 4).
Table 4.
Low-frequency variation at clinically relevant sites in tumor samples
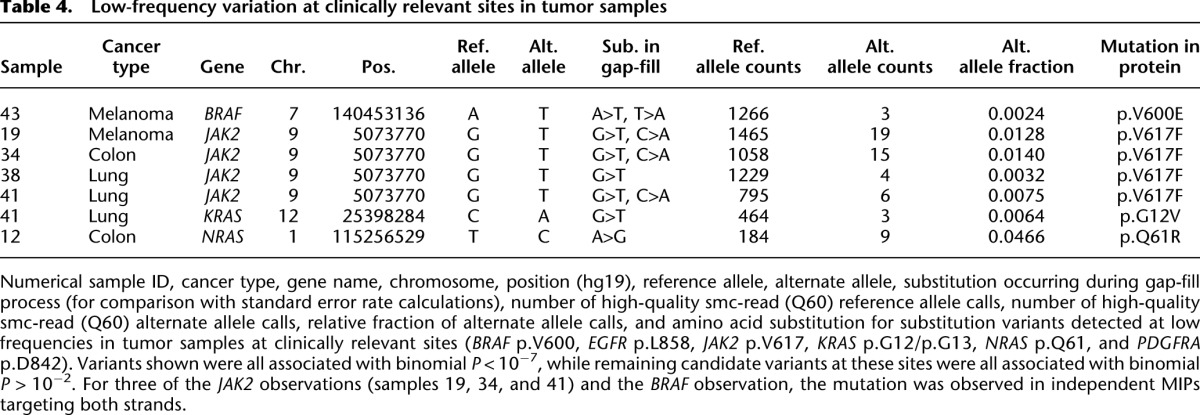
These seven subclonal variants consisted of low-frequency JAK2 p.V617F mutations (n = 4) in two lung cancers, a melanoma, and a colon cancer; a BRAF p.V600E mutation in a melanoma; a KRAS p.G12V mutation in one of the lung tumors that also harbored a low-frequency JAK2 p.V617F mutation; and an NRAS p.Q61R mutation in a colon cancer. To exclude the possibility of artifactual low-frequency variant detection due to sample cross-contamination or index cross-talk, we subjected independent DNA aliquots from the four specimens with low-frequency JAK2 mutations to confirmatory clinical testing using an allele-specific PCR assay; all four mutations were confirmed. Furthermore, JAK2 mutations have been reported at low frequency in a previous study in non-small cell lung cancer samples (Lipson et al. 2012). Additionally, index sequence cross-talk, which would be predicted to give rise to mixed read groups, is not a likely explanation for these low-frequency calls; we required very high-quality smc-read base-calls, and mixed read groups are not a general phenomenon in our data (Supplemental Fig. S8). We note that, prior to our study, the melanoma sample was genotyped clinically for BRAF p.V600, and this mutation was not detected (as expected given that assay's limited sensitivity); additionally, polyclonality in melanomas with respect to BRAF p.V600 mutation status has been previously observed (Lin et al. 2009, 2011; Yancovitz et al. 2012).
We therefore expect, based on experimental and biological evidence, that these seven candidate variants are bona fide. However, based on the FDR analysis performed using the HapMap samples, the P-value cutoff of 10−7 is expected to yield a nontrivial FDR of ∼40% for variants near 0.1% frequency (Supplemental Fig. S9), so it remains possible that one or more of these variants is artifactual.
Rapid workflow characterization
Finally, we sought to develop a rapid smMIP workflow using the Illumina MiSeq platform to enable return of results on a clinically useful timescale (Supplemental Table S6). In addition to using the more rapid sequencing instrument, we further streamlined the experimental protocol. To assess the performance of this revised workflow, we applied it to eight of the clinical samples that we had already characterized. Five of these samples harbored low-frequency variation at clinically relevant sites as described above; the other three were selected at random. Briefly, we observed excellent agreement between genotype calls using the slow workflow (high-coverage) and rapid workflow (low-coverage) approaches (Supplemental Table S7). A more thorough discussion of these results can be found in Supplemental Note S4.
Discussion
Here, we combined technologies for single molecule tagging and molecular inversion probes toward the development of a practical and highly multiplexed method for ultrasensitive detection and precise quantitation of subclonal genetic variation. Smc-reads represent the consensus of reads derived from the same progenitor molecule in genomic DNA, and the molecular tagging inherent to the smMIP assay facilitates error correction down to a substitution rate of 8.4 × 10−6 per base. Errors in smc-reads predominantly exhibit patterns consistent with DNA damage, namely, the oxidative adduct 8-oxo-guanine and deamination of cytosine or 5-methyl-cytosine, and this pattern is observed in DNA from cell lines as well as fresh and FFPE clinical specimens, but is especially elevated in FFPE specimens. Furthermore, the smMIP assay is highly quantitative for alternate allele frequencies as low as ∼0.2% and enables sensitive and specific detection of variation present down to at least 1%.
When we applied this method using probes targeting 33 clinically informative cancer genes to a diverse panel of genomic DNAs including 45 clinical cancer specimens (40 of which were FFPE), we observed strong concordance with expected mutations based on clinical single-gene tests, and discovered as many new mutations at clinically informative sites as we missed. Overall, we detected 134 putatively somatic coding mutations across the 45 clinical samples, and also identified seven high-confidence low-frequency variants at clinically informative sites. Finally, we established and validated a simple and rapid smMIP workflow that is capable of going from DNA sample to analyzed result in less than 72 h.
The smMIP assay has important advantages over alternative approaches, including hybrid capture (Lipson et al. 2012; Wagle et al. 2012) and highly multiplexed PCR (Tewhey et al. 2009; Harismendy et al. 2011; Forshew et al. 2012). Compared with hybrid capture, smMIP offers very low per-sample reagent costs and a substantially simpler and more rapid workflow. Furthermore, the capture reagent is modular, meaning new probes can be added “on-the-fly.” Alternatively, the reagent could be split into single gene pools and combined as desired for small batches of samples to most efficiently leverage the rapid turnaround and lower throughput of the bench-top sequencing platforms. Finally, molecular tagging facilitates single molecule consensus base-calling without relying on pseudo-random fragmentation breakpoints, which may not be informative at high-sequencing depths. However, a smMIP-based approach will not likely scale as well to very large targets (i.e., thousands of genes), and may be less sensitive to large-scale genomic rearrangements. Compared with highly multiplexed droplet (Tewhey et al. 2009; Harismendy et al. 2011) or microfluidic (Forshew et al. 2012) PCR, smMIP does not rely on sophisticated instrumentation, and, because of molecular tagging, is more sensitive and quantitative for low-frequency variation. However, smMIP may not be as compatible with very low sample inputs (i.e., less than ∼10 ng), because the initial enrichment step is nonexponential.
There are a number of ways in which the smMIP assay can likely be improved. Coverage of poorly captured sites and capture uniformity from probe to probe will be improved by further probe rebalancing and supplementation of the probe set to more densely tile problematic regions. Further optimization of the capture protocol directed toward reducing formation of undesired low-molecular weight artifacts should improve mapping rates, increasing sensitivity and reducing or eliminating the need for time- and labor-intensive size-selection steps during sample preparation. Another opportunity is the development of higher resolution models to prioritize variants, potentially using dinucleotide or even site-specific error rates. Application of the smMIP method to larger numbers of samples is expected to facilitate the development of such models. Finally, improved algorithms and/or probe content may also facilitate the detection of loss-of-heterozygosity and copy-number changes.
A single multiplex assay that is capable of accurately and sensitively identifying subclonal variation in a large panel of genes has the potential to enable new avenues of research. For example, in one colon cancer sample, we identified a KRAS p.G13D mutation and a low-frequency NRAS p.Q61R mutation. NRAS mutations are infrequent (∼5% overall; ∼3% p.Q61; ∼1% p.Q61R) in KRAS wild-type colon cancers (Vaughn et al. 2011), but have similar implications, i.e., reduced response to the targeted anti-EGFR monoclonal antibodies cetuximab and panitumumab (Maughan et al. 2011), and KRAS mutations have been detected in ∼20% of cancers also harboring NRAS mutations (Maughan et al. 2011). Furthermore, colon cancers harboring KRAS p.G13D, unlike those harboring other KRAS mutations, may remain responsive to cetuximab/panitumumab (De Roock et al. 2010), although this finding was not replicated in a subsequent study (Maughan et al. 2011). Based on our observation, one possible explanation for the disagreement between those studies is that some subset of tumors harboring KRAS p.G13D also harbor NRAS mutations at clonal or subclonal frequencies, and that NRAS mutation status is also influencing response to antibody therapy. Further study will be needed to better establish the prevalence of co-occurring KRAS p.G13D and clonal or subclonal NRAS mutations and the relationship between mutational status and response to therapy. Characterization of large panels of archival clinical specimens using the smMIP assay could be used to address this and other questions.
Genetic heterogeneity in populations of cells derived from a clonal origin is a fundamental aspect of biology and has important implications in fields ranging from evolution to cancer. However, DNA sequencing methods have been largely blind to subclonal variation because of limitations in throughput and/or sequencing error rate. In the long term, researchers will require genomic characterization methods that include the reliable detection and quantitation of low-frequency mutations. For example, genetic heterogeneity is emerging as a common attribute of human cancers (Navin et al. 2011; Carter et al. 2012; Gerlinger et al. 2012; Nickel et al. 2012; Nik-Zainal et al. 2012) and the rapid emergence of resistance to therapy in some cancers may be explained in part by the pre-existence of subclonal resistance mutations (Diaz et al. 2012; Misale et al. 2012). We anticipate that a practical, sensitive, and accurate method for targeted subclonal variation detection will enable the design and execution of many more and much larger studies than have been previously possible. The speed, simplicity, parallelizability, and very low substitution error rate (≤3 × 10−5) of the smMIP assay also raises the possibility of processing multiple independent samplings of a given specimen over time and space. In cancer, the smMIP assay could be applied to multiple biopsies from the same tissue mass or multiple independent metastases (Gerlinger et al. 2012; Nickel et al. 2012), while in the case of somatic mosaicism, many tissue specimens of diverse embryological origin could be analyzed. Finally, the smMIP assay could be useful in other scenarios where the sensitive and precise quantification of low-frequency variation is relevant, e.g., the detection of extremely low-frequency cancer-related mutations in circulating cell or cell-free DNA (Diehl et al. 2008; Forshew et al. 2012), or as a complementary approach for noninvasively assaying fetal DNA from maternal plasma (Fan et al. 2012; Kitzman et al. 2012) at clinically relevant sites.
Methods
Preparation of smMIP capture reagent
MIPs were designed as described elsewhere (O'Roak et al. 2012a) against the coding exons of 33 cancer-related genes (Supplemental Table S1) with 50 nt of “splash” on either side of each exon. We designed probes with targeting arms summing to 40 nt in length, with extension arms ranging in length from 16 to 20 nt and ligation arms ranging in length from 20 to 24 nt. The gap-fill length was fixed to 112 nt. Targeting arms were joined by a constant 40-mer “backbone” sequence (common oligo sequences can be found in Supplemental Table S9) containing a stretch of 12 random nucleotides, such that each probe could exist in ∼412 = 1.67 × 106 unique sequences. After adding probes to accommodate sites of common variation in the genome that fell in targeting arms, we had a set of 1312 probes targeting ∼88 kb of coding sequence and ∼125 kb overall. These 80-mer probes were procured individually as column-synthesized oligos at 25 nanomole scale in 96-well plate format without any modifications or purification at a cost of $7.20 per probe. While a nontrivial up-front cost, this represents an effectively infinite supply, as each capture reaction consumes less than one 10-millionth of the supply of a given probe. Aliquots of each probe were pooled at equimolar ratios and 85 μL of this pool was 5′-phosphorylated using 50 units of T4 Polynucleotide Kinase (NEB) and 1× T4 DNA ligase buffer in a total volume of 100 μL for 45 min at 37°C, followed by 20 min at 80°C to inactivate the kinase. Test captures using cell line genomic DNA were then carried out as described below, using the equimolar probe pool at a 1000-fold probe-to-target molar excess. Based on sequencing results from these test capture reactions, probes were ranked with respect to capture efficiency and the worst-performing ∼30% of probes were spiked into the main probe pool at a 100-fold relative molar excess.
Capture and library construction
Genomic DNA for HapMap cell line samples (NA12892 and NA19239) was purchased (Coriell). Clinical specimens consisted of DNA prepared from formalin-fixed paraffin-embedded (FFPE) tissue, peripheral blood, or bone-marrow aspirates from patients with sporadic colorectal cancer (n = 18), melanoma (7), non-small cell lung cancer (11), bladder cancer (1), ovarian cancer (1), gastrointestinal stromal tumor (4), acute myeloid leukemia (2), and myeloproliferative disorders (3). De-identified residual clinical specimens were obtained from the University of Washington molecular diagnostics laboratory in accordance with the declaration of Helsinki and ethics guidelines of the local institutional review board. Specifically, the de-identified samples were determined by the institutional review board to be nonhuman subjects research. Hematoxylin and Eosin-stained slides were used as a guide to manually dissect areas of tumor tissue from unstained slide sections for FFPE tissue samples. Genomic DNA was prepared with the Gentra Puregene DNA Isolation Kit (Qiagen). A 3-h to overnight proteinase K digestion step was included for FFPE samples. Genomic DNA from the HapMap sample NA12892 was serially diluted twofold and added to 500 ng of genomic DNA from HapMap samples NA19239 at six relative ratios ranging from 1:8 to 1:256. Captures of the six cell line mixtures, two pure cell line samples, and 47 clinical samples were then performed using ∼500 ng of each genomic DNA.
Captures were performed as previously described (Shen et al. 2011) with some modifications. A total of 500 ng of genomic DNA, 330 femtomoles of probe mixture (ignoring the contribution of the spiked-in poor performers), and 1 μL of 10× Ampligase DNA ligase buffer (Epicentre) were added to molecular biology-grade water for a total of 10 μL. For the probe hybridization phase, these mixtures were incubated in a thermocycler (Bio-Rad) with a heated lid at 98°C for 3 min, 85°C for 30 min, 60°C for 60 min, and 56°C for 120 min. For the gap-fill and ligation phase, we added 300 picomoles each dNTPs (NEB), 7.5 micromoles betaine (Sigma), 20 nanomoles NAD+ (NEB), 1 μL of 10× Ampligase buffer, 5 units of Ampligase DNA ligase (Epicentre), 3.2 units of Phusion DNA polymerase (NEB), and molecular biology grade water to 10 mL for a total reaction volume of 20 μL. The gap-fill and ligation phase was carried out at 56°C for 60 min and 72°C for 20 min.
Following the gap-fill and ligation phase, the reactions were cooled to 37°C, and to each reaction we added 20 units of Exonuclease I (NEB) and 100 units of Exonuclease III (NEB) to degrade uncircularized probe and genomic DNA. The digestion was incubated at 37°C for 45 min, heated to 80°C, and incubated for 20 min to inactivate the exonucleases.
After exonuclease treatment and heat-inactivation, the samples were cooled on ice and, optionally, stored at −20°C. For each capture reaction, two PCR reactions were prepared, each with Phusion HF buffer to 1× (Fermentas), forward primer and indexed reverse PCR primers to 500 nM, SYBR green (Invitrogen) to 0.5×, dNTPs to 200 μm each (NEB), 2 units of Phusion Hot-Start II polymerase, 10 μL of capture reaction, and nuclease-free water to 50 μL. PCR cycling conditions were an initial denaturation step for 2 min at 95°C, followed by 26 cycles of: 15 sec at 98°C, 15 sec at 65°C, and 45 sec at 72°C. A subset of samples was run on a real-time PCR instrument (Bio-Rad MiniOpticon) to estimate the required number of cycles; the remaining samples were run without real-time monitoring (Bio-Rad DNA Engine Tetrad 2).
Library purification and pooling
PCR products were purified individually using Ampure XP beads (Agencourt) at 1.8× according to the manufacturer's instructions. Purified PCR products were then pooled naively (i.e., equal volumes) for initial quality control or based on MiSeq sequence data considering the number of reads mapping on target per sample. To remove a low molecular-weight artifact, the PCR product pool was split across 8 wells of a 10-well pre-cast 6% polyacrylamide TBE gel (Invitrogen), run at 140 V for 50 min, and stained with 5 μL of SYBR Gold (Invitrogen). The capture product band at ∼280 bp was excised, crushed, soaked in 800 μL of Tris-EDTA (pH 8.0), and recovered from the supernatant using 100 μL of Ampure XP beads and 700 μL of home-made Ampure buffer (20% PEG 8000, 2.5M NaCl) according to the manufacturer's instructions. Pool concentration was assessed using Qubit (Invitrogen).
As an alternative strategy to reduce the time, labor, and cost required for library construction, purified PCR products were subjected to an Ampure-based size enrichment and normalization step. Twenty microliters of each purified PCR product was purified using 16 μL of a mixture of one part Ampure XP bead solution and four parts homemade Ampure buffer, and eluted in 20 μL of Buffer EB (Qiagen). Two microliters of each sample was then run on a diagnostic precast 6% polyacrylamide TBE gel as described above to assess relative concentrations of capture product. The gel image was analyzed for band intensity (ImageJ), and the purified PCR products were pooled according to relative band intensity and the pool was quantified via Qubit.
Sequencing and primary analysis
Samples were sequenced using the HiSeq 2000 and MiSeq (Illumina) platforms according to the manufacturer's instructions using custom sequencing primers (Supplemental Table S9). On the HiSeq platform, we collected two 101-nt reads to determine the sequence of the gap-fill and the molecular tag and one 8-nt read to determine the sequence of the sample index. On the MiSeq platform, we collected two 152-nt reads and one 8-nt read. Initial quality control and capture performance of an equivolume, non-size selected pool of all 55 samples was assessed using one run of the MiSeq platform. Purified PCR products were then repooled according to MiSeq data, size-selected using a PAGE gel as described above, and subjected to 2.75 lanes of HiSeq 2000 sequencing (two lanes with no other samples mixed in and one lane with 25% by moles of an unrelated library). For the establishment of a rapid workflow, eight samples were processed as described above and subjected to one run of the MiSeq platform.
Read-pairs were assigned to samples requiring an exact match to the expected 8-nt sample index sequence (Supplemental Table S9) and the first 12 nt of the reverse read (corresponding to the molecular tag sequence) were stripped out and placed in the header. Read-pairs with molecular tags with homopolymers longer than 4 nt were discarded. Overlapping regions of read-pairs were then reconciled to form single “fr-reads” using a custom Smith-Waterman-based strategy. For positions where the read-pairs did not overlap, quality scores from the individual reads were retained. For positions where the read-pairs did overlap, quality scores for the resulting consensus calls were estimated as below for smc-reads. Only successfully overlapped fr-reads were retained for downstream analysis; read-pairs that failed to merge were discarded, although subsequent implementations aimed at greater sensitivity toward large insertions such as those found in FLT3 will retain and analyze these reads, and smMIP does not explicitly require collecting overlapping read-pairs. Fr-reads were aligned to the human reference genome (hg19) using the bwasw alignment mode of the aligner bwa (Li and Durbin 2010) (v0.5.9) with non-default parameters “-r 1”. Based on expected alignment positions according to the probe design, fr-reads were then assigned to individual probes, allowing 1 nt of tolerance in each direction for the beginning of the read, which primarily accommodates insertion and deletion mutations during probe synthesis. Then, for each sample and each probe, fr-reads were grouped by molecular tag sequence to form tag-defined read groups (TDRGs).
Single molecule consensus read calling
Alignments to the reference genome were used to call a consensus sequence (i.e., a single molecule consensus read or “smc-read”) for each TDRG. Positions expected to be derived from probe targeting arms (and therefore synthetic DNA) were excluded from consideration at this step. We adopted a likelihood ratio framework to incorporate both the abundance and associated quality-scores of fr-read base-calls supporting each possible nucleotide at each position. Briefly, we calculated the log-likelihood Lx of consensus nucleotide x as the difference of the log-likelihood of a model in which a given nucleotide x was underlying none of the calls and the log-likelihood of a model in which x was underlying all of the calls. This can be represented as in the equation below, where a given observation is associated with a nucleotide call o and a “phred-like” quality score Qo.
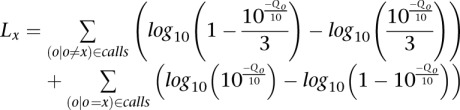 |
This value (Lx) is computed for each possible consensus call (A, C, G, T, N, and deletion) at each position in the alignment based on the set of base-calls and associated quality scores in the TDRG at that position in the alignment. The consensus call is then determined as the call with the minimum (i.e., most negative) log-likelihood value, as this indicates the consensus nucleotide where the model assuming that nucleotide did not underlie any of the fr-readcalls was the least likely relative to the model assuming that nucleotide underlay all of the fr-read calls. The final phred-like quality score is calculated as the integer casting of −10*Lx. For example, in the event that a model against a given consensus nucleotide is 10−3 times as likely as a model for a given consensus nucleotide, the associated quality score was calculated to be 30. These scores were capped at 60 as we did not observe substitution rates substantially below 1 × 10−6 in practice. We note that a smc-read base-call that is derived from a single Q60 fr-read base-call (which may have been derived from two Q40 calls at an overlapping position, for example) will be assigned an estimated quality score of 59 because of the integer casting step. In practice, therefore, only smc-read base-calls that were supported by at least two independent fr-read base-calls can attain quality 60.
For reference positions where at least one read in the alignment indicated a deletion and at least one a match, or where at least one read contained an insertion relative to reference and at least one read lacked that insertion, we applied the same framework, assigning deletion calls a phred-like quality score of 40. We note that this framework makes simplifying assumptions: that a given nucleotide, in the event of a sequencing error, is equally likely to give rise to any of the other three substitution nucleotides, and that a single nucleotide truly underlies all calls across all reads in the TDRG. Furthermore, for interpretability, we used three as the denominator to distribute the probability when the observed nucleotide was not the candidate consensus nucleotide, though we computed this value for a total of six possible consensus calls and not four.
This strategy was also used to determine quality scores for consensus calls at overlapping positions in fr-reads, which represents the simpler case of two and only two calls.
Variant calling and classification
To accommodate variants present across a wide range of frequencies, we adopted a two-pronged variant-calling strategy. First, to detect variants present at higher frequencies (i.e., ∼10% or higher) we used alignments of fr-reads and smc-reads for each sample individually (i.e., single sample calling) as inputs to the Genome Analysis Toolkit (GATK, v1.6-5-g557da77) (McKenna et al. 2010) variant caller “UnifiedGenotyper” with non-default command line options as follows:
-U ALLOW_UNSET_BAM_SORT_ORDER\
–output_mode EMIT_ALL_SITES\
–downsampling_type NONE\
–genotype_likelihoods_model BOTH\
–read_filter BadCigar\
–min_base_quality_score 20
Variants were then filtered using the GATK tool “VariantFiltration” using the following non-default command line options:
–filterExpression “QD < 10.0”\
–filterName “LowQD”\
–filterExpression “DP < 30”\
–filterName “LowDP”
Variants flagged as “LowDP” were ignored in all analyses. Variants with low “quality-by-depth” according to GATK (i.e., QD < 10, “LowQD”) were ignored when attempting to discover germline variation (i.e., for the unmixed HapMap samples) and retained when attempting to discover somatic variation. Variants called by GATK were used to determine concordance between smMIP genotypes and 1000 Genomes genotypes to exclude sites from consideration when quantifying substitution rates and for the detection of somatic variation in clinical samples. Variants were annotated using the SeattleSeq webserver (http://snp.gs.washington.edu/SeattleSeqAnnotation134/).
To detect low-frequency variation and quantify substitution rates, we adopted a distinct strategy. Alignments of fr-reads and smc-reads were considered directly and base-calls at putative homozygous reference sites (according to genotypes called by GATK) were tabulated, considering only very high-quality calls (at least Q41 for fr-reads, Q60 for smc-reads). We note that some, but not all positions in fr-reads can attain higher-quality scores via the merging process (up to a maximum of 60 as specified by the quality score estimation process described above). To estimate confidence in and prioritize subclonal variant calls, we used empirically computed error rates for each of the 12 possible single-nucleotide substitutions and assumed a binomial error process to estimate the probability of observing a given number of variant calls against a background of reference calls for each position. We then adjusted these P-values for multiple testing in R using the function p.adjust() with method=”BH”.
To categorize variation as putative germline or somatic, we performed several filtering steps. First, we obtained a list of sites (“ESP5400”) that had been detected as variant in at least one of 5400 exomes sequenced at the University of Washington as part of the NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/). Because we observed that some sites in this list were also present in the COSMIC database (CosmicMutantExport_v59_230512.tsv obtained from ftp://ftp.sanger.ac.uk/pub/CGP/cosmic/data_export/), we first filtered the ESP5400 list to remove these positions (e.g., JAK2 p.V617F). Next, variants were categorized as putative germline if they occurred at a site present in the ESP5400 list that had been filtered of COSMIC variant sites. Remaining variant sites were then compared with the COSMIC list and categorized thusly.
Data access
All raw sequencing data collected for this study have been deposited in the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under accession number SRA064171. All custom analysis scripts are available by request.
Competing interest statement
J.S. is a member of the scientific advisory board or serves as a consultant for Ariosa Diagnostics, Stratos Genomics, Good Start Genetics, and Adaptive Biotechnologies. A patent application has been filed for aspects of the methods disclosed here (“Subassembly of Short Sequencing Reads”; 12/559124).
Acknowledgments
We thank C. Lee for assistance with sequencing, and members of the Shendure Lab for helpful discussions. We thank the NHLBI GO Exome Sequencing Project and its ongoing studies that produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010). We thank C. Smith, K. Koehler, M. Murillo, and H-S. Yi for help genotyping clinical samples. Our work was supported by a gift from the Washington Research Foundation; grant CA160080 from the National Cancer Institute (J.S.); grant AG039173 from the National Institute on Aging (J.B.H.); a fellowship from Achievement Rewards for College Scientists (J.B.H.); and the Department of Laboratory Medicine, University of Washington Medical Center.
Author contributions: J.B.H., C.C.P, S.J.S., and J.S. conceived and designed the study. J.B.H. and B.J.O. designed the smMIPs and developed protocols. C.C.P. and S.J.S. obtained anonymized clinical samples, coordinated or oversaw single mutation genotyping, and aided with interpretation of results. J.B.H. performed all other experiments and all data analysis. J.B.H. and J.S. wrote and revised the manuscript with substantial input and revisions from all other authors. J.S. supervised all aspects of the study.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.147686.112.
References
- The 1000 Genomes Project Consortium 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielas JH, Loeb LA 2005. Quantification of random genomic mutations. Nat Methods 2: 285–290 [DOI] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. 2012. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30: 413–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casbon JA, Osborne RJ, Brenner S, Lichtenstein CP 2011. A method for counting PCR template molecules with application to next-generation sequencing. Nucleic Acids Res 39: e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, Lamba S, Arena S, Frattini M, Piessevaux H, et al. 2010. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 304: 1812–1820 [DOI] [PubMed] [Google Scholar]
- Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al. 2012. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486: 537–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, et al. 2008. Circulating mutant DNA to assess tumor dynamics. Nat Med 14: 985–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B 2003. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci 100: 8817–8822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druley TE, Vallania FL, Wegner DJ, Varley KE, Knowles OL, Bonds JA, Robison SW, Doniger SW, Hamvas A, Cole FS, et al. 2009. Quantification of rare allelic variants from pooled genomic DNA. Nat Methods 6: 263–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan HC, Gu W, Wang J, Blumenfeld YJ, El-Sayed YY, Quake SR 2012. Non-invasive prenatal measurement of the fetal genome. Nature 487: 320–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty P, Natsoulis G, Muralidharan O, Winters M, Buenrostro J, Bell J, Brown S, Holodniy M, Zhang N, Ji HP 2012. Ultrasensitive detection of rare mutations using next-generation targeted resequencing. Nucleic Acids Res 40: e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. 2011. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 39: D945–D950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, Dawson SJ, Piskorz AM, Jimenez-Linan M, Bentley D et al. 2012. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 4: 136ra168. [DOI] [PubMed] [Google Scholar]
- Fu GK, Hu J, Wang PH, Fodor SP 2011. Counting individual DNA molecules by the stochastic attachment of diverse labels. Proc Natl Acad Sci 108: 9026–9031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. 2012. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366: 883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstung M, Beisel C, Rechsteiner M, Wild P, Schraml P, Moch H, Beerenwinkel N 2012. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat Commun 3: 811. [DOI] [PubMed] [Google Scholar]
- Greagg MA, Fogg MJ, Panayotou G, Evans SJ, Connolly BA, Pearl LH 1999. A read-ahead function in archaeal DNA polymerases detects promutagenic template-strand uracil. Proc Natl Acad Sci 96: 9045–9050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harismendy O, Schwab RB, Bao L, Olson J, Rozenzhak S, Kotsopoulos SK, Pond S, Crain B, Chee MS, Messer K, et al. 2011. Detection of low prevalence somatic mutations in solid tumors with ultra-deep targeted sequencing. Genome Biol 12: R124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiatt JB, Patwardhan RP, Turner EH, Lee C, Shendure J 2010. Parallel, tag-directed assembly of locally derived short sequence reads. Nat Methods 7: 119–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabara CB, Jones CD, Roach J, Anderson JA, Swanstrom R 2011. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc Natl Acad Sci 108: 20166–20171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B 2011. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci 108: 9530–9535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzman JO, Snyder MW, Ventura M, Lewis AP, Qiu R, Simmons LE, Gammill HS, Rubens CE, Santillan DA, Murray JC et al. 2012. Noninvasive whole-genome sequencing of a human fetus. Sci Transl Med 4: 137ra176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivioja T, Vaharautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, Taipale J 2012. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods 9: 72–74 [DOI] [PubMed] [Google Scholar]
- Lasken RS, Schuster DM, Rashtchian A 1996. Archaebacterial DNA polymerases tightly bind uracil-containing DNA. J Biol Chem 271: 17692–17696 [DOI] [PubMed] [Google Scholar]
- Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, et al. 2012. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet 44: 941–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26: 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang L, Mamon H, Kulke MH, Berbeco R, Makrigiorgos GM 2008. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nat Med 14: 579–584 [DOI] [PubMed] [Google Scholar]
- Lin J, Takata M, Murata H, Goto Y, Kido K, Ferrone S, Saida T 2009. Polyclonality of BRAF mutations in acquired melanocytic nevi. J Natl Cancer Inst 101: 1423–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Goto Y, Murata H, Sakaizawa K, Uchiyama A, Saida T, Takata M 2011. Polyclonality of BRAF mutations in primary melanoma and the selection of mutant alleles during progression. Br J Cancer 104: 464–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, Curran JA, Balasubramanian S, Bloom T, Brennan KW, et al. 2012. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med 18: 382–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacConaill LE, Campbell CD, Kehoe SM, Bass AJ, Hatton C, Niu L, Davis M, Yao K, Hanna M, Mondal C, et al. 2009. Profiling critical cancer gene mutations in clinical tumor samples. PLoS ONE 4: e7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamanova L, Coffey AJ, Scott CE, Kozarewa I, Turner EH, Kumar A, Howard E, Shendure J, Turner DJ 2010. Target-enrichment strategies for next-generation sequencing. Nat Methods 7: 111–118 [DOI] [PubMed] [Google Scholar]
- Maughan TS, Adams RA, Smith CG, Meade AM, Seymour MT, Wilson RH, Idziaszczyk S, Harris R, Fisher D, Kenny SL, et al. 2011. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: Results of the randomised phase 3 MRC COIN trial. Lancet 377: 2103–2114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milbury CA, Correll M, Quackenbush J, Rubio R, Makrigiorgos GM 2012. COLD-PCR enrichment of rare cancer mutations prior to targeted amplicon resequencing. Clin Chem 58: 580–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, et al. 2012. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 486: 532–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628 [DOI] [PubMed] [Google Scholar]
- Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, et al. 2011. Tumour evolution inferred by single-cell sequencing. Nature 472: 90–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, et al. 2010. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 42: 790–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel GC, Barnholtz-Sloan J, Gould MP, McMahon S, Cohen A, Adams MD, Guda K, Cohen M, Sloan AE, LaFramboise T 2012. Characterizing mutational heterogeneity in a glioblastoma patient with double recurrence. PLoS ONE 7: e35262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, et al. 2012. The life history of 21 breast cancers. Cell 149: 994–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, et al. 2012a. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338: 1619–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. 2012b. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485: 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan RP, Hiatt JB, Witten DM, Kim MJ, Smith RP, May D, Lee C, Andrie JM, Lee SI, Cooper GM, et al. 2012. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol 30: 265–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riviere JB, Mirzaa GM, O'Roak BJ, Beddaoui M, Alcantara D, Conway RL, St-Onge J, Schwartzentruber JA, Gripp KW, Nikkel SM, et al. 2012. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet 44: 934–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen P, Wang W, Krishnakumar S, Palm C, Chi AK, Enns GM, Davis RW, Speed TP, Mindrinos MN, Scharfe C 2011. High-quality DNA sequence capture of 524 disease candidate genes. Proc Natl Acad Sci 108: 6549–6554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibutani S, Takeshita M, Grollman AP 1991. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 349: 431–434 [DOI] [PubMed] [Google Scholar]
- Shiroguchi K, Jia TZ, Sims PA, Xie XS 2012. Digital RNA sequencing minimizes sequence-dependent bias and amplification noise with optimized single-molecule barcodes. Proc Natl Acad Sci 109: 1347–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, et al. 2012. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337: 64–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewhey R, Warner JB, Nakano M, Libby B, Medkova M, David PH, Kotsopoulos SK, Samuels ML, Hutchison JB, Larson JW, et al. 2009. Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nat Biotechnol 27: 1025–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner EH, Lee C, Ng SB, Nickerson DA, Shendure J 2009. Massively parallel exon capture and library-free resequencing across 16 genomes. Nat Methods 6: 315–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn CP, Zobell SD, Furtado LV, Baker CL, Samowitz WS 2011. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer 50: 307–312 [DOI] [PubMed] [Google Scholar]
- Wagle N, Berger MF, Davis MJ, Blumenstiel B, Defelice M, Pochanard P, Ducar M, Van Hummelen P, Macconaill LE, Hahn WC et al. 2012. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov 2: 82–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancovitz M, Litterman A, Yoon J, Ng E, Shapiro RL, Berman RS, Pavlick AC, Darvishian F, Christos P, Mazumdar M, et al. 2012. Intra- and inter-tumor heterogeneity of BRAFV600E mutations in primary and metastatic melanoma. PLoS ONE 7: e29336. [DOI] [PMC free article] [PubMed] [Google Scholar]