

Abstract
Although biofilms have been shown to be reservoirs of pathogens, our knowledge of the microbial diversity in biofilms within critical areas, such as health care facilities, is limited. Available methods for pathogen identification and strain typing have some inherent restrictions. In particular, culturing will yield only a fraction of the species present, PCR of virulence or marker genes is mainly focused on a handful of known species, and shotgun metagenomics is limited in the ability to detect strain variations. In this study, we present a single-cell genome sequencing approach to address these limitations and demonstrate it by specifically targeting bacterial cells within a complex biofilm from a hospital bathroom sink drain. A newly developed, automated platform was used to generate genomic DNA by the multiple displacement amplification (MDA) technique from hundreds of single cells in parallel. MDA reactions were screened and classified by 16S rRNA gene PCR sequence, which revealed a broad range of bacteria covering 25 different genera representing environmental species, human commensals, and opportunistic human pathogens. Here we focus on the recovery of a nearly complete genome representing a novel strain of the periodontal pathogen Porphyromonas gingivalis (P. gingivalis JCVI SC001) using the single-cell assembly tool SPAdes. Single-cell genomics is becoming an accepted method to capture novel genomes, primarily in the marine and soil environments. Here we show for the first time that it also enables comparative genomic analysis of strain variation in a pathogen captured from complex biofilm samples in a healthcare facility.
Ongoing efforts to understand the genomic diversity of microbes in nature and in human health are hampered by the limited availability of cultivated organisms and their genomes (The Human Microbiome Jumpstart Reference Strains Consortium 2010). Only 1%–10% of known bacterial species (Rappe and Giovannoni 2003) are thought to be currently cultivated, although great progress is being made for some bacterial communities; for example, about half of bacterial species within the human oral cavity have been cultivated (Dewhirst et al. 2010). The recent advancements in DNA sequencing of single bacterial cells (Raghunathan et al. 2005) have accelerated the discovery of uncultivated microbes (Lasken 2012), providing genomic assemblies for species previously known only from 16S rRNA clone libraries and metagenomic data (Marcy et al. 2007; Podar et al. 2007; Binga et al. 2008; Eloe et al. 2011; Youssef et al. 2011; Dupont et al. 2012). This newly developed methodology provides a culture-independent approach to capture the genomes of uncultivated organisms, which can then be integrated into many intensive genomics-based studies. A high-throughput strategy was recently established to sequence and assemble single-cell genomes of bacteria (Chitsaz et al. 2011) and viruses (Allen et al. 2011), including novel uncultivated bacteria from environmental samples (Chitsaz et al. 2011; Eloe et al. 2011; Dupont et al. 2012). The workflow consists of (1) delivery of single bacterial cells into 384-well microtiter wells by fluorescence activated cell sorting (FACS); (2) use of a robotic platform to perform 384-well automated cell lysis and amplification of DNA by the multiple displacement amplification (MDA) method (Dean et al. 2001, 2002; Hosono et al. 2003) to create libraries of genomic DNA derived from single cells; (3) PCR and cycle sequencing of 16S rRNA genes to profile the taxonomy and diversity of the libraries; (4) selection of candidate amplified genomes for whole-genome sequencing; and (5) sequencing and assembly of selected genomes using assembly tools designed specifically for MDA-amplified single cells (Chitsaz et al. 2011; Bankevich et al. 2012). A highly integrated robotic platform, described in this study for the first time, was used to increase the throughput, ease, and overall cost of processing single cells.
Here we have focused this approach on the indoor environment. Despite the fact that a typical person spends ∼90% of their time indoors (Klepeis et al. 2001), there is little known about the microbial diversity of this environment. Of particular interest is the prevalence of species affecting human health, including both opportunistic and primary pathogens. Recent studies of indoor environments using culture-independent molecular methods indicate an unexpectedly high bacterial diversity on surfaces within daycare facilities and public bathroom facilities (Lee et al. 2007; Flores et al. 2011), where the majority of organisms in the latter environment were human associated (Flores et al. 2011). Another study shows that bacterial diversity is lower in indoor air at a healthcare facility compared with outdoor air; however, the indoor air contained a higher number of potential human pathogens as shown by 16S rRNA gene sequence analyses (Kembel et al. 2012). Biofilms in particular are thought to be reservoirs of disease-causing organisms in both outdoor and indoor environments. Several pathogens, including Escherichia coli, Vibrio cholerae (Shikuma and Hadfield 2010), and Helicobacter pylori (Percival and Thomas 2009; Linke et al. 2010), have been detected in biofilms within water distribution systems. In addition, the long-term persistence of Legionella pneumophila, the causative agent of Legionnaire's disease, in biofilms within natural and human-impacted freshwater environments is well known (Walker et al. 1993; Murga et al. 2001; Declerck 2010; Giao et al. 2011). Recent 16S rRNA gene molecular surveys have revealed a significant load of Mycobacterium avium in showerhead biofilms (Feazel et al. 2009), and studies on biofilms growing on shower curtains suggest that these communities also harbor potential opportunistic pathogens that can threaten immune-compromised patients (Kelley et al. 2004). In another study, the source of a deadly outbreak of a multidrug-resistant strain of Pseudomonas aeruginosa was traced to biofilms in hand hygiene sink drains, where its viable cells could be identified (Hota et al. 2009).
There is great interest, therefore, to investigate biofilms as reservoirs of pathogens at higher resolution than allowed by the most commonly used detection and identification methodologies. Culture-independent surveys using the 16S rRNA gene as a marker are currently the most widely used approach; however, genetic strain differences reflecting pathogenicity are often difficult to resolve due to this gene being highly conserved among many bacterial strains. Quantitative PCR and direct culturing are focused on either a handful of predetermined pathogens or what can be readily cultivated. Metagenomic surveys are becoming common, but so far, our ability is limited to accurately predicting taxonomic affiliation at species or strain levels from highly diverse and complex data sets. Additionally, a whole-genome comparative genomic study on the evolution and transmission of a pathogen requires substantial amounts of DNA or a cultured strain, which often cannot be obtained. It has been demonstrated in a controlled experiment with 10 pg of extracted DNA provided as a template that MDA-amplified genotyping call and accuracy rates were only slightly lower than those for genomic DNA isolated directly from cultured cells (Giardina et al. 2009). Using single-cell genomic approaches, partial to near complete genomes should be obtainable without cultivation, from difficult samples within critical indoor environments such as healthcare facilities. In-depth analyses of these genomic data can then provide accurate and detailed information of strain-specific pathogen-gene signatures and other virulence factors.
The aim of this study was to investigate for the first time the bacteria present in a healthcare facility with a high-throughput single-cell genomics approach. Based on the known prevalence of pathogens in biofilms, we focused on a sink drain biofilm from a public restroom adjoining an emergency waiting room. Sequencing 16S rRNA genes PCR-amplified from 416 single-cell MDA reactions, we found 18 candidate commensal and potentially pathogenic species that were selected for 454 shallow sequencing. Initial read mapping and de novo assembly of the low-coverage 454 sequence data confirmed that we had obtained genomic sequences for the pathogen Streptococcus pneumoniae as well as bacterial species highly similar to and those reported to be potentially pathogenic, including Sphingobacterium spiritivorum (Tronel et al. 2003; Kampfer et al. 2005), Leptotrichia buccalis (Hammann et al. 1993; Hot et al. 2008), as well as the host-associated oral bacteria, Streptococcus mitis and Veillonella parvula. Of particular note, we found three MDA products with sequences for the oral pathogen Porphyromonas gingivalis, which is a periodontal pathogen involved in periodontal bone loss that has also been linked to progression of atherosclerotic disease (Pussinen et al. 2007; Yilmaz 2008). P. gingivalis possesses many virulence factors, including functions that allow it to survive intracellularly and to be transmitted between different types of host cells (Li et al. 2008). Despite being detected at a very low abundance in the oral cavity, P. gingivalis can strongly disrupt the host–microbial homeostasis (Hajishengallis et al. 2011). As with many pathogens, the environmental reservoirs and mode(s) of transmission of P. gingivalis are not fully understood, yet it is a globally important pathogen with only three sequenced genomes available at the time of this report. It was recently stated by a CDC report that nearly 50% of American adults have mild, moderate, or severe periodontitis, and this percentage rises to 70% in adults greater than age 65 (Eke et al. 2012). To our knowledge, there are no previous reports detecting P. gingivalis outside of a host.
Three MDA-amplified genomes with 16S rRNA gene sequences identified as P. gingivalis were chosen for additional deep sequencing on the Illumina GA IIx platform, and the resulting reads were mapped to P. gingivalis genomes. One MDA-read data set had ∼90% sequence coverage to P. gingivalis strain TDC60, which was isolated from a patient in Japan with severe periodontitis (Watanabe et al. 2011). A new single-cell de novo assembly algorithm, SPAdes (Bankevich et al. 2012), was used to generate contigs of the highest-coverage MDA product, which produced a 2.35-Mb draft genome (PG JCVI SC001). Comparative genomics and pangenome analyses were performed with the three other available P. gingivalis genomes; virulent strains W83 (Nelson et al. 2003) and TDC60 (Watanabe et al. 2011), and the less virulent strain ATCC 33277 (Naito et al. 2008). We demonstrate that single-cell genomics is a powerful approach that can produce highly accurate sequence data, enabling comparative genomic studies of pathogens obtained from a complex heterogeneous environmental sample.
Results
Sampling and sorting cells from biofilms
Seawater-derived samples contain relatively high bacterial counts and were a rich source for sorting single cells by FACS (Chitsaz et al. 2011; Dupont et al. 2012) with ∼20%–30% of single-cell amplifications yielding amplified genomic DNA based on sequencing of PCR-amplified 16S rRNA genes (Methods). In contrast, attempts to randomly sort single bacterial cells from the indoor environment, such as from surface swabs (data not shown) and sink drains, yielded <1% amplified bacterial genomes due to failure to lyse cells or due to noise from fluorescence background signals of nonbacterial particles during sorting. To reduce the background noise of nonbacterial events, which obscure bacterial cells stained with SYBR Green, the biofilm sample was vortexed, filtered through a 5-μm filter, and processed through a Nycodenz cushion (Methods). After processing, the percentage of positive stained events that were in the bacterial size range approached 20% of the total particle count (Supplemental Fig. S1). This positive gate was used to sort single events into 384-well microtiter plates.
Microtiter plates containing single sorted events were then processed on the highly integrated high-throughput single-cell platform (Methods; Supplemental Fig. S2). In total, 78 MDAs of 416 sorted wells were identified as candidates based on the taxonomy of their 16S rRNA gene sequence. This 19% overall success rate does not include 16S rRNA gene sequences that can be attributed to common bacterial MDA reagent contaminants detected in control MDA reactions without a sorted cell (NTC, no template control). NTCs were always run in parallel with each sort of single cells to determine the relative amount and identity of contaminating bacterial DNA in the MDA reagents; this is a necessary standard practice in single-cell genomics due to the highly processive strand displacement activity of the phi29 DNA polymerase and the near-ubiquitous presence of bacterial DNA in reagents (Allen et al. 2011; Blainey and Quake 2011; Woyke et al. 2011). Based on 16S rRNA gene analysis, a wide diversity of genera was found in this random sort from the sink biofilm community (Fig. 1).
Figure 1.

Distribution of 78 candidate 16S rRNA sequences found in single-cell sorted wells from the sink biofilm sample. 16S rRNA sequences from single-cell amplifications observed for this FACS sorted sample.
Screening MDA products using multiplexed 454 sequencing
A total of 18 candidate MDA products from the sink biofilm sample were of interest in terms of their relationship to human health being reported as potentially pathogenic or a commensal species. These were each sequenced as barcoded libraries on one-fourth of a 454 plate. A range of 5500–13,500 reads was obtained for the 18 libraries, with overall average read length of 321 bp. Complex environmental samples, such as a sink biofilm, may be difficult to lyse, had poorer amplification, and are more likely to contaminate single-cell MDA reactions with free DNA from other organisms. The 454 data sets for the 18 candidate MDAs were therefore screened for quality, contamination, and overall suitability for analysis by several criteria: (1) the presence of reads having BLAST matches (NCBI nr database) to the same genera indicated by the 16S rRNA gene; (2) confirmation of identity for contigs generated from de novo assembly; and (3) successful mapping of reads to a representative sequenced genome. A total of nine of the original 18 MDAs met these criteria. These comprise species previously reported to be pathogenic or potentially pathogenic, including S. pneumoniae (1 MDA product), S. spiritivorum (1), L. buccalis (1), P. gingivalis (3), S. mitis (2), and the commensal oral bacteria V. parvula (1). These 454 reads were assembled, and the open reading frames were annotated to determine the genes captured from these species, as well as to assess potential for full genome sequencing (Supplemental Table S1). Although many of these products were very intriguing, P. gingivalis was chosen for detailed analysis because there was more than one MDA representing a genome, they passed all the quality criteria, and all three of the MDAs contained a high proportion of genomic sequences from P. gingivalis (labeled MDA 1, 2, and 3). These were chosen for deep sequencing using the Illumina GAIIx platform on one-third of a lane each.
Read-based analyses of the P. gingivalis MDA products
Although Porphyromonas is a widespread and well recognized pathogen, only three P. gingivalis genomes have been sequenced: strain W83 (virulent) (Nelson et al. 2003), ATCC 33277 (less virulent than W83) (Naito et al. 2008), and the most recent, TDC60 (virulent) (Watanabe et al. 2011). A majority of the raw 100-bp paired-end reads for each of the three MDA products yielded BLAST hits to the newly completed genome of a virulent P. gingivalis strain (TDC60) from a severe periodontal lesion in a Japanese patient (Watanabe and Frommel 1993), confirming the 16S rRNA gene PCR sequence matches. The total reads passing quality filtering, along with the results of mapping the reads of the single cells to strain TDC60, are summarized in Table 1. Coverage of the reference genome varied with 41%, 87%, and 91% for MDA 1, 2, and 3, respectively. As with many MDA-derived single-cell genomes, the coverage depth varied widely across the genome: The average coverage for MDA3 was 237×, with ∼90% of the genome covered at 10× or greater (Supplemental Fig. S3). Single nucleotide polymorphism (SNP) and deletion insertion and polymorphism (DIP) analyses revealed 847 shared SNPs between the three amplified genomes with respect to TDC60, with 791 in coding regions (202 missense) (Supplemental Fig. S4). There were 75 shared DIPs in total. Although MDA1 had the highest number of reads, it had the lowest reference genome coverage. Mapping of reads from all three MDAs did not increase the percentage of the reference genome mapped compared with coverage by MDA3 alone.
Table 1.
Individual amplified single cells and combined single-cell read mapping against P. gingivalis TDC60 reference

De novo assembly of P. gingivalis genomes
All three MDA sequence data sets were assembled de novo. Our assembly analysis described here is restricted to MDA3, which corresponds to the MDA having the highest genome coverage to TDC60. Two newly developed de novo assembly tools specifically designed to assemble reads generated from single-cell MDA reactions, Euler correction with Velvet-Single Cell E+V-SC (Chitsaz et al. 2011) and SPAdes (Bankevich et al. 2012), were compared (Table 2) with the Velvet assembly tool (Zerbino and Birney 2008). For single-cell projects, these significantly improve on traditional assemblers (designed for mono-species cultured samples), since they are able to cope with the wide variations in coverage and elevated numbers of chimeric reads and read pairs characteristic of MDA reactions. Completeness of the assemblies was assessed by using Plantagora (Barthelson et al. 2011) to compare with the reference. Plantagora determines “misassembly breakpoints” with the assumption that the data set being assembled is a sample of the reference genome. It is emphasized here that since contigs are compared against a reference from a similar but nonidentical strain, the term “misassembly breakpoints” does not apply and the term “breakpoints versus TDC60” is used instead. Assemblers were compared based on the fraction of the genome covered by the assembled contigs, and an adjusted N50, in which assembly contigs are broken into multiple contigs at the breakpoints determined by Plantagora. Sequence in the contigs that does not align to the TDC60 reference is not counted. Note that the adjusted N50 is likely to eliminate misassemblies from the N50 computation (which result in incorrectly overestimating N50), but this is at the expense of underestimating the true N50. While assembly of sample MDA3 with Velvet resulted in only 52% coverage of the TDC60 reference genome with a very small adjusted N50 (Table 2), assemblies with Velvet-SC and SPAdes resulted in 88% and 90% coverage, respectively, which are close to the 91% coverage by reads (Table 1). The adjusted N50 was 10,732 for Velvet-SC and 13,589 for SPAdes. For MDA2, SPAdes yielded a much better assembly than Velvet-SC: 58% coverage of TDC60 for Velvet-SC versus 78% for SPAdes (while the reads have 87% coverage) (see Table 1), and adjusted N50 of 1495 for Velvet-SC versus 5887 for SPAdes. Based on this benchmarking, SPAdes assemblies only were used for further analyses.
Table 2.
Comparison of assemblies of single-cell P. gingivalis MDA3

Following assembly, contigs resulting from nontarget environmental sequences, as well as MDA contaminants, were identified and removed from the three MDA assemblies using a combination of BLAST and the Automated Phylogenetic Inference System (APIS) (Badger et al. 2006), as described previously (Chitsaz et al. 2011; Dupont et al. 2012). APIS performs a BLAST analysis of each open reading frame (ORF) against reference genomes and, when possible, generates a phylogenetic tree for each ORF. APIS analysis confirmed that the majority of ORFs on the contigs were placed within the genera Porphyromonas. APIS also helped to identify contaminant contigs as those containing ORFs that were phylogenetically similar to each other and distinct from the rest of the assembly (Chitsaz et al. 2011; Dupont et al. 2012). We also assessed the relative proportion and phylogenetic association of target and nontarget contigs by MGTAXA (http://mgtaxa.jcvi.org) (Supplemental Fig. S5), which performs taxonomic classification of metagenomic sequences with machine learning techniques. This process also confirmed that the majority of contigs for MDA3 belonged to P. gingivalis.
We investigated the three assembled genomes by remapping the three sets of MDA reads to the final contigs generated by MDA3. We reported SNPs using the same criteria as described above, revealing an insignificant number of SNPs in a few genes between the data sets (Supplemental Table S2). We also confirmed that the three shared identical 16S rRNA gene sequences based on 938 bp of alignment from initial PCR-amplified 16S rRNA gene results as well as identical full-length 16S rRNA gene sequences in the three assemblies. Although the data may provide support that these three cells are likely the same strain and the assemblies represent the same genome, it is not conclusive, so we chose to treat them as separate genomes rather than combine reads to attempt to generate a better assembly. Given the fact that MDA3 had the highest mapped coverage of the reference genome and the largest number of contigs classified as P. gingivalis, we further restricted our analyses to MDA3. In total, 288 contigs from MDA3 were chosen for further annotation and genomic comparisons.
General genomic features
The single-cell genome assembled from MDA3, designated PG JCVI SC001, was 2.35 Mb in size and is similar to the other three P. gingivalis genomes for a number of genome parameters (Table 3). A total of 1869 genes (86%) were found to have some level of homology in ATCC33277, W83, or TDC60 genomes (Supplemental Table S3), with a total of 1500 genes making up the pangenome encompassing all three genomes (Supplemental Fig. S6). The 524 genes unique to PG JCVI SC001, i.e., no ortholog to reference genomes, were primarily annotated as hypothetical proteins (Table 4; see full table in Supplemental Table S4). Contigs were then ordered based on the TDC60 reference genome and concatenated to provide a scaffold for further comparative genomics. The circular representation of the genome (Fig. 2) displays the ordered contigs and predicted CDSs, as well as BLASTN analyses to the three P. gingivalis genomes and a distant relative, Prevotella buccae ATCC33574.
Table 3.
General features of the PG JCVI SC001 genome and comparisons with sequenced P. gingivalis genomes

Table 4.
JCVI SCOO1 specific CDS (top 12 of 524) identified via reciprocal best BLAST analysis
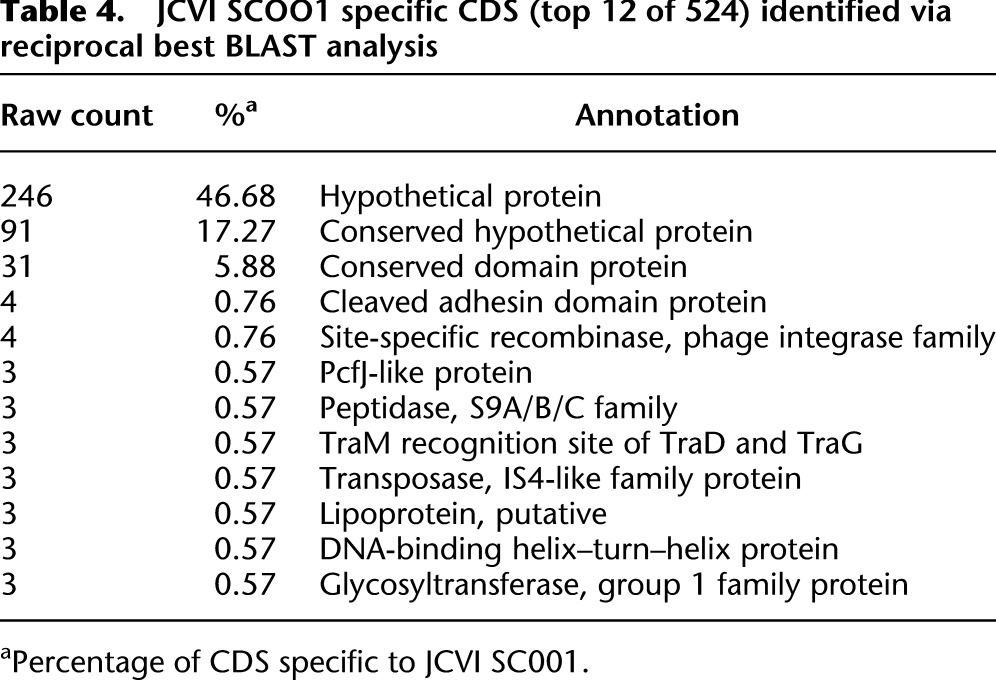
Figure 2.

Circular representation of the draft JCVI SC001 genome. The assembled draft genome is the SPAdes assembly of MDA3 with the contigs ordered to the TDC60 reference genome. From the inner to the outer ring: coordinates in the assembled and concatenated JCVI SC001genome, G+C content, GCskew, ordered contigs, predicted CDS, TBLASTN alignment showing percent identity against P. gingivalis TDC60, W83, ATCC 33277, and Prevotella buccae ATCC33574 (near neighbor) reference genomes.
Multiple locus sequence typing
We used the seven gene multiple locus sequence typing (MLST) scheme for P. gingivalis (Jolley et al. 2004; Enersen et al. 2008a) and sequence types (STs) from human periodontitis isolates of P. gingivalis in the MLST database (http://www.pubmlst.org/pgingivalis) (Jolley et al. 2004) to type the PG JCVI SC001 genome. MLST detects allelic variation at multiple housekeeping loci accumulating slowly in bacterial populations. This database has been used to investigate the clonality of P. gingivalis, which was previously reported to have a weakly clonal population structure comparable with Neisseria menigitidis (Enersen 2011). Using the PG JCVI SC001 contig sequences as input, the MLST database searches revealed that the sequenced single cell has five exact matches to previously sequenced genes of the seven MLST loci (ftsQ, gpdxJ, hagB, mcmA, pepO, pga, recA). Strain TDC60 also had only five exact matches to the database, indicating that they have a unique allele pattern for hagB (SC001), gpdxJ (TDC60), and pga (both) absent in the database. Using existing MLST database tools that generate trees based on the allelic profiles of the 138 sequence types (http://www.pubmlst.org), we confirmed that the nearest sequence type to PG JCVI SC001 is ST-68, having three identical matches (pepO, pga, and recA) (Supplemental Fig. S7).
Analysis of virulence factors
Diversity in P. gingivalis is reported to arise by genetic recombination rather than mutation (Frandsen et al. 2001; Koehler et al. 2003; Nadkarni et al. 2004). In addition to the MLST data, the structure and function of major virulence factors, such as fimbriae (fimA) and the capsular polysaccharide biosynthesis locus (CPS), provide insight into strain variation and degrees of pathogenicity (Lamont and Jenkinson 1998). The single-cell read data and de novo–assembled contigs provide the opportunity to examine strain variation and pathogenicity of this uncultivated strain. In some cases, mapping reads to the reference TD60 genome provided deep coverage sufficient to detect SNPs, whereas in other highly variable regions, such as the CPS region, reads could not be accurately mapped. The CPS region could, however, be recovered in the assembled contigs and used for comparative genome analyses.
Coverage and analysis of fimbriae gene
It is becoming evident that the fimbriae A gene (fimA), encoding the major fimbrial subunit of P. gingivalis, is one of the main virulent factors of this organism. Based on fimA, P gingivalis is classified into six genotypes (genotype I, Ib, II, III, IV, and V). Epidemiological studies have shown that advanced periodontitis patients harbor fimA type II (Enersen et al. 2008a,b; Enersen 2011). It should be noted that fimA type II P. gingivalis is most frequently detected in cardiovascular disease patients (Nakagawa et al. 2006) and has been shown to invade epithelial cells (Nakagawa et al. 2006). TDC60 has type II fimbriae, and our analysis of read mapping and de novo assemblies confirms that our single-cell fimA sequences are related to the fimA of TDC60. A total of six SNPS in fimA are shared by all three MDAs with >10× coverage (Fig. 3), giving further confidence that these six SNPs are valid and not assembly error or MDA artifacts.
Figure 3.

Single nucleotide polymorphisms and read coverage across fimA of the reference strain TDC60. (Row 1) Reference gene fimA; (row 2) shared SNPs across the three single-cell genomes with 100% frequency at a coverage of 10×; (row 3) shared SNPs at a coverage of 30×; (rows 4–8) SNPs at 10× and 30× for each single-cell amplification; (rows 9–10) mapped deletions; (rows 11–13) mapped reads; (row 14) mapped 454 reads.
Variation in the capsular polysaccharide biosynthesis locus
The CPS locus has been described as a virulence factor of various pathogenic bacteria involved in evasion of the host immune system. Encapsulated P. gingivalis strains such as W83 have been shown to be more virulent than nonencapsulated strains (e.g., ATCC 33277 in the mouse infection model) (Laine and van Winkelhoff 1998; Brunner et al. 2010a,b). The CPS loci were poorly covered by reads of all three MDA data sets, suggesting that these may be highly variable regions within the genome. The arrangement of the genes between the three sequenced genomes and JCVI SC001 single-cell genome from MDA3 (Fig. 4) reveals how the region is bounded by shared homology in the gene encoding glycosyl transferase, group 4 family protein, and epsC (UDP-N-acetyl-D-mannosaminuronic acid dehydrogenase) upstream and a pair of downstream genes encoding UDP-N-acetylglucosamine 2-epimerase (epsD) and DNA-binding protein HU. The synteny of genes in this region was more similar between the virulent TDC60 (Watanabe et al. 2011) and the less virulent ATCC 33277 than between the two more virulent strains W83 and TDC60. Our MDA3 assembly has only five ORFs between epsD and epsC and appears very distinct compared with the reference sequences. A recent report has shown that a loss in the ability to produce a capsule, by deletion of the glycosyl transferase, group 4 family protein, increases biofilm formation by W83 and ATCC 33277. Although we do not have an isolate to test if these single cells would produce a capsule, one may speculate based on the genomic data that PG JCVI SC001 lacks a capsule.
Figure 4.

Comparison of the polysaccharide capsule locus found in MDA3 (bottom) with W83, ATCC 33277, and TDC60. Genes of the same color are from the same orthologous group.
Evidence of unique CRISPR region
Differences between species can be observed in clustered regularly interspaced short palindromic repeats (CRISPR) sequences, which are noncontinuous direct repeats separated by variable (spacer) sequences that have been shown to confer immunity to phage (Jansen et al. 2002; Barrangou et al. 2007). Amplification and de novo assembly was successful for the previously identified CRISPR (designated 36-30) within W83; this CRISPR contains eight repeats of 36 bp. Comparisons of this region with other genomes (Fig. 5) reveal identical repeat sequences between all genomes. This region, however, varies in the number of repeats, number of spacer sequences, and spacer identity. Additional confirmed CRISPR repeat sequences of 46 bp (five repeats and four spacers) as well as putative CRISPR direct repeat sequences of 30, 38, 45, and 52 bp, and associated spacers were found in PG JCVI SC001 using the CRIPSR finder database (http://crispr.u-psud.fr/crispr/). The 36-30 region is not flanked by CRISPR-associated (cas) genes in W83 or TDC60, and all three MDA assemblies also failed to capture any cas genes or any homologous genes related to the cas system. Zegans et al. (2009) noted that loss or disruption of five of the six cas genes results in a restoration of biofilm formation in Pseudomonas aeruginosa strains that were infected with a lysogenic phage. It is interesting to speculate that a lack of cas genes in PG JCVI SC001 may also provide an advantage for this strain to integrate into a biofilm community, although no prophage regions were detected in the MDA products.
Figure 5.

Comparison of a clustered regularly interspaced short palindromic repeat region (CRISPR). Successful multiple displacement amplification and de novo assembly of the repeats in CRISPR region 36-30. This region was first identified in strain W83, and all three genomes have 100% identical repeat sequences. The regions vary in the number of repeats, number of spacer sequences, and spacer identity.
Discussion
A vast majority of bacteria in the environment as well as those associated with the human microbiome have eluded standard culturing approaches, and therefore their physiology and their gene content are unknown. This leaves a large gap in our knowledge of the potential roles for these organisms in the environment and also in human health and disease. This is the first report describing the recovery of genomes of bacterial pathogens from single cells out of an environmental sample. We demonstrate that a single-cell approach enables analysis of the genetic diversity between the captured environmental cells and sequenced pathogen genomes, permits identification of variations in virulence factors, and supports discovery of variant genes in the genome. Single-cell genomics comparing multiple single cells enables analysis of genetic diversity within a population and, although it has inherent biases of its own, may potentially be free from biasing effects that can occur during subculturing, such as gene loss (Karch et al. 1992; Nair et al. 2004). Based on the work reported here, capturing genomes from environmental samples using single-cell approaches could support studies on the prevalence and genotype of pathogens from environmental sources and may ultimately help reveal their possible modes of transmission between the host and environment.
Methods
Sample collection and isolation of bacterial cells from sink material
Sink drain samples were collected with sterile cotton-tipped swabs from a publicly accessible restroom adjacent to an emergency waiting room within a Medical School Hospital (University of California, San Diego, Medical Center, Hillcrest, San Diego, CA, USA). The initial sample was fixed with ethanol and vortexed briefly for 20 sec (Supplemental Fig. S1). The sample was filtered through a 35-μm filter. A 2-mL cushion of prechilled Nycodenz gradient solution (Nycoprep Universal, Axis Shield) was placed in a 17-mL ultracentrifuge tube, and 6 mL of supernatant was placed gently over the Nycodenz cushion. The pair of balanced tubes was centrifuged at 9000 rpm for 20 min at 4°C in an ultracentrifuge SW32.1 rotor. The visible cloudy interface containing the bacterial cells was collected gently, mixed by inversion to create a suspension, and diluted 1000-fold for flow cytometry. For further details, see the Supplemental Methods.
FACS detection and single-cell sorting
FACS detection was performed on the Nycodenz fractionated bacteria. Filter-sterilized (0.2 μm) phosphate-buffered saline (PBS, 1×) was used as sheath fluid and for sample dilution. Unstained and SYBR Green I (0.5×) stained material was 35-μm filtered, and a 1:1000 dilution was assessed for event rate at low flow rate (<2000 total events/sec) and adjusted if necessary. The low flow rate is critical to reduce the likelihood of sorting coincident events. Fluorescent events were collected using gating parameters FSC-PMT versus SSC and FSC-PMT versus SYBR Green (Supplemental Fig. S1). One thousand events from each gate were sorted at a lower purity setting onto glass slides for viewing with fluorescent microscopy on an Olympus I×70 inverted fluorescence microscope at 60× magnification to confirm the presence of cellular morphologies. A bead targeting strategy (see Supplemental Methods) was used to ensure that only single cells were sorted from the sample into each well of a 384-well PCR plate (FrameStar). Single-cell events were sorted into 4 μL of a low EDTA TE (10 mM Tris, 0.1 mM EDTA at pH 8.0) and immediately frozen on dry ice and held there until transfer to −80°C for storage prior to processing. For further details, see the Supplemental Methods.
Multiple displacement amplification
MDA of single-cell genomes was performed in a 384-well format using the GenomiPhi HY kit (GE Healthcare) with a custom Agilent BioCel robotic system (see detailed schematic in Supplemental Fig. S2). Briefly, cells were lysed by addition of 2 μL of alkaline lysis solution (645 mM KOH, 265 mM DTT, 2.65 mM EDTA at pH 8.0), then incubated for 10 min at 4°C. After lysis, 7 μL of a neutralization solution (2.8 μL of 1290 mM Tris-Cl at pH 4.5 and 4.2 μL of GE Sample Buffer) was added, followed by 12 μL of GenomiPhi master mix (10.8 μL of GE Reaction Buffer and 1.2 μL of GE Enzyme Mix) for a reaction volume of 25 μL. Reactions were incubated for 16 h at 30°C, followed by a 10-min inactivation step at 65°C. MDA yield was determined by Picogreen assay. Three hundred eighty-four no template control (NTC) MDA reactions were included to reveal any contaminating sequences and processed in parallel through 16S PCR analysis. These negative controls lacking a sorted cell were run in parallel to determine the relative amount and identity of contaminating bacterial DNA in the MDA reagents, a necessary standard practice in single-cell genomics due to the highly processive strand displacement activity of the phi29 DNA polymerase (Allen et al. 2011; Blainey and Quake 2011; Woyke et al. 2011). For further details, see the Supplemental Methods.
PCR and analysis of 16S rRNA genes
16S rRNA genes were amplified from diluted MDA product using universal bacterial primers 27f and 1492r (Lane 1991) according to previously established protocols (Chitsaz et al. 2011; Eloe et al. 2011; Dupont et al. 2012) (Supplemental Methods). BLASTN analysis against the SILVA SSU ref NR 102 database (Pruesse et al. 2007) was performed to classify the 16S rRNA gene sequences taxonomically and to determine their relationship to sequenced bacterial genomes and 16S rRNA gene sequences. An additional BLASTN analysis was performed against a curated database of near full-length to full-length 16S sequences (at least 900 bp) from human fecal sample 16S rRNA sequences (at least 900 bp) from several survey studies (Eckburg et al. 2005; Gill et al. 2006; Dethlefsen et al. 2008; Tap et al. 2009; Turnbaugh et al. 2009) as well as 16S rRNA sequences from the Human Oral Microbiome Database (HOMD). In both cases, sequences were clustered by cd-hit at the 99% level to remove redundancy. This reduced the combined data set of 57,894 sequences to 28,335 OTU representatives at 99% identity. Because the survey sequences had no taxonomy assigned to them, they were classified using the SILVA taxonomy via the classification feature of mothur (Schloss et al. 2009). MDA products with 16S rRNA gene taxonomy similar to those NTC reactions were excluded from further analysis. For further details, see the Supplemental Methods.
Sequencing
Nextera transposition-based 454 compatible fragment libraries were constructed for each MDA as per the manufacturer's instructions (Epicentre Technologies) using MDA genomic DNA as template, including incorporation of the 48 Nextera 454 barcodes and use of Zymo DNA Clean & Concentrator-5 columns (Zymo Research) following the transposition and PCR amplification steps. Nextera transposition-based Illumina fragment libraries were constructed for each MDA as per the manufacturer's instructions (Epicentre Technologies). The library concentrations were determined by absorbance on a NanoDrop spectrophotometer. The column-purified barcoded fragment libraries were pooled at ∼1:1, without size selection, and emulsion PCR and sequencing were performed at JTC. Illumina sequencing (Bentley 2006) was performed on the MDAs using the Genome Analyzer II System according to the manufacturer's specifications. The three MDAs assigned to P. gingivalis were barcoded and pooled on a half-lane; this generated 11.5 GB of data and 47 million reads that passed quality score >20.
Reference mapping
Reference mapping was conducted using the CLC Genomics Workbench, with the reference P. gingivalis (NC-015571). Mapping parameters were as follows: local alignment with mismatch cost 2, insertion cost 3, deletion cost 3, length fraction 0.9, and similarity 0.9 (90% of the read length needed to be aligned at 90% similarity).
Single nucleotide polymorphism (SNP) and deletion insertion and polymorphism (DIP) analysis
Parameters for SNP analysis using the CLC Genomics Workbench were max # of gaps and mismatches 2, minimum average of quality of surrounding bases 30, minimum quality of central base 30, minimum coverage 10, minimum paired coverage = 10, minimum variant frequency 80%. DIP analysis parameters using the CLC Genomics Workbench were minimum coverage 4, minimum variant frequency 35%. Coverage above 30× with a minimum count of 10 reads with a frequency of 80% cutoff was considered for the detection of shared SNPs.
Single-cell assemblies
Assemblies were produced using Velvet-SC (Chitsaz et al. 2011) and SPAdes (Bankevich et al. 2012). Both of these assemblers are based on the de Bruijn graph, and both have been adapted for uneven coverage found in single-cell MDA data sets. SPAdes was also adapted for the elevated numbers of chimeric reads and read pairs in MDA-amplified data sets. For Velvet-SC, we assembled the data with k-mer size k = 55. For SPAdes, we iterated over k-mer sizes k = 21, 33, and 55. As described in Results, we selected the SPAdes assembly of MDA3 for detailed analysis.
Contig analyses
Assemblies were manually curated using a conservative approach for single-cell MDA data as described in Chitsaz et al. (2011), Eloe et al. (2011), and Dupont et al. (2012). Briefly, all contigs <150 bp in length were removed. Taxonomic affiliations of the predicted protein sequences on the contigs were assigned using APIS (Badger et al. 2006), and contigs that contained a majority of proteins with taxonomic affiliations other than the genus-level classification Porphyromonas were removed. Further verification of correct taxonomic classification of the contigs was performed using MGTAXA software, which performs taxonomic classification of metagenomic sequences and is fundamentally based on the frequency of k-mers (http://mgtaxa.jcvi.org). Although the use of combined approaches for enrichment of target contigs of interest can remove a fraction of potentially new genomic information, it also provided confidence in the final data sets since taxonomic affiliation of the sequences are in accordance with the 16S rRNA phylogeny.
Gene annotation
The assembly from MDA3 that represented most of the genome was annotated using the JCVI prokaryotic annotation pipeline (http://www.jcvi.org/cms/research/projects/annotation-service/overview/). In addition, the XBASE rapid annotation service (http://www.xbase.ac.uk/annotation/) was used to annotate homologs in this genome to P. gingivalis strain W83, which XBASE provides as the nearest reference genome for the purpose of rapid annotation.
Orthology
Reciprocal best BLASTp analysis of genes for reference genomes was P. gingivalis TDC60, ATCC33277, W83, and predicted genes of PG JCVI SCOO1 at a cutoff of 1 × 10−9. Annotations for genes that were found only in PG JCVI SC001 (524, 22.8%) were from the JCVI prokaryotic annotation pipeline.
Multi Locus Sequence Typing
This publication made use of the P. gingivalis Multi Locus Sequence Typing website (http://pubmlst.org/pgingivalis/) developed by Keith Jolley and sited at the University of Oxford.
Data access
The genome data have been submitted to the NCBI GenBank (http://www.ncbi.nlm.nih.gov/genbank/) under accession number PRJNA167667 ID:167667.
Acknowledgments
This work was supported by grants to R.F., J.C.V., and R.S.L. by the Alfred P. Sloan Foundation (Sloan Foundation-2007-10-19); to J.S.M. from the National Institutes of Health (1R01GM095373 and 1R01DE020102); to P.P. and G.T. from the National Institutes of Health (NIH 3P41RR024851-02S1); to P.A.P. from the Government of the Russian Federation (grant 11.G34.31.0018); and to M.G.Z. from the National Institutes of Health (UL1TR000100). We thank Mark Adams for helpful discussions and Mathangi Thiagaran (J. Craig Venter Institute) for bioinformatics support.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.150433.112.
Freely available online through the Genome Research Open Access option.
References
- Allen LZ, Ishoey T, Novotny MA, McLean JS, Lasken RS, Williamson SJ 2011. Single virus genomics: A new tool for virus discovery. PLoS ONE 6: e17722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badger JH, Hoover TR, Brun YV, Weiner RM, Laub MT, Alexandre G, Mrazek J, Ren Q, Paulsen IT, Nelson KE, et al. 2006. Comparative genomic evidence for a close relationship between the dimorphic prosthecate bacteria Hyphomonas neptunium and Caulobacter crescentus. J Bacteriol 188: 6841–6850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD et al. 2012. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19: 455–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315: 1709–1712 [DOI] [PubMed] [Google Scholar]
- Barthelson R, McFarlin AJ, Rounsley SD, Young S 2011. Plantagora: Modeling whole genome sequencing and assembly of plant genomes. PLoS ONE 6: e28436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR 2006. Whole-genome re-sequencing. Curr Opin Genet Dev 16: 545–552 [DOI] [PubMed] [Google Scholar]
- Binga EK, Lasken RS, Neufeld JD 2008. Something from (almost) nothing: The impact of multiple displacement amplification on microbial ecology. ISME J 2: 233–241 [DOI] [PubMed] [Google Scholar]
- Blainey PC, Quake SR 2011. Digital MDA for enumeration of total nucleic acid contamination. Nucleic Acids Res 39: e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner J, Scheres N, El Idrissi NB, Deng DM, Laine ML, van Winkelhoff AJ, Crielaard W 2010a. The capsule of Porphyromonas gingivalis reduces the immune response of human gingival fibroblasts. BMC Microbiol 10: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner J, Wittink FR, Jonker MJ, de Jong M, Breit TM, Laine ML, de Soet JJ, Crielaard W 2010b. The core genome of the anaerobic oral pathogenic bacterium Porphyromonas gingivalis. BMC Microbiol 10: 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitsaz H, Yee-Greenbaum JL, Tesler G, Lombardo MJ, Dupont CL, Badger JH, Novotny M, Rusch DB, Fraser LJ, Gormley NA, et al. 2011. Efficient de novo assembly of single-cell bacterial genomes from short-read data sets. Nat Biotechnol 29: 915–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean FB, Nelson JR, Giesler TL, Lasken RS 2001. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res 11: 1095–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean FB, Hosono S, Fang L, Wu X, Faruqi AF, Bray-Ward P, Sun Z, Zong Q, Du Y, Du J, et al. 2002. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci 99: 5261–5266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Declerck P 2010. Biofilms: The environmental playground of Legionella pneumophila. Environ Microbiol 12: 557–566 [DOI] [PubMed] [Google Scholar]
- Dethlefsen L, Huse S, Sogin ML, Relman DA 2008. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 6: e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, Lakshmanan A, Wade WG 2010. The human oral microbiome. J Bacteriol 192: 5002–5017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont CL, Rusch DB, Yooseph S, Lombardo MJ, Richter RA, Valas R, Novotny M, Yee-Greenbaum J, Selengut JD, Haft DH, et al. 2012. Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6: 1186–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA 2005. Diversity of the human intestinal microbial flora. Science 308: 1635–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eke PI, Dye BA, Wei L, Thornton-Evans GO, Genco RJ 2012. Prevalence of periodontitis in adults in the United States: 2009 and 2010. J Dent Res 91: 914–920 [DOI] [PubMed] [Google Scholar]
- Eloe EA, Fadrosh DW, Novotny M, Zeigler Allen L, Kim M, Lombardo MJ, Yee-Greenbaum J, Yooseph S, Allen EE, Lasken R, et al. 2011. Going deeper: Metagenome of a hadopelagic microbial community. PLoS ONE 6: e20388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enersen M 2011. Porphyromonas gingivalis: A clonal pathogen?: Diversities in housekeeping genes and the major fimbriae gene. J Oral Microbiol 3. doi: 10.3402/jom.v3i0.8487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enersen M, Olsen I, Caugant DA 2008a. Genetic diversity of Porphyromonas gingivalis isolates recovered from single “refractory” periodontitis sites. Appl Environ Microbiol 74: 5817–5821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enersen M, Olsen I, Kvalheim O, Caugant DA 2008b. fimA genotypes and multilocus sequence types of Porphyromonas gingivalis from patients with periodontitis. J Clin Microbiol 46: 31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feazel LM, Baumgartner LK, Peterson KL, Frank DN, Harris JK, Pace NR 2009. Opportunistic pathogens enriched in showerhead biofilms. Proc Natl Acad Sci 106: 16393–16399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores GE, Bates ST, Knights D, Lauber CL, Stombaugh J, Knight R, Fierer N 2011. Microbial biogeography of public restroom surfaces. PLoS ONE 6: e28132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frandsen EV, Poulsen K, Curtis MA, Kilian M 2001. Evidence of recombination in Porphyromonas gingivalis and random distribution of putative virulence markers. Infect Immun 69: 4479–4485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giao MS, Azevedo NF, Wilks SA, Vieira MJ, Keevil CW 2011. Interaction of Legionella pneumophila and Helicobacter pylori with bacterial species isolated from drinking water biofilms. BMC Microbiol 11: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardina E, Pietrangeli I, Martone C, Zampatti S, Marsala P, Gabriele L, Ricci O, Solla G, Asili P, Arcudi G, et al. 2009. Whole genome amplification and real-time PCR in forensic casework. BMC Genomics 10: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE 2006. Metagenomic analysis of the human distal gut microbiome. Science 312: 1355–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, et al. 2011. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10: 497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammann R, Iwand A, Brachmann J, Keller K, Werner A 1993. Endocarditis caused by a Leptotrichia buccalis–like bacterium in a patient with a prosthetic aortic valve. Eur J Clin Microbiol Infect Dis 12: 280–282 [DOI] [PubMed] [Google Scholar]
- Hosono S, Faruqi AF, Dean FB, Du Y, Sun Z, Wu X, Du J, Kingsmore SF, Egholm M, Lasken RS 2003. Unbiased whole-genome amplification directly from clinical samples. Genome Res 13: 954–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hot A, Coppere B, Ninet J, Thiebault A 2008. Lemierre syndrome caused by Leptotrichia buccalis in a neutropenic patient. Int J Infect Dis 12: 339–340 [DOI] [PubMed] [Google Scholar]
- Hota S, Hirji Z, Stockton K, Lemieux C, Dedier H, Wolfaardt G, Gardam MA 2009. Outbreak of multidrug-resistant Pseudomonas aeruginosa colonization and infection secondary to imperfect intensive care unit room design. Infect Control Hosp Epidemiol 30: 25–33 [DOI] [PubMed] [Google Scholar]
- The Human Microbiome Jumpstart Reference Strains Consortium 2010. A catalog of reference genomes from the human microbiome. Science 328: 994–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R, Embden JD, Gaastra W, Schouls LM 2002. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43: 1565–1575 [DOI] [PubMed] [Google Scholar]
- Jolley KA, Chan MS, Maiden MC 2004. mlstdbNet—distributed multi-locus sequence typing (MLST) databases. BMC Bioinformatics 5: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampfer P, Engelhart S, Rolke M, Sennekamp J 2005. Extrinsic allergic alveolitis (hypersensitivity pneumonitis) caused by Sphingobacterium spiritivorum from the water reservoir of a steam iron. J Clin Microbiol 43: 4908–4910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch H, Meyer T, Russmann H, Heesemann J 1992. Frequent loss of Shiga-like toxin genes in clinical isolates of Escherichia coli upon subcultivation. Infect Immun 60: 3464–3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley ST, Theisen U, Angenent LT, St Amand A, Pace NR 2004. Molecular analysis of shower curtain biofilm microbes. Appl Environ Microbiol 70: 4187–4192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kembel SW, Jones E, Kline J, Northcutt D, Stenson J, Womack AM, Bohannan BJM, Brown GZ, Green JL 2012. Architectural design influences the diversity and structure of the built environment microbiome. ISME J 6: 1469–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klepeis NE, Nelson WC, Ott WR, Robinson JP, Tsang AM, Switzer P, Behar JV, Hern SC, Engelmann WH 2001. The National Human Activity Pattern Survey (NHAPS): A resource for assessing exposure to environmental pollutants. J Expo Anal Environ Epidemiol 11: 231–252 [DOI] [PubMed] [Google Scholar]
- Koehler A, Karch H, Beikler T, Flemmig TF, Suerbaum S, Schmidt H 2003. Multilocus sequence analysis of Porphyromonas gingivalis indicates frequent recombination. Microbiology 149: 2407–2415 [DOI] [PubMed] [Google Scholar]
- Laine ML, van Winkelhoff AJ 1998. Virulence of six capsular serotypes of Porphyromonas gingivalis in a mouse model. Oral Microbiol Immunol 13: 322–325 [DOI] [PubMed] [Google Scholar]
- Lamont RJ, Jenkinson HF 1998. Life below the gum line: Pathogenic mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev 62: 1244–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DJ. 1991. 16S/23S rRNA sequencing. In Nucleic acid techniques in bacterial systematics (ed. Stackebrandt E, Goodfellow M), pp. 115–175. Wiley, New York. [Google Scholar]
- Lasken RS 2012. Genomic sequencing of uncultured microorganisms from single cells. Nat Rev Microbiol 10: 631–640 [DOI] [PubMed] [Google Scholar]
- Lee L, Tin S, Kelley ST 2007. Culture-independent analysis of bacterial diversity in a child-care facility. BMC Microbiol 7: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Michel R, Cohen J, Decarlo A, Kozarov E 2008. Intracellular survival and vascular cell-to-cell transmission of Porphyromonas gingivalis. BMC Microbiol 8: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke S, Lenz J, Gemein S, Exner M, Gebel J 2010. Detection of Helicobacter pylori in biofilms by real-time PCR. Int J Hyg Environ Health 213: 176–182 [DOI] [PubMed] [Google Scholar]
- Marcy Y, Ouverney C, Bik EM, Losekann T, Ivanova N, Martin HG, Szeto E, Platt D, Hugenholtz P, Relman DA, et al. 2007. Dissecting biological “dark matter” with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc Natl Acad Sci 104: 11889–11894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga R, Forster TS, Brown E, Pruckler JM, Fields BS, Donlan RM 2001. Role of biofilms in the survival of Legionella pneumophila in a model potable-water system. Microbiology 147: 3121–3126 [DOI] [PubMed] [Google Scholar]
- Nadkarni MA, Nguyen KA, Chapple CC, DeCarlo AA, Jacques NA, Hunter N 2004. Distribution of Porphyromonas gingivalis biotypes defined by alleles of the kgp (Lys-gingipain) gene. J Clin Microbiol 42: 3873–3876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S, Alokam S, Kothapalli S, Porwollik S, Proctor E, Choy C, McClelland M, Liu SL, Sanderson KE 2004. Salmonella enterica serovar Typhi strains from which SPI7, a 134-kilobase island with genes for Vi exopolysaccharide and other functions, has been deleted. J Bacteriol 186: 3214–3223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito M, Hirakawa H, Yamashita A, Ohara N, Shoji M, Yukitake H, Nakayama K, Toh H, Yoshimura F, Kuhara S, et al. 2008. Determination of the genome sequence of Porphyromonas gingivalis strain ATCC 33277 and genomic comparison with strain W83 revealed extensive genome rearrangements in P. gingivalis. DNA Res 15: 215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa I, Inaba H, Yamamura T, Kato T, Kawai S, Ooshima T, Amano A 2006. Invasion of epithelial cells and proteolysis of cellular focal adhesion components by distinct types of Porphyromonas gingivalis fimbriae. Infect Immun 74: 3773–3782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson KE, Fleischmann RD, DeBoy RT, Paulsen IT, Fouts DE, Eisen JA, Daugherty SC, Dodson RJ, Durkin AS, Gwinn M, et al. 2003. Complete genome sequence of the oral pathogenic bacterium Porphyromonas gingivalis strain W83. J Bacteriol 185: 5591–5601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival SL, Thomas JG 2009. Transmission of Helicobacter pylori and the role of water and biofilms. J Water Health 7: 469–477 [DOI] [PubMed] [Google Scholar]
- Podar M, Abulencia CB, Walcher M, Hutchison D, Zengler K, Garcia JA, Holland T, Cotton D, Hauser L, Keller M 2007. Targeted access to the genomes of low-abundance organisms in complex microbial communities. Appl Environ Microbiol 73: 3205–3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO 2007. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188–7196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pussinen PJ, Tuomisto K, Jousilahti P, Havulinna AS, Sundvall J, Salomaa V 2007. Endotoxemia, immune response to periodontal pathogens, and systemic inflammation associate with incident cardiovascular disease events. Arterioscler Thromb Vasc Biol 27: 1433–1439 [DOI] [PubMed] [Google Scholar]
- Raghunathan A, Ferguson HR Jr, Bornarth CJ, Song W, Driscoll M, Lasken RS 2005. Genomic DNA amplification from a single bacterium. Appl Environ Microbiol 71: 3342–3347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappe MS, Giovannoni SJ 2003. The uncultured microbial majority. Annu Rev Microbiol 57: 369–394 [DOI] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. 2009. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikuma NJ, Hadfield MG 2010. Marine biofilms on submerged surfaces are a reservoir for Escherichia coli and Vibrio cholerae. Biofouling 26: 39–46 [DOI] [PubMed] [Google Scholar]
- Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet JP, Ugarte E, Munoz-Tamayo R, Paslier DL, Nalin R, et al. 2009. Towards the human intestinal microbiota phylogenetic core. Environ Microbiol 11: 2574–2584 [DOI] [PubMed] [Google Scholar]
- Tronel H, Plesiat P, Ageron E, Grimont PA 2003. Bacteremia caused by a novel species of Sphingobacterium. Clin Microbiol Infect 9: 1242–1244 [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. 2009. A core gut microbiome in obese and lean twins. Nature 457: 480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JT, Sonesson A, Keevil CW, White DC 1993. Detection of Legionella pneumophila in biofilms containing a complex microbial consortium by gas chromatography-mass spectrometry analysis of genus-specific hydroxy fatty acids. FEMS Microbiol Lett 113: 139–144 [DOI] [PubMed] [Google Scholar]
- Watanabe K, Frommel TO 1993. Detection of Porphyromonas gingivalis in oral plaque samples by use of the polymerase chain reaction. J Dent Res 72: 1040–1044 [DOI] [PubMed] [Google Scholar]
- Watanabe T, Maruyama F, Nozawa T, Aoki A, Okano S, Shibata Y, Oshima K, Kurokawa K, Hattori M, Nakagawa I, et al. 2011. Complete genome sequence of the bacterium Porphyromonas gingivalis TDC60, which causes periodontal disease. J Bacteriol 193: 4259–4260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woyke T, Sczyrba A, Lee J, Rinke C, Tighe D, Clingenpeel S, Malmstrom R, Stepanauskas R, Cheng JF 2011. Decontamination of MDA reagents for single cell whole genome amplification. PLoS ONE 6: e26161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O 2008. The chronicles of Porphyromonas gingivalis: The microbium, the human oral epithelium and their interplay. Microbiology 154: 2897–2903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef N, Blainey P, Quake S, Elshahed M 2011. Partial genome assembly for a candidate division OP11 single cell from an anoxic spring (Zodletone Spring, Oklahoma). Appl Environ Microbiol 77: 7804–7818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegans ME, Wagner JC, Cady KC, Murphy DM, Hammond JH, O'Toole GA 2009. Interaction between bacteriophage DMS3 and host CRISPR region inhibits group behaviors of Pseudomonas aeruginosa. J Bacteriol 191: 210–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E 2008. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]