

Abstract
The human genome encodes 1500–2000 different transcription factors (TFs). ChIP-seq is revealing the global binding profiles of a fraction of TFs in a fraction of their biological contexts. These data show that the majority of TFs bind directly next to a large number of context-relevant target genes, that most binding is distal, and that binding is context specific. Because of the effort and cost involved, ChIP-seq is seldom used in search of novel TF function. Such exploration is instead done using expression perturbation and genetic screens. Here we propose a comprehensive computational framework for transcription factor function prediction. We curate 332 high-quality nonredundant TF binding motifs that represent all major DNA binding domains, and improve cross-species conserved binding site prediction to obtain 3.3 million conserved, mostly distal, binding site predictions. We combine these with 2.4 million facts about all human and mouse gene functions, in a novel statistical framework, in search of enrichments of particular motifs next to groups of target genes of particular functions. Rigorous parameter tuning and a harsh null are used to minimize false positives. Our novel PRISM (predicting regulatory information from single motifs) approach obtains 2543 TF function predictions in a large variety of contexts, at a false discovery rate of 16%. The predictions are highly enriched for validated TF roles, and 45 of 67 (67%) tested binding site regions in five different contexts act as enhancers in functionally matched cells.
The complex spatiotemporal regulation of gene expression is a critical component in vertebrate development, evolution, and disease (Visel et al. 2009; Levine 2010). Understanding this regulation involves unraveling the cis-regulatory architecture, namely, the biological roles of transcription factors, their target genes in different biological contexts, and the regulatory elements such as promoters and enhancers through which they exert their effect (Michelson 2002).
The recent coupling of chromatin immunoprecipitation with deep sequencing (ChIP-seq) is allowing unprecedented and mostly unbiased access to the whole genome landscape of transcription factor (TF) binding (Bernstein et al. 2012). Hundreds of such experiments for different TFs under different conditions have revealed a few general phenomena. A typical TF reproducibly binds thousands of genomic regions in any given context. The majority of bound sites are distal, located 10–1000 kb upstream of or downstream from the nearest transcription start site. Transcription factors almost invariably are found to bind near a large number (dozens to hundreds) of target genes involved in a shared biological function, with most of these binding sites also being distal (McLean et al. 2010). Interestingly, TFs also often bind not once but multiple times next to some of their best-known functional target genes. We have recently incorporated all of these observations into a new statistical test used to reveal the functions of a ChIP-seq data set, which we call GREAT (for genomic regions enrichment of annotations tool) (McLean et al. 2010).
GREAT and similar analyses reveal yet another key property of ChIP-seq experiments—their context dependence. While TFs are often pleiotropic, playing key roles in multiple independent cellular contexts, a ChIP-seq experiment reveals only the subset of functions relevant to the assayed cell population. For example, when SRF—an important regulator of muscle development—is assayed by ChIP-seq in immune cells, its role in muscle developmental is not readily apparent (Valouev et al. 2008). To examine the function of SRF in muscle cells, muscle cells must be assayed. Although ChIP-seq is a high-throughput approach, the required expense, time, and technical skill result in it being only rarely used as an exploratory tool to ask whether a TF has a role in a newly hypothesized cellular context. Almost invariably, a TF ChIP-seq is attempted in a given context only after the TF has already been shown to be important in said context. Yet, the human genome encodes 1500–2000 different transcription factors, and recent progress shows that many factors play important roles in biological contexts that remain to be discovered. Moreover, the genome itself encodes the transcriptional response of all cells in our body under numerous different cellular conditions.
Motivated by these observations, we aimed to apply transcription factor binding site prediction to produce novel hypotheses for transcription factor function in a wide range of contexts, as a guide for further experimental exploration. Recent technologies including protein binding microarrays (Berger et al. 2008), high-throughput SELEX (Jolma et al. 2010), and ChIP-seq itself have facilitated the quantification of the binding preferences of hundreds of different TFs. Meanwhile, decades of protein research have been collected into biological ontologies and large gene annotation repositories. And lastly, while single sequence/species prediction of transcription factor binding sites results in far too many erroneous predictions, the subset of binding site predictions that shows cross-species sequence conservation exhibits much higher specificity as measured by ChIP-seq occupancy of predicted sites (Xie et al. 2009) and a higher likelihood of residing within active enhancers (Cheng et al. 2008; Rada-Iglesias et al. 2012). Although a conservation-based assay misses many species-specific functional binding sites (Blow et al. 2010; Schmidt et al. 2010), it is not limited to a single cellular context and allows exploratory questions about the roles of a transcription factor.
Previous approaches to this challenge have focused on small numbers of TFs, gene promoters, and specific biological processes, thus ignoring the vast majority of binding events (Das et al. 2006; Down et al. 2007; Sinha et al. 2008). To extend this work, we here develop the PRISM (predicting regulatory information from single motifs) method, which combines genome-wide conserved binding site prediction with transcription factor and binding site function prediction. We introduce the excess conservation score, an improved measurement of binding site conservation that favors sites that are more conserved than neighboring nucleotides. We compile a nonredundant, high-quality library from more than 800 public transcription factor motifs, covering all major DNA binding domains, and predict 3.3 million binding sites for all factors across the human and mouse genomes. We then place GREAT (McLean et al. 2010), a tool for functional analysis of a set of cis-regulatory regions, in a novel statistical framework that lets us predict transcription factor and binding site functions en masse. In total, we infer more than 2500 transcription factor functions, covering nearly 7700 different target genes. We show that our inferences include hundreds of transcription factor function predictions directly supported by existing literature and annotations, for each of which we implicate tens to hundreds of novel binding sites. We validate a subset of our predictions experimentally in a variety of functional contexts. Lastly, we present novel hypothesized transcription factor functions with supporting evidence. We offer the PRISM predictions to the community through a web portal at PRISM.stanford.edu.
Results
Improving transcription factor binding site prediction using excess conservation
A long line of previous works (culminating in Xie et al. 2009) has defined a methodology for predicting conserved binding sites from a genome-wide multiple alignment and the position weight matrix, or motif, representation of transcription factor binding specificity. However, focusing on the motif alone ignores the surrounding sequence in which prediction is done. Mammalian genomes are full of long (100–1000 bp), highly conserved noncoding regions (Waterston et al. 2002; Bejerano et al. 2004). The more conserved a longer genomic stretch is, the more likely it is to include conserved binding site–like patterns in it by chance (see Fig. 1B). Accounting for this differential likelihood of false predictions has been valuable in improving earlier methods (Kheradpour et al. 2007).
Figure 1.
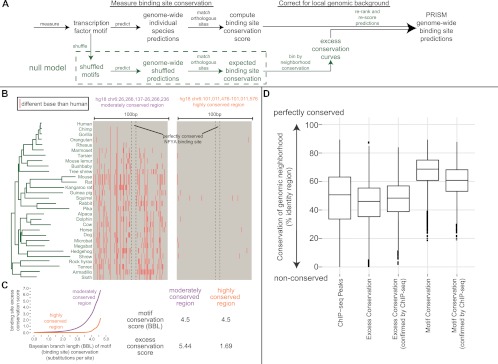
PRISM excess conservation rescoring favors predicted transcription factor binding sites conserved above their local environment. (A) Excess conservation uses the abundance of conserved binding site predictions for shuffled versions of the input motif, in similarly conserved 100-bp genomic neighborhoods, to rescore conserved binding site predictions (framework in green). (B) The NFYA binding site motif is equally conserved in the two shown loci. Yet it is intuitively appealing to consider the left, less conserved 100-bp neighborhood, more likely to conserve an actual NFYA site. (C, left) Excess conservation plots made from all 100-bp neighborhoods conserved like the two loci in panel B. The y-axis is −log10 of the likelihood of a shuffled NFYA motif to achieve the motif conservation score on the x-axis or higher by chance. (Right) Because shuffled versions of NFYA more easily achieve high motif conservation scores in loci like the right locus of panel B, the excess conservation score of this NFYA prediction is lower. (D) Excess conservation rescoring is shown to correct motif conservation-only binding site predictions toward the conservation profile observed in real ChIP-seq peaks. It also outperforms it in area-under-the-curve analysis of 44 (94%) of 47 analyzed ChIP-seq sets (see text).
We have developed an adjustment to the latest conservation metrics that accounts for the conservation level of the predicted binding site's immediate genomic vicinity (Fig. 1A–C). We assign each binding site prediction an “excess conservation” score, which measures how unlikely it is for the binding site to be conserved by chance to the observed depth in a particular region of the genome based on the behavior of shuffled versions of its motif in similarly conserved regions of the genome (see Methods). Shuffled motifs have previously been shown to be the most realistic null model for motif-based prediction methods (Lewis et al. 2003; Kheradpour et al. 2007). The method explicitly favors binding sites that are conserved more strongly than surrounding sequence, which suggests evolutionary constraint aimed at transcription factor binding site preservation (Fig. 1C). While motif conservation-only metrics gravitate toward prediction in deeply conserved regions, the excess conservation adjustment produces predictions with a conservation profile matching closely to that of actual ChIP-seq binding sites (Fig. 1D). The excess conservation method also more accurately identifies binding sites, as measured by area under the curve analysis of overlap with ChIP-seq, for 44 of 47 (94%) examined transcription factors (Supplemental Table 1).
Genome-wide binding site prediction reveals trends in mammalian transcription regulation
We obtained 389 motifs covering 289 factors from UniPROBE (Newburger and Bulyk 2009), 133 motifs covering 90 factors from JASPAR (Bryne et al. 2008), and 294 motifs covering 151 factors from TransFac (Matys et al. 2006). Careful semiautomated curation to select only high-quality, nonredundant motifs (see Methods) resulted in 332 motifs, covering at least one member from every major DNA binding domain family (Fig. 2A).
Figure 2.

Genome-wide excess conservation binding site predictions reveal fundamental properties of mammalian transcription regulation. (A) The curated library of 332 nonredundant high-quality transcription factor (TF) motifs includes members of all major DNA binding domain (DBD) families. (B) Distributions of all genomic bases (red) and all conserved binding site predictions (blue) as a function of distance from the transcription start site (TSS). While predictions are 2.3-fold enriched in the proximal promoter, >90% of them are distal. (C) Different DNA binding domain families exhibit different binding distance preferences relative to the TSS. (Black ticks) Median distances per motif; (green dot) the family median; random is the median of 332 uniform shuffles. (D) Number of predicted target genes for the different TF DBD families. Black ticks, green dots, and random are as in panel C. POUs and Homeodomains cluster the most around target genes, while CTCF is at the opposite extreme. (E) Distance to TSS and number of target genes have a strong inverse correlation. (F) Transcription factors (blue) are the most densely regulated gene families in the human genome, as measured by the fraction of base pairs in the gene's regulatory domain covered by a binding site prediction. Shown are all nonredundant significant terms after Bonferroni correction (see text).
For each motif, we identified the approximately 5000 instances genome-wide with the highest excess conservation scores, for a total of nearly 3.3 million predicted binding sites for the human and mouse genomes across all motifs at a false-positive rate of 0.6 (see Methods). While our predictions are encouragingly enriched in the proximal promoter (2.3-fold compared with genome-wide expectation), >90% of binding site predictions lie outside of proximal promoters (Fig. 2B).
The predictions reveal interesting trends in the propensity of certain DNA binding domain families to target genes more proximally or distally than others (Fig. 2C). We associate binding sites to target genes using default GREAT gene regulatory domains (basal domain: 5 kb upstream + 1 kb downstream; distal domain: up to 1 Mb in each direction to the nearest basal domain), which we previously have shown is ideal for analysis of distal binding sites from ChIP-seq (McLean et al. 2010). For the most proximal family, the E2F/TDF genes, >47% of binding site to target gene associations are within 5 kb of the transcription start site (TSS). Many fewer associations are within 5 kb for the most distal families: HMG (4.2%), Homeodomain (3.6%), and POU (3.2%). In fact, >93% of the predictions for the HMG, Homeodomain, and POU families are >100 kb from the TSS of the associated target gene.
Interestingly, the transcription factor families with the most distal predictions have the fewest downstream targets, showing a tendency to cluster around a relatively small number of target genes (Fig. 2D). In fact, a clear inverse relationship between distance to TSS and number of predicted target genes holds across the full set of motifs, with a Pearson correlation of −0.75 (Fig. 2E). No family has a markedly wider set of target genes than random expectation, but the C2H2 zinc-finger CTCF motif is a clear outlier (Fig. 2D,E), reflecting its special role in genome structure organization (Phillips and Corces 2009).
To examine which genes and gene families are most densely regulated, we calculated the fraction of base pairs in a gene's regulatory domain that are covered by a binding site prediction. Not surprisingly, HOX genes are among the most densely regulated genes (Supplemental Table 3). When we grouped all target genes into families using Interpro domain composition and performed a Wilcoxon rank-sum test, numerous transcription factor families rose to the top, suggesting that the regulators themselves have the most upstream regulation (Fig. 2F; Supplemental Table 4).
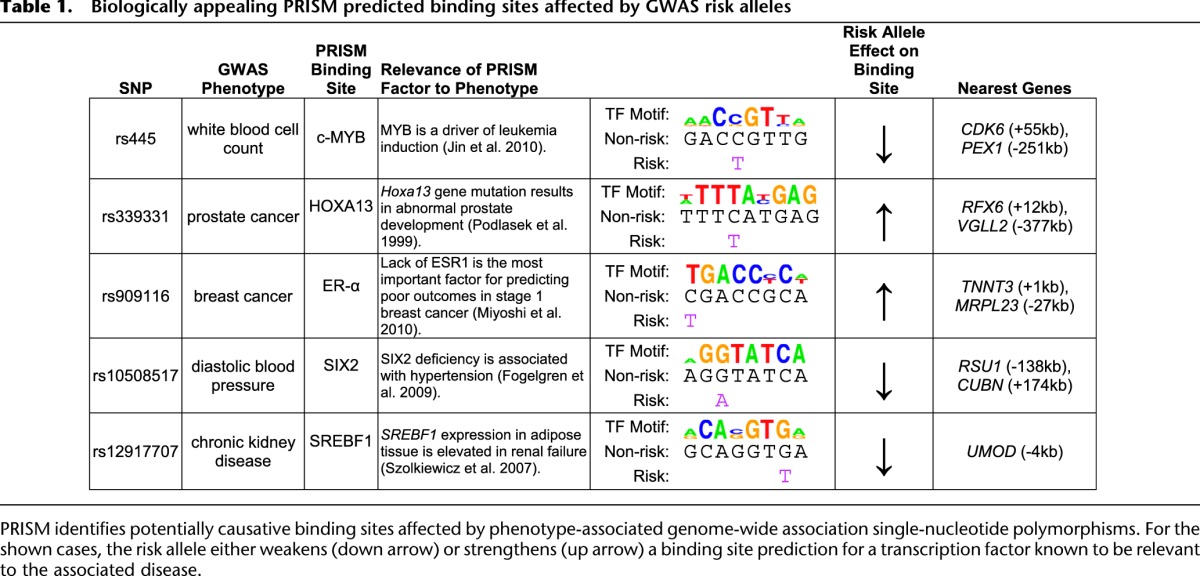
Excess conservation binding site predictions overlapping GWAS SNPs
The NHGRI maintains a catalog of the most significant simple nucleotide polymorphisms (SNPs) associated with a growing number of diseases and traits, discovered using genome-wide association studies (GWAS) (Hindorff et al. 2009). Our excess conservation binding site predictions overlap 15 of these phenotype-associated SNPs (Supplemental Table 5), a significant overlap (1.86-fold enriched compared with dbSNP overlap, P-value < 0.018, Fisher's exact test) (see Methods). For at least five of these SNPs, the transcription factor that we predict to bind has itself been associated with the phenotype in question (Table 1). For example, rs445 is associated (P < 10−7) with white blood cell count (Kamatani et al. 2010). The risk allele weakens a predicted binding site for c-MYB, a key player in the onset of leukemia, a cancer characterized by an abnormal increase in white blood cells (Jin et al. 2010). In other cases, such as rs339331, associated (P < 10−11) with prostate cancer (Takata et al. 2010), the risk allele strengthens a potential binding site for HOXA13, a key factor in prostate gland development (Podlasek et al. 1999).
Table 1.
Biologically appealing PRISM predicted binding sites affected by GWAS risk alleles
Predicting transcription factor functions from binding site predictions
To analyze ChIP-seq using microarray/gene list–based tools, researchers would often ignore distal binding sites, convert proximal sites into a gene list, and test this gene list against the full list of genes in the genome for any enriched function. GREAT (the genomic regions enrichment of annotations tool) never converts peaks to genes. Instead, each gene is assigned a putative “regulatory domain,” which always contains 5 kb upstream of and 1 kb downstream from its transcription start site and an extension up to the basal regulatory domain of the nearest upstream and downstream genes within 1 Mb. Given a list of genes for a particular term (e.g., actin cytoskeleton), GREAT computes the fraction of the genome covered by the regulatory domains of the genes in the list and the number of peaks hitting these regulatory domains. From this a binomial P-value is computed (see Fig. 1 in McLean et al. 2010).
We have previously shown that GREAT outperforms gene list–based or microarray-based tools at revealing biologically meaningful enrichments in ChIP-seq data sets (McLean et al. 2010). Our extensive comparisons featured four transcription factors—REST (NRSF), GABPA, SRF, and STAT3—in both human and mouse contexts, for which GREAT leverages distal binding sites to reveal accurate and specific function predictions (McLean et al. 2010). To compare our transcription factor and binding site predictions from motif and genome sequence alone to those obtained via antibody ChIP-seq in a particular cellular context, we predicted binding sites from high-quality motifs for the same four factors and analyzed the predictions with GREAT (Table 2).
Table 2.
A comparison of GREAT transcription factor function predictions from PRISM binding site predictions versus ChIP-seq peaks for four different factors
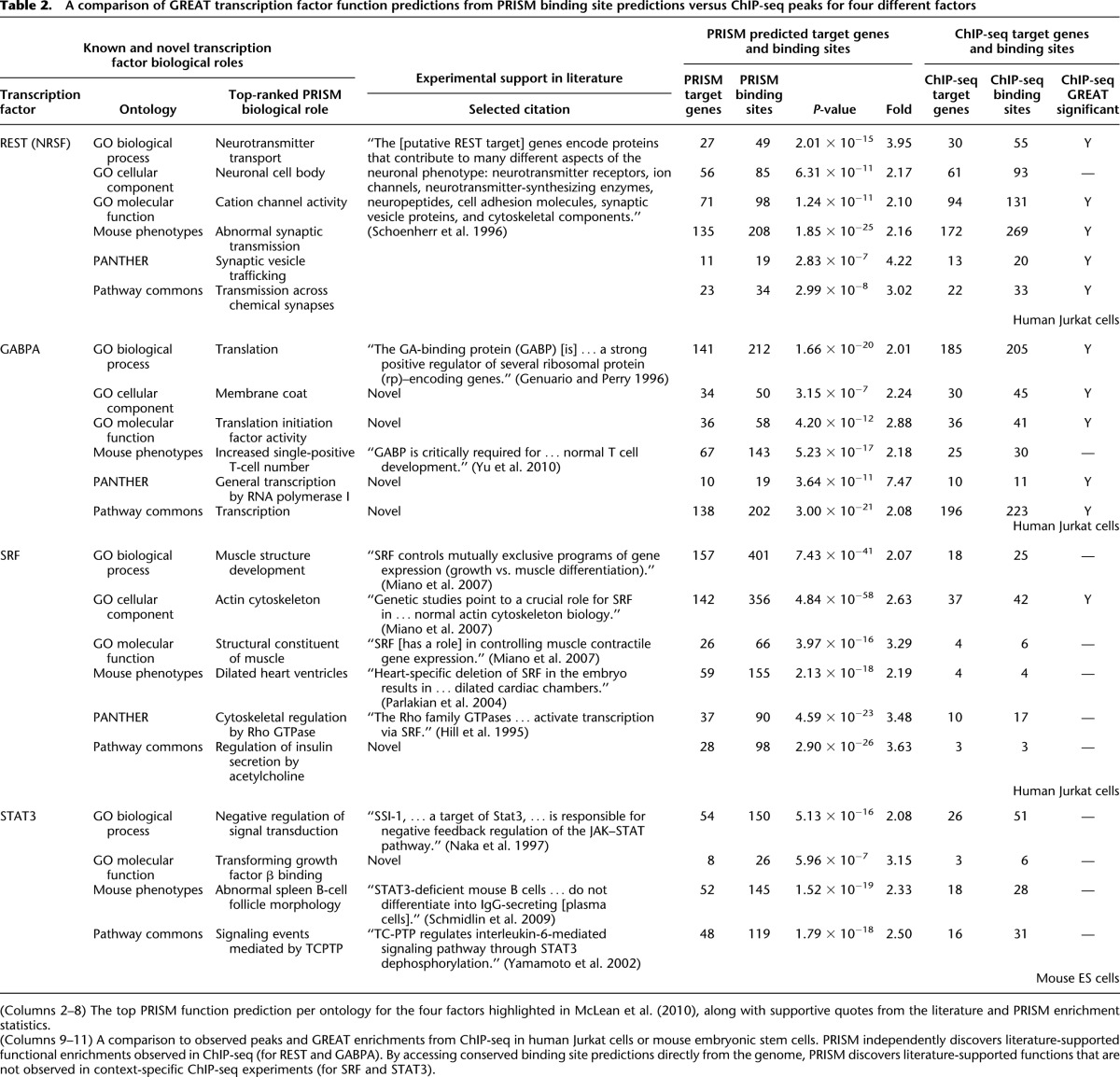
GREAT analysis of REST and GABPA binding site predictions substantially agrees with analysis of ChIP-seq peaks for these factors, with ChIP-seq peaks overlapping 31%–71% of implicated binding site predictions when the enrichments agree (Table 2; Supplemental Tables 6, 7). GREAT analysis of SRF ChIP-seq data from human Jurkat cells generates enrichments that reflect the known role of SRF as the master regulator of actin (42 peaks, P < 10−8) (Supplemental Table 8; McLean et al. 2010). Using our binding site predictions for SRF, we see this same result (P < 10−57) from a broad set of 356 binding sites for 142 target genes (false discovery rate = 38%), the majority of which are not identified in this particular ChIP-seq set (Table 2). In addition, 155 of our binding site predictions for SRF are strongly associated with genes that cause a dilated heart phenotype when knocked out (P < 10−17; binding site FDR = 46%). SRF is well known for its role in heart development, and a conditional knockout of Srf itself in the developing mouse heart leads to a dilated heart phenotype (Parlakian et al. 2004). This experimentally supported result is not found when analyzing the SRF ChIP-seq data, which was generated using Jurkat cells, a T-cell-derived cell line unlikely to reflect the biology of the developing heart.
The enrichments for STAT3 differ markedly between the ChIP-seq and binding site prediction sets. The top enrichments for the STAT3 ChIP-seq data set reflect the context of the experiment, mouse embryonic stem cells (mESC) (see Supplemental Table 9). In contrast, GREAT analysis of genome-wide conserved binding site predictions for STAT3 highlights its well-known role in signaling (P < 10−15; 150 predicted binding sites; binding site FDR = 48%) and the immune system (P < 10−18; 145 sites; binding site FDR = 43%), two functions with no overlapping peaks in the mESC ChIP-seq data (Table 2). Conserved STAT3 binding sites and ChIP-seq data thus produce distinct yet complementary enrichments, which are equally supported by experimental literature.
GREAT analysis of our binding site predictions also produces novel, plausible hypotheses. For example, 98 predicted SRF binding sites show an association with target genes related to the regulation of insulin secretion (P < 10−25; binding site FDR = 28%) (Table 2). While, to our knowledge, this association has not yet been experimentally verified, a recent paper shows that insulin resistance in humans and mice is marked by increased SRF activity (Jin et al. 2011). Similarly, GREAT analysis implicates GABPA in regulating “general transcription by RNA polymerase I” (P < 10−11; 19 binding sites at FDR = 13%), an enzyme that transcribes ribosomal RNA. GABPA is known to regulate transcription of ribosomal proteins (Genuario and Perry 1996). Our predictions suggest that GABPA may function as a regulator of multiple facets of ribosome synthesis.
The PRISM framework: Predicting biological roles, target genes, and enhancers for hundreds of transcription factors
Motivated by the biological function predictions obtained for the four different factors, we set out to analyze the predicted binding sites from each of our 332 curated motifs using GREAT (McLean et al. 2010). We examined nine GREAT ontologies that provide more than 2.4 million facts about human and mouse gene roles in different biological processes, molecular functions, cellular components, phenotypes, molecular pathways, and gene families (Supplemental Table 10; see Methods).
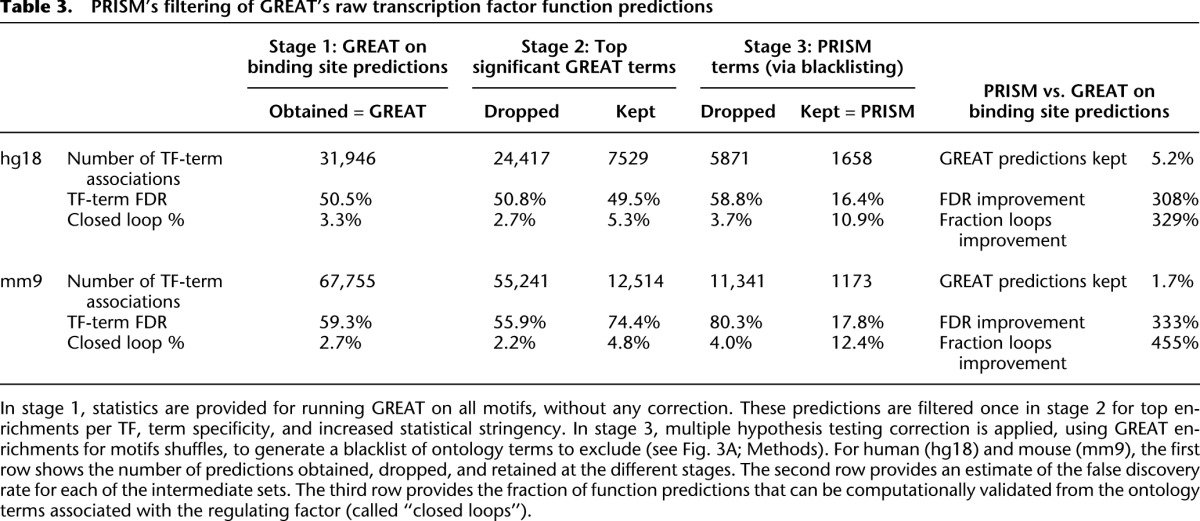
Applying GREAT to binding site predictions from hundreds of transcription factors results in many TF function predictions (Table 3, stage 1). While GREAT accounts for multiple hypothesis test correction for multiple ontology terms against a single set of genomic regions, here we repeatedly apply GREAT to hundreds of sets, one for each motif. To control for multiple hypothesis testing in this framework, we used two filtering stages. First, we focused our attention on only up to the top 20 predictions per motif using a more stringent P-value, and removing broad terms that annotate many genes (see Methods). This resulted in keeping only 23% of the human, and 18% of the mouse GREAT predictions (Table 3, stage 2).
Table 3.
PRISM's filtering of GREAT's raw transcription factor function predictions
To properly account for multiple hypothesis testing, we then applied our entire method to the 2857 shuffled versions of transcription factor motifs used as null models in calculating the excess conservation score (Fig. 3A) (see Methods). We expect such shuffled motifs, by and large, to lack real functional signals, although the method is conservative because some shuffles may capture whole or partial binding preferences of uncharacterized factors and complexes.
Figure 3.
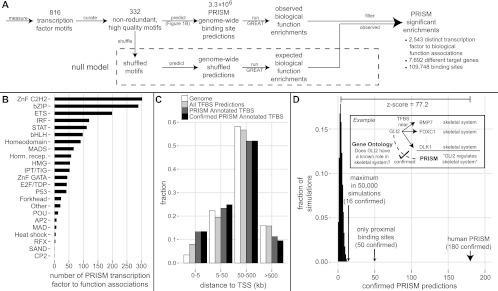
PRISM transcription factor and binding site function predictions. (A) PRISM combines excess conservation binding site prediction (Fig. 1) with GREAT function prediction from proximal and distal sites into a novel statistical framework to arrive at thousands of transcription factor (TF) function predictions, at a false discovery rate of 16%. Numbers are summed over human and mouse (see text). (B) Distribution of PRISM human TF function prediction across the major DNA binding families. (C) Most of the binding sites that support PRISM predictions—including high confidence confirmed predictions—are distal from putative target genes. (D) PRISM predictions are highly enriched for support by previous literature. The GREAT ontologies tag the transcription factor itself with the function predicted by PRISM as enriched among its target genes 180 (11%) times, Z-score = 77, P < 1/50,000 simulation runs (red); 3.6-fold enriched over using only proximal binding sites.
For each biological role (annotation term), we used the total fraction of shuffled motifs for which the term satisfies the GREAT significance thresholds to estimate the expected number of times the term would be falsely called as significant for a set of 332 motifs. Any term expected to occur falsely once or more was excluded (see Methods). Following this very stringent pruning, only 22% of human and 9% of mouse TF function predictions were retained (Table 3, stage 3).
Our shuffled motif TF function predictions can also be used to compute the false discovery rate (FDR) of our original and filtered set, by harshly assuming that all shuffled enrichments are false (see Methods). We see that while the TF function FDR of the human original GREAT predictions is 50.5%, the filtered predictions have a much more appealing FDR of 16.4%. Similarly, for mouse the FDR improves from 59.3% to 17.8%, a more than threefold improvement for both species (Table 3).
In summary, we predicted binding sites using the excess conservation method in the human and mouse genomes, analyzed the predictions with GREAT, and controlled for multiple hypothesis testing using shuffled versions of the same motifs. We term this combined approach PRISM (for predicting regulatory information from single motifs) (Fig. 3A). For each transcription factor, PRISM predicts: (1) biological roles, (2) target genes, (3) binding sites, and implicitly (4) cis-regulatory elements through which the factor regulates each target gene in each biological role.
For the human genome, PRISM predicts 1658 associations between a transcription factor and a biological role (Fig. 3B; Supplemental Figs. 10A, 12A). In all, the predictions connect 178 transcription factors with 5340 target genes via a wide range of 883 different biological roles (captured as a word cloud in Supplemental Fig. 10B) and 59,135 role-specific binding sites, >85% of which are distal (>5 kb from TSS) (see Fig. 3C). The approach produces a similar breadth and quality of coverage for the mouse genome—1173 associations connecting 168 factors with 4993 target genes and 61,437 binding sites through 640 biological roles (Supplemental Figs. 11, 12B). Combining the human and mouse sets and counting identical orthologous predictions only once, PRISM predicts 2543 transcription factor–biological role associations, connecting 217 distinct transcription factors with 7692 distinct target genes (see Methods).
PRISM offers both breadth and depth of biological role predictions
PRISM predictions offer not only breadth (as reflected in Supplemental Fig. 10B), but can also offer depth and accuracy in terms of specific function and perturbation predictions. For example, five genes have been previously identified as key master regulators of muscle differentiation: MYOD1, MYOG, MYF5, MYF6 (MRF4), and the MEF2 family (Pownall et al. 2002). PRISM predicts muscle-related roles for all five (Supplemental Table 13). However, the actual function prediction differs between the factors, reflecting their different biological roles in muscle formation. PRISM correctly implicates MYF5 in regulating the myosin complex (P < 10−7; 59 sites, binding site FDR = 45%; human), MEF2A in broader regulation of contractile fiber (P < 10−12; 128 sites, binding site FDR = 47%; human), and MYOD1 in broad regulation of striated muscle tissue development (P < 10−22; 236 sites, binding site FDR = 50%; mouse). These different functional roles have all been validated experimentally (Supplemental Table 13). PRISM also offers different perturbation predictions. For example, it predicts that both MYOG (P < 10−24; 146 sites, binding site FDR = 37%) and MYF6 (P < 10−10; 110 sites, binding site FDR = 50%) disruption results in general abnormal muscle development. Both predictions have been validated in mouse (Supplemental Table 13). Furthermore, in humans, MYF6 mutations have been associated with Becker muscular dystrophy (Kerst et al. 2000). For MEF2A, PRISM predicts that disruption results specifically in abnormal cardiac output (P < 10−5; 47 sites, binding site FDR = 47%). Indeed, Mef2a knockout mice suffer from severe heart phenotypes resulting in sudden death associated with heart failure and cardiac arrest (Naya et al. 2002).
Objective experimental support for PRISM biological role predictions
The same ontologies used by PRISM to infer biological roles of a transcription factor from its predicted binding sites can sometimes be used to directly confirm a role prediction in an objective, unbiased manner (and thus further support our mostly novel binding site predictions). Because PRISM makes its predictions based solely on the annotations of the predicted target genes, the ontologies themselves can provide such support when the transcription factor itself is tagged with the same function that PRISM identifies as enriched among its downstream target genes. For example, the PRISM prediction that GLI2 is involved in “skeletal system development” (because a surprisingly high number of its predicted target genes are labeled as such) is confirmed by the Gene Ontology, which tags GLI2 itself with the same term and provides a supporting reference (Mo et al. 1997).
This objective test was used to optimize PRISM's design. We have performed an extensive search in parameter space, varying multiple values and design choices (see Methods). The approach we describe here obtains an optimum between computational validation rate and prediction breadth. Indeed, in the unfiltered set of human GREAT predictions, only 3.3% of predictions can be confirmed computationally, a validation rate that improves 3.3-fold in the PRISM subset. Similarly, for the unfiltered mouse GREAT predictions, only 2.7% can be validated computationally, improving 4.5-fold in the PRISM subset (Table 3). In total, 180 (11%) of the 1658 human-based predictions of biological roles for transcription factors and 145 (12%) of the 1173 mouse-based predictions are confirmed this way (Supplemental Tables 11, 12). The number of observed confirmations is highly significant (P < 2 × 10−5, Z-score = 77.2), because no more than 16 matches (1%) were observed in 50,000 simulations that apply a transcription factor's annotations to its shuffled motifs (Fig. 3D).
Distal binding sites contribute greatly to the PRISM approach. With only proximal binding sites (−5 kb to +1 kb from TSS), PRISM in human only predicts 50 (3.6-fold less) biological roles that are confirmed by the ontologies (Fig. 3D), and only 23 (6.3-fold less) mouse predictions confirmed by the ontologies.
While direct ontology support can confirm function predictions, the lack of such support does not imply an incorrect prediction. For example, as discussed above, PRISM predicts that SRF regulates genes that compose the “actin cytoskeleton.” Although SRF is known as the master regulator of the actin cytoskeleton (Miano et al. 2007), it acts in the nucleus and is not involved in building the cytoskeleton itself; thus, it is appropriately not annotated with the Gene Ontology Cellular Component term “actin cytoskeleton.” Other missing confirmations are due to the incompleteness of annotation. For example, PRISM predicts 91 GATA6 binding sites near 23 genes whose mutations lead to abnormal pancreas development (P < 10−12; binding site FDR = 43%). While GATA6 currently lacks the same annotation, a very recent study identified inactivating mutations in GATA6 as the most common cause of pancreatic agenesis in humans (Allen et al. 2011). Similarly, other unconfirmed PRISM predictions may well represent accurate novel predictions.
Overlap of PRISM annotated binding site predictions with ChIP-seq
To evaluate the accuracy and comprehensiveness of individual PRISM binding site predictions, we examined the overlap of binding site predictions with ChIP-seq peaks for four transcription factors with literature-confirmed PRISM biological function predictions. For all four factors, a single ChIP-seq experiment in a single context confirms a considerable fraction of the predicted sites: from 7% for CRX (mouse) to 56% for REST (human). Importantly, this represents a lower bound on the accuracy of binding site prediction, because other ChIP-seq experiments in the same or different contexts likely will support even more binding sites (Fig. 4A).
Figure 4.
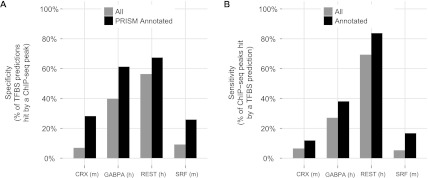
Overlap of PRISM binding site predictions with ChIP-seq. PRISM binding site predictions for CRX (m = mouse), GABPA (h = human), REST (human), and SRF (mouse) were overlapped with ChIP-seq binding sites for the same four factors. Overlap was observed both for the full set of binding site predictions and a subset annotated by PRISM in a particular functional role (“sensory perception of light stimulus” for CRX, “translation” for GABPA, “neurotransmitter transport” for REST, and “actin cytoskeleton” for SRF). (A) Percent of PRISM binding site predictions hit by a ChIP-seq peak. (B) Percent of ChIP-seq peaks hit by a PRISM binding site prediction. Only ChIP-seq peaks with a match to the transcription factor motif in the reference species were considered (CRX: 74%; GABPA: 81%; REST: 60%; SRF: 34%).
From all the binding site predictions, PRISM annotates a subset with specific biological roles. The overlap with ChIP-seq for the annotated subset is significantly larger than for the full set of predictions: >25% for CRX and SRF (mouse), and >60% for REST and GABPA (human). Again, this provides a lower bound on accuracy. It demonstrates that the accuracy of the PRISM-annotated subset of the binding site predictions is often much better than estimated for the full set of predictions (Fig. 4A).
To evaluate the comprehensiveness of PRISM, we examined which fraction of the ChIP-seq peaks for a transcription factor is identified by a PRISM binding site prediction. Interestingly, a number of ChIP-seq peaks for each of the four examined factors lack a match to the transcription factor motif in the genome of the assayed species, ranging from 66% of SRF ChIP-seq peaks to 19% of GABPA peaks. Of the peaks with a motif match in the assayed species, PRISM hits between 5.3% (for SRF) and 69% (for REST) of the experimentally identified peaks. The comprehensiveness significantly improves when examining only those ChIP-seq peaks in the regulatory domains of genes with a relevant biological function. For instance, 16.8% of the SRF peaks near actin cytoskeleton genes are identified by PRISM, compared with 5.3% of all peaks (3.2-fold increase). For REST, PRISM identifies >83% of the ChIP-seq peaks near neurotransmitter transport genes. Thus, while PRISM does not identify every ChIP-seq binding site, it does discover a sizeable fraction, particularly when considering the most confident ChIP-seq peaks that are connected to a specific biological role (Fig. 4B).
Enhancer assays support a role for MYF6 in pancreas as predicted by PRISM
In addition to its known role in muscle development, PRISM predicts a role for myogenic factor 6 (MYF6) in pancreas development (P-value = 1.67 × 10−10; 85 binding sites; binding site FDR = 46%). MYF6 is indeed expressed in the pancreas (Kutlu et al. 2009), but to our knowledge no role in pancreas development has yet been characterized. To examine whether the predicted MYF6 target enhancers drive activity and are responsive to MYF6 in pancreas cells, 15 elements were tested in luciferase enhancer assays in the mPAC cell line, which is derived from pancreatic ductal cells.
Six of the 15 tested elements function as enhancers (luciferase activity ≥2 × empty vector) in the pancreatic cell line (Fig. 5A). All six of the positive elements respond significantly when MYF6 is ectopically expressed via cDNA cotransfection. Two other elements (elt4, which putatively regulates HES1, and elt13, which putatively regulates INSM1) are not enhancers in the standard mPAC cell line but do drive activity in response to ectopically expressed MYF6 (Fig. 5A).
Figure 5.

Enhancer assays support the accuracy of PRISM predictions. (A) Fifteen predicted MYF6 targets that PRISM implicates in pancreas development (estimated binding site FDR = 46%) were tested for enhancer activity and responsiveness to MYF6 in mPAC cells, which are derived from pancreatic ductal cells. Firefly luciferase to Renilla luciferase ratios were normalized to empty vector. Error bars show standard error of the mean over three replicates. (*) A significant (unpaired t-test, P-value < 0.05) response to MYF6 cotransfection (for 8/15 elements = 53%). (B) Predicted PRISM targets were tested across four cell lines matched in context to the PRISM prediction. Across all sets, 67% of the targets respond significantly to the transcription factor predicted by PRISM.
Enhancer assays support the accuracy of PRISM predictions
Four other transcription factor to function predictions were tested using luciferase enhancer assays. Specifically, we examined 20 putative targets of RUNX1 in lung inflammation (P-value = 2.71 × 10−15; 153 binding sites; binding site FDR = 50%) using NHBE cells, 14 targets of STAT1 in regulation of angiogenesis (P-value = 1.22 × 10−10; 96 binding sites; binding site FDR = 49%) using HUVEC cells, eight targets of MYF6 in abnormal muscle development (P-value = 3.27 × 10−11; 110 binding sites; binding site FDR = 50%) using UaSMC cells, and 10 targets of MEF2A in myofibril (P-value = 1.31 × 10−12; 122 binding sites; binding site FDR = 49%) using UaSMC cells. The majority of the enhancers predicted by PRISM to drive activity are responsive to the predicted transcription factor in the appropriate context. Across all examined elements, 67% successfully drive activity and are responsive to the predicted transcription factor (Fig. 5B).
Discussion
As we (Table 2) and others have shown, TF binding is cell type and condition dependent. Here we develop PRISM, a novel approach to predict broad TF functions directly from the genome. It is important to stress that PRISM does not attempt to predict TF occupancy in any particular context (i.e., to offer an alternative to ChIP-seq). It is clear that our understanding of the rules that govern gene regulation is not sufficient (e.g., Fig. 4). Rather, we show that cross-species conserved binding site prediction has become powerful enough to allow us to obtain a subset of binding sites for the predicted factor that is accurate enough and large enough to allow us to correctly predict transcription factor functions.
The general approach has been applied successfully in the past (Das et al. 2006; Down et al. 2007; Sinha et al. 2008). Our main contributions here are:
Unprecedented scope—we use more than 300 different motifs and test them against a vast body of gene function annotation, far more vast than has ever been done before. Hundreds of additional motifs will soon become available, and the body of gene function annotation is constantly on the rise.
Distal binding sites are accounted for—distal binding sites make the majority of observed and predicted binding events (Fig. 2; McLean et al. 2010). They contribute markedly toward our ability to make accurate TF function predictions (Fig. 3D). Using the GREAT test, we let them pull their full weight, whereas other screens before have discarded all but proximal promoter events alone.
We develop a rigorous, nontrivial, and purposefully conservative framework to ensure the quality of our TF function prediction. We improve conserved binding site prediction, we use a harsh null model of shuffled versions of our motifs, and we exclude function predictions that arise from our null.
The results we obtain are in line with our expectations: Our conserved binding site predictions have a relatively high FDR (60%). The FDR, however, markedly improves to 40% when one considers only the subset over which we make TF function predictions, supported by our experimental results (Fig. 5). Most importantly for the goal at hand, our TF function prediction FDR of 16% is appealingly low.
Our rigorous pruning leaves in PRISM only 5.2% of the human transcription factor function predictions that GREAT makes from conserved binding site predictions. This harsh pruning, however, markedly improves by a factor of more than 3 both our FDR and the fraction of predictions we can validate computationally from the annotations of the regulating TF (Table 3). To learn more about the nature of the ontology terms that PRISM blacklists, we used a set of conserved nonexonic genomic elements (CNEs). If we pick 10,000 random subsets of CNEs and create a blacklist from them the same way done in PRISM from motif shuffles, we obtain 2279 terms to blacklist (with E-value > 1). One thousand seven hundred thirty-three (76%) of these make up a full 70% of PRISM's shuffled motifs–based blacklist. This suggests that the majority of PRISM's blacklist derives from the nonrandom distribution of CNEs, well known to be rich in binding sites and their predictions.
Vertebrate transcription regulation is proving to be a complex affair (Bernstein et al. 2012). Large empty gene deserts are now appreciated to be packed with conserved noncoding and active cis-regulatory sequence. Transcription factors thought to form narrow cascades, directly binding a handful of targets, are found to bind thousands of loci in a single context, some conserved, others not, many with no obvious effect on their target genes in perturbation experiments. Adding to this emerging picture, our own analysis suggests that the transcription factors themselves are among the most densely regulated gene families in the mammalian genome. Here we provide windows to this complex system by designing a comprehensive framework for binding site and transcription factor function prediction for a wide range of human and mouse transcription factors and offering them to the community for further exploration. With the recent advent of high-throughput genome editing technologies, PRISM makes a particularly timely contribution (Joung and Sander 2012; Pennisi 2012).
Methods
Multiple genome alignments and phylogenetic tree
All comparative genomic analyses with a human reference used the human-anchored MULTIZ alignment of 44 vertebrates available from UCSC for the hg18 assembly along with the corresponding phylogenetic tree and branch lengths (Kent et al. 2002). Mouse analyses used an extension of the mm9-based MULTIZ alignment of 30 vertebrates from UCSC that includes the same 44 species as the human alignment. Only the eutherian mammals were considered for binding site prediction, and exon and repeat regions were ignored using UCSC annotations. Similar UCSC hg18-based MULTIZ alignments of 17 and 28 vertebrates were used for comparison to evaluate trends in multiple alignments as new species are added.
Transcription factor motif library curation
To obtain a nonredundant set of high-quality motifs, we combined publicly available motifs from UniPROBE (Newburger and Bulyk 2009), JASPAR (Bryne et al. 2008), and TransFac public version 7.0 (Matys et al. 2006). We associated each motif with the gene or genes it describes. Because of high redundancy between and within the different resources and low sample sizes for older entries, we clustered all motifs for a given gene, and used semiautomated curation to identify the highest-quality motif(s) for each factor. Among highly similar motifs for the same gene, we favored motifs derived from larger sample sizes, and higher information content respecting general expectations from related family members. This reduced our library from 816 to a high-quality nonredundant subset of 332 motifs, sampling all major DNA binding domains (Fig. 2A; Supplemental Fig. 5).
Single genome transcription factor binding site prediction
We predicted binding sites in a single genome or region using position weight matrix models of transcription factor binding specificity. Position frequency matrices (fi,j) were converted to position weight matrices (PWMs) (p[i,j]) by weighting each column by its information content (Kel et al. 2003).
Information content of column i: IC(i) = 2 + Σj∈(A,C,G,T) fi,j · log2 fi,j
Weight of base j in column i: p[i,j] = IC(i) · fi,j
Position weight matrix (motif) sequence scores were normalized by dividing by the maximum attainable score. Sequences with a score of at least 0.8 (i.e., matching at least 80% of the possible information content) were considered matches to the motif.
Motif shuffling
For each transcription factor motif, we generated up to 10 null model motifs by shuffling its columns. In shuffling, we preserved adjacent CpG columns, ensured that the shuffles did not resemble any known transcription factor motif or each other, and maintained the “information content profile” (by only swapping high/low information columns with other columns in the same class defining 0.7 bits as the minimum information of a “high” column) (Supplemental Fig. 6).
We defined the similarity of two motifs in a functional manner as the fraction of binding site predictions that overlap. We predicted binding sites with the two factors (see above) over a subset of human genomic gene deserts likely depleted for functional binding events (Ovcharenko et al. 2005). For each offset, i, at which the two motifs overlap, we counted the number of overlapping predictions, picked the highest, and normalized:
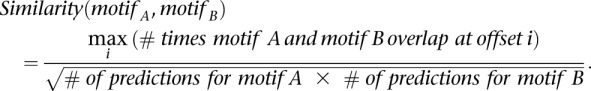 |
In generating shuffled motifs, a similarity threshold of 0.2 was used to reject motifs that resemble known transcription factor motifs or other shuffles of the same motif. This process resulted in 2857 shuffles for the 332 motifs (Supplemental Fig. 7).
Robust binding site prediction across a multiple alignment
The inclusion of more species in a comparative analysis improves detection of conserved regions (Margulies et al. 2006), but it also fragments multiple alignments into smaller blocks (Supplemental Fig. 1). The fragmentation separates nearby genomic bases in alignment space, falsely splitting or distancing binding sites across alignment blocks (Supplemental Fig. 2). To quantify the effect of alignment fragmentation on prediction sensitivity, we considered the subset of binding site predictions confirmed by overlap with an ENCODE ChIP-seq peak from Supplemental Table 2 (see below). In a 17-way multiple alignment, 11% of confirmed binding site predictions would be lost due to alignment fragmentation without corrective measures, with the loss rate increasing to 16.7% in a 44-way alignment, and projected to grow linearly to nearly 30% of confirmed predictions in a forthcoming alignment of 100 species (Supplemental Fig. 3).
To overcome this artifact and recover all lost predictions, we padded alignment blocks with 30 bp (longer than the longest analyzed motif) of adjacent sequence from the genomes of all aligned species, collapsed binding site predictions to their single start coordinate, and placed them on their respective genome (Supplemental Fig. 2A). To robustly predict conserved binding sites, the distance between motif matches was defined as the maximum of the distance measured in the reference and nonreference species genomic coordinates, with the multiple alignment used only to map start positions back to the genome (Supplemental Fig. 2B). We associated motif matches at a distance of up to 20 bp upstream or downstream, previously shown to be optimal for providing robustness to biological or artifactual binding site shifting (Kheradpour et al. 2007).
After associating binding sites in reference and all aligning species, we calculated for every binding site: (1) the number of species with a matching binding site prediction; (2) the total branch length (BL) of the tree over which the binding site is conserved (Kheradpour et al. 2007); and (3) the weighted Bayesian branch length (BBL), which weights phylogenetic distance between species with the binding site match probability (or quality) in each species. BBL was previously shown to outperform BL for motif conservation score and is extensively discussed in Xie et al. (2009).
Efficient conserved binding site prediction
To predict binding sites, we slide a cursor column-by-column in the multiple alignment, scoring every reference position with all motifs and retaining all reference binding site predictions in a window. Given the objective to predict binding sites in the reference genome, one may avoid scoring sequences in an aligning species if the reference does not contain a corresponding binding site nearby, within the allowed local misalignment window (Supplemental Fig. 4A).
Implementing this optimization eliminated the need to predict at 90.4% of aligning positions (Supplemental Fig. 4B), reducing the prediction computation time of the human set from 822 h to 131 h (6.3-fold speed-up) on a cluster of Dell PowerEdge 1950 computers with 2.66 GHz Intel Xeon processors and 16 GB RAM (Supplemental Fig. 4C).
Excess conservation score
The excess conservation framework (Fig. 1A) rescores every motif binding site prediction according to a null distribution of scores of shuffled versions of the motif in genomic windows of 100 bp of similar conservation level. Formally:
 |
To compute it, we partition the reference genome into genomic windows of similar conservation: First, every base in the reference genome is given a weighted “% identity” score from 0% (found only in the reference species) to 100% (same base across the eutherian phylogeny) by calculating the total branch length over which the reference base pair is matched in the multiple alignment as a fraction of the complete branch length in the phylogeny. We then smooth the single position values by averaging over a 100-bp window centered on it, and group into 1% bins.
Next, for every motif m, we generate a set of shuffles Mm (as above). We predict over the reference genome using all shuffled motifs and bin the scores for shuffled motifs into frequency histograms according to the genomic conservation bins just described (Supplemental Fig. 8). We then go back, conceptually, through the reference genome and predict using the motif itself. Every prediction has a certain motif score and is done in a genomic 100-bp neighborhood of certain conservation. We use the frequency curves for that particular genomic neighborhood value to derive the empirical P-value of observing the motif score in that conservation neighborhood. The excess conservation score is −log10 of this P-value (see formula above).
Note that the excess conservation framework can be applied to any motif scoring method. Here it is applied to the Bayesian branch length (BBL) score (above). As anticipated, when the same motif is equally conserved in two different genomic locations, its excess conservation score is higher in less conserved genomic windows (Fig. 1B,C; Supplemental Fig. 8A). In addition, when two different motifs are equally conserved in equally conserved genomic windows, the motif with higher information content has a higher excess conservation score, reflecting the higher specificity of its shuffles (Supplemental Fig. 8B).
Binding site predictions in the reference genome were retained and ranked by excess conservation score, if they were supported by at least four additional species, with a branch length (BL) score of at least two substitutions per site and an excess conservation score of at least 1.3 (i.e., the P-value of the observed motif score in similarly conserved windows ≤ 0.05).
Evaluating accuracy of binding site prediction using ChIP-seq data
We used the UCSC Table Browser to download transcription factors ChIP-seq peaks (binding sites) assayed in human cells by the ENCODE Consortium (Bernstein et al. 2012). When multiple ChIP-seq experiments or replicates were available for the same factor, we selected the one with the largest number of peaks, yielding 56 distinct transcription factor sets. All 47 transcription factors for which we had a motif in our library were used (Supplemental Table 2).
The accuracy of binding site prediction before and after the excess conservation adjustment is summarized by area under the curve of precision-recall curves (Supplemental Table 1). We considered a binding site to overlap a ChIP-seq peak if at least one base pair overlapped. The conservation neighborhood for ChIP-seq peaks in Figure 1D is measured at the peak center.
To evaluate the overlap for functionally annotated binding sites (Fig. 4), four ChIP-seq sets from appropriate functional contexts were identified: CRX in mouse retina (Corbo et al. 2010), GABPA in human epithelial cells (Bernstein et al. 2012), REST in human Jurkat cells (Valouev et al. 2008), and SRF in mouse cardiomyocytes (He et al. 2011). To evaluate sensitivity (fraction of ChIP-seq peaks hit by a binding site prediction), only the ChIP-seq peaks with a match to the motif in the reference species (PWM threshold = 0.8) were considered.
Identifying binding site overlap with GWAS SNPs
The NHGRI GWAS catalog of disease-associated SNPs (Hindorff et al. 2009) was obtained from the UCSC Genome Browser “gwasCatalog” track (hg18 assembly). All PRISM binding site predictions that overlap the SNP by at least one base pair were identified (Table 1; Supplemental Table 5). The association of ESR1 (ER-alpha) (ER-α) with rs909116 is through a predicted binding site for the paralogous factor ESRRA (ERR-alpha) (ERR-α) (protein similarity BLASTP E-value <10−45). Statistical enrichment of overlap of binding site predictions with GWAS SNPs was calculated using a one-tailed Fisher's exact test: dbSNP build 130 has 14,985,544 single nucleotide SNPs; the NHGRI GWAS catalog associates 3776 of these SNPs with a phenotype; PRISM binding site predictions overlap 32,069 SNPs of which 15 are connected to a phenotype.
PRISM en masse transcription factor function prediction
PRISM function predictions were obtained in three stages (Fig. 3A): In stage 1 (Table 3) for each of our 332 transcription factor motifs, the top 5000 excess conservation binding site predictions in human and mouse were analyzed using GREAT v1.7 (McLean et al. 2010). Binding sites were associated with target genes using the GREAT default “Basal plus extension” association rule with default distances of 5 kb upstream, 1 kb downstream, and up to 1 Mb extension, or up to the next gene. We used the default GREAT filters for significant terms: region-based fold enrichment ≥2, and a region-based and gene-based false discovery rate (FDR) Q-value ≤0.05, with the additional requirement that at least five genes with the term were hit. Analysis was done over nine GREAT ontologies—the three Gene Ontology domains (Ashburner et al. 2000), Mouse Phenotypes (Blake et al. 2009), PANTHER Pathway (Mi et al. 2007), Pathway Commons (Cerami et al. 2006), BioCyc Pathway (Caspi et al. 2008), TreeFam (Ruan et al. 2008), and HGNC Gene Families (Supplemental Table 10; Bruford et al. 2008).
In stage 2 (Table 3), we pruned our results to focus attention on the top enrichments. We limited to the top 20 terms per ontology for each motif, ignored broad terms annotated to more than 500 genes, and required an uncorrected region-based GREAT P-value ≤ 10−5.
In stage 3 (Table 3), to provide multiple testing correction for running GREAT 332 times per genome, we used our shuffled motifs. We separately repeated the entire GREAT analysis with all 2857 shuffled motifs. For each term, we then computed its Expected value (E-value), or number of times it would appear (by chance) in 332 runs of shuffled motifs:
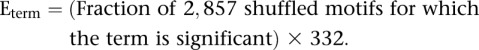 |
When Eterm > 1, the term was removed from the predicted enrichments for real motifs. This resulted in blacklisting 9% of the 27,956 human ontology terms and 13% of the 26,656 mouse ontology terms (Supplemental Fig. 9), resulting in the human and mouse sets reported in Figure 3B and Supplemental Figures 10 and 11.
To estimate the false discovery rate (Table 3), we compared the number of PRISM enrichments for real and shuffled transcription factor motifs. We applied the same PRISM pipeline to shuffled motifs as to real motifs. To avoid unfairly having a motif contribute to blacklisting its own terms, each shuffled motif was excluded when calculating its Eterm values. To calculate FDR, we assume that (i) the number of false positive enrichments is the same for real and shuffled motifs and (ii) all enrichments for shuffled motifs are false:
(i) FPreal = FPshuffle
(ii) FPshuffle = Pshuffle
→ (iii) FPreal = Pshuffle
(iv) FDR = FPreal/Preal = Pshuffle/Preal
For human, the library motifs average 4.99 associations per motif (Preal = 4.99), while the shuffled motifs average 0.74 associations (Pshuffle = 0.82), for an FDR of 0.82/4.99 = 16.4%. For mouse, the library motifs average 3.53 associations per motif (Preal = 3.53), and the shuffled motifs average 0.63 (Pshuffle = 0.63) for an FDR of 17.8%. Similar computation was performed to compute the FDR of intermediate sets in Table 3.
To evaluate PRISM with only proximal binding sites (Fig. 3D), we analyzed the set of top binding site predictions for each motif with GREAT v1.7 using only basal regulatory domains (5 kb upstream of and 1 kb downstream from the TSS). Shuffled motifs were used to calculate proximal-specific Expected (E) values for terms, which were then filtered as explained above.
To rank target genes, we created a GREAT ontology with each gene as its own term. Genes were ranked by the region-based binomial P-value, thus prioritizing genes with an unexpectedly high number of binding sites in the regulatory domain given the size of the assigned domain.
To unify human and mouse predictions, we first manually verified all mappings of motifs to human and mouse transcription factors. We then mapped orthologous genes and binding sites between species. Human and mouse orthologous target genes were defined as top BLASTP hits from the UCSC Table Browser hg18.mmBlastTab table, collapsing transcripts into loci through UCSC gene clusters (Kent et al. 2002). Binding site predictions were considered orthologous if they were identified as nearest matches in the multiple alignment in binding site prediction (see above).
Optimality of PRISM parameters
The PRISM method requires multiple parameter and threshold choices for binding site prediction and then inference of TF function. To optimize our approach, we evaluate multiple parameter and design choice combinations using the objective measure of the number of PRISM function predictions (derived from target gene annotations) confirmed by the ontologies (from an identical annotation for the regulating TF itself) (see Fig. 3D). We optimized parameter values using coordinate descent (in which one parameter is varied with all others fixed, iterating through the different parameters), starting from different initial parameter settings as seed. To test our design choices, we also varied the ways in which certain operations were performed, including an information content versus log likelihood interpretation of PWMs, passing a fixed number of binding sites to GREAT versus passing a variable number of sites meeting a uniform FDR threshold, defining the E-value for the functional enrichments as the fraction observed in any shuffles or as the fraction observed in shuffles that had a P-value at least as significant as the one observed in the real data set, the choice of top 20 terms per ontology before or after applying the E-value filter, changing the order in which our filters are applied and more. The approach and parameter settings we describe maximize the fraction of annotations with direct ontology support, from the large number of alternatives tested (data not shown). Note that this approach does not explicitly optimize the FDR of TF–term associations; thus, the TF function FDR provides an independent measure of the quality of the selected parameters.
Enhancer assays
Cell line/primary cell culture
The mPAC cell line was grown in Dulbecco's Modified Eagle's Medium (DMEM), supplemented with 10% Fetal Bovine Serum (FBS; Life Technologies). Normal Human Bronchial/Tracheal Epithelial Cells (NHBE; Lonza Walkersville, Inc.) were grown in Clonetics Bronchial/Tracheal Epithelial Growth Medium (BEGM). Normal Human Umbilical Vein Endothelial Cells (HUVEC; Lonza Walkersville, Inc.) were grown in Clonetics Endothelial Growth Medium (EGM; Lonza Walkersville, Inc.). Human Umbilical artery Smooth Muscle Cells (UaSMC; Lonza Walkersville, Inc.) were grown in Clonetics Smooth Muscle Growth Medium (SmGM; Lonza Walkersville, Inc.).
Plasmids
MYF6 (EMSV-MRF4 (puro), rat myf6) was a gift of Michael Rudnicki (Sabourin and Rudnicki 2000) (Addgene plasmid 14713). RUNX1 (pCMV5-AML1B [human runx1]) was a gift of Scott Hiebert (Meyers et al. 1995) (Addgene plasmid 12426). STAT1 (Stat1 alpha Flag pRc/CMV [human stat1 alpha]) was a gift of Jim Darnell (Horvath et al. 1995) (Addgene plasmid 8691). MEF2A (pCGN-MEF2A [human mef2a]) was a gift of Ron Prywes (Han and Prywes 1995) (Addgene plasmid 32958).
Cloning
The firefly luciferase reporter vector pGL4.23 (Promega) was modified for ligation independent cloning (Du et al. 2011) by cloning the annealed oligos:
LIC fwd site: 5′- cGCTCTTCGGGATGGAGGGATATCCACCTTACCCGAAGAGCa-3′
LIC rev site: 5′-agcttGCTCTTCGGGTAAGGTGGATATCCCTCCATCCCGAAGAGCggtac-3′
into the KpnI and HindIII sites of the pGL4.23 vector. The vector was prepped by digesting with EcoRV and Nt.BspQI (New England Biolabs). Human genomic DNA (Clonetech) was amplified using target-specific primers with the sequence ggatggaggatc on the forward primer 5′ end and ggtaaggtggatc on the reverse primer 5′ end. Primers were synthesized by Elim Biopharm. Inserts were treated with T4 DNA polymerase in the presence of 10 mM GTP, annealed to the treated vector, and transformed according to published methods (Du et al. 2011). The primers used for cloning are listed in Supplemental Table 14.
Transfections and luciferase assay
Cells were cultured to 80%–90% confluence and transfected using Lipofectomine LTX and PLUS reagent according to the manufacturer's instructions (Life Technologies). Transfections were done in a 96-well format using a 1-μL LTX:250 ng plasmid ratio. The plasmids transfected consisted of 200 ng of reporter construct and 50 ng of TF expression plasmid or control plasmid pCAG-DsRED (a gift of Connie Cepko) (Matsuda and Cepko 2004) (Addgene plasmid 11151). Media was changed 4–6 h after transfection, and luciferase assays were done 24 h after transfection. Luciferase assays were done using a DLR 100 kit (Promega) according to the manufacturer's instructions and read using a Promega Glomax luminometer.
Data access
PRISM predictions for the human and mouse genomes are available at http://PRISM.stanford.edu. The PRISM portal offers an interface to explore our predictions from the perspective of transcription factors, biological roles, target genes, or target binding sites/regions (Supplemental Fig. 13). PRISM integrates with GREAT (McLean et al. 2010) and the UCSC Genome Browser (Kent et al. 2002).
Acknowledgments
We thank Seung Kim for providing us mPAC cells; Tom Cramer for freeing the PRISM Stanford domain name; Ravi Parikh for improving the user interface of the PRISM resource; Michael Hiller for the mouse 44-way alignment; and Will Talbot, Nadav Ahituv, Betty Booker, and the Bejerano laboratory for helpful comments. This work was supported by a Stanford Graduate Fellowship (A.M.W.), a Bio-X Stanford Interdisciplinary Graduate Fellowship (A.M.W.), an HHMI Gilliam Fellowship (S.L.C.), a National Science Foundation Fellowship DGE-1147470 (H.G.), a Bio-X Graduate Fellowship (C.Y.M.), NIH grants R01HG005058 and R01HD059862, NSF Center for Science of Information (CSoI) grant CCF-0939370, and KAUST (all to G.B.). G.B. is a Packard Fellow and Microsoft Research Fellow.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.139071.112.
Freely available online through the Genome Research Open Access option.
References
- Allen HL, Flanagan SE, Shaw-Smith C, De Franco E, Akerman I, Caswell R, Ferrer J, Hattersley AT, Ellard S 2011. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat Genet 44: 20–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. 2000. Gene Ontology: Tool for the unification of biology. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D 2004. Ultraconserved elements in the human genome. Science 304: 1321–1325 [DOI] [PubMed] [Google Scholar]
- Berger MF, Badis G, Gehrke AR, Talukder S, Philippakis AA, Pena-Castillo L, Alleyne TM, Mnaimneh S, Botvinnik OB, Chan ET, et al. 2008. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 133: 1266–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake JA, Bult CJ, Eppig JT, Kadin JA, Richardson JE 2009. The Mouse Genome Database genotypes∷phenotypes. Nucleic Acids Res 37: D712–D719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow MJ, McCulley DJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. 2010. ChIP-Seq identification of weakly conserved heart enhancers. Nat Genet 42: 806–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruford EA, Lush MJ, Wright MW, Sneddon TP, Povey S, Birney E 2008. The HGNC Database in 2008: A resource for the human genome. Nucleic Acids Res 36: D445–D448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryne JC, Valen E, Tang MH, Marstrand T, Winther O, da Piedade I, Krogh A, Lenhard B, Sandelin A 2008. JASPAR, the open access database of transcription factor-binding profiles: New content and tools in the 2008 update. Nucleic Acids Res 36: D102–D106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R, Foerster H, Fulcher CA, Kaipa P, Krummenacker M, Latendresse M, Paley S, Rhee SY, Shearer AG, Tissier C, et al. 2008. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res 36: D623–D631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami EG, Bader GD, Gross BE, Sander C 2006. cPath: Open source software for collecting, storing, and querying biological pathways. BMC Bioinformatics 7: 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, King DC, Dore LC, Zhang X, Zhou Y, Zhang Y, Dorman C, Abebe D, Kumar SA, Chiaromonte F, et al. 2008. Transcriptional enhancement by GATA1-occupied DNA segments is strongly associated with evolutionary constraint on the binding site motif. Genome Res 18: 1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbo JC, Lawrence KA, Karlstetter M, Myers CA, Abdelaziz M, Dirkes W, Weigelt K, Seifert M, Benes V, Fritsche LG, et al. 2010. CRX ChIP-seq reveals the cis-regulatory architecture of mouse photoreceptors. Genome Res 20: 1512–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das D, Nahle Z, Zhang MQ 2006. Adaptively inferring human transcriptional subnetworks. Mol Syst Biol 2: 2006–0029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Down TA, Bergman CM, Su J, Hubbard TJ 2007. Large-scale discovery of promoter motifs in Drosophila melanogaster. PLoS Comput Biol 3: e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du R, Li S, Zhang X 2011. A modified plasmid vector pCMV-3Tag-LIC for rapid, reliable, ligation-independent cloning of polymerase chain reaction products. Anal Biochem 408: 357–359 [DOI] [PubMed] [Google Scholar]
- Fogelgren B, Yang S, Sharp IC, Huckstep OJ, Ma W, Somponpun SJ, Carlson EC, Uyehara CF, Lozanoff S 2009. Deficiency in Six2 during prenatal development is associated with reduced nephron number, chronic renal failure, and hypertension in Br/+ adult mice. Am J Physiol Renal Physiol 296: F1166–F1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genuario RR, Perry RP 1996. The GA-binding protein can serve as both an activator and repressor of ribosomal protein gene transcription. J Biol Chem 271: 4388–4395 [DOI] [PubMed] [Google Scholar]
- Han TH, Prywes R 1995. Regulatory role of MEF2D in serum induction of the c-jun promoter. Mol Cell Biol 15: 2907–2915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He A, Kong SW, Ma Q, Pu WT 2011. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc Natl Acad Sci 108: 5632–5637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CS, Wynne J, Treisman R 1995. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 81: 1159–1170 [DOI] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA 2009. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci 106: 9362–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath CM, Wen Z, Darnell JE Jr 1995. A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes Dev 9: 984–994 [DOI] [PubMed] [Google Scholar]
- Jin S, Zhao H, Yi Y, Nakata Y, Kalota A, Gewirtz AM 2010. c-Myb binds MLL through menin in human leukemia cells and is an important driver of MLL-associated leukemogenesis. J Clin Invest 120: 593–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Goldfine AB, Boes T, Henry RR, Ciaraldi TP, Kim EY, Emecan M, Fitzpatrick C, Sen A, Shah A, et al. 2011. Increased SRF transcriptional activity in human and mouse skeletal muscle is a signature of insulin resistance. J Clin Invest 121: 918–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolma A, Kivioja T, Toivonen J, Cheng L, Wei G, Enge M, Taipale M, Vaquerizas JM, Yan J, Sillanpaa MJ, et al. 2010. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res 20: 861–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung JK, Sander JD 2012. TALENs: A widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 14: 49–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamatani Y, Matsuda K, Okada Y, Kubo M, Hosono N, Daigo Y, Nakamura Y, Kamatani N 2010. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat Genet 42: 210–215 [DOI] [PubMed] [Google Scholar]
- Kel AE, Gossling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E 2003. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res 31: 3576–3579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D 2002. The human genome browser at UCSC. Genome Res 12: 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerst B, Mennerich D, Schuelke M, Stoltenburg-Didinger G, von Moers A, Gossrau R, van Landeghem FK, Speer A, Braun T, Hubner C 2000. Heterozygous myogenic factor 6 mutation associated with myopathy and severe course of Becker muscular dystrophy. Neuromuscul Disord 10: 572–577 [DOI] [PubMed] [Google Scholar]
- Kheradpour P, Stark A, Roy S, Kellis M 2007. Reliable prediction of regulator targets using 12 Drosophila genomes. Genome Res 17: 1919–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutlu B, Burdick D, Baxter D, Rasschaert J, Flamez D, Eizirik DL, Welsh N, Goodman N, Hood L 2009. Detailed transcriptome atlas of the pancreatic β cell. BMC Med Genomics 2: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M 2010. Transcriptional enhancers in animal development and evolution. Curr Biol 20: R754–R763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB 2003. Prediction of mammalian microRNA targets. Cell 115: 787–798 [DOI] [PubMed] [Google Scholar]
- Margulies EH, Chen CW, Green ED 2006. Differences between pair-wise and multi-sequence alignment methods affect vertebrate genome comparisons. Trends Genet 22: 187–193 [DOI] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL 2004. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci 101: 16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. 2006. TRANSFAC and its module TRANSCompel: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34: D108–D110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28: 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers S, Lenny N, Hiebert SW 1995. The t(8;21) fusion protein interferes with AML-1B-dependent transcriptional activation. Mol Cell Biol 15: 1974–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Guo N, Kejariwal A, Thomas PD 2007. PANTHER version 6: Protein sequence and function evolution data with expanded representation of biological pathways. Nucleic Acids Res 35: D247–D252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miano JM, Long X, Fujiwara K 2007. Serum response factor: Master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol 292: C70–C81 [DOI] [PubMed] [Google Scholar]
- Michelson AM 2002. Deciphering genetic regulatory codes: A challenge for functional genomics. Proc Natl Acad Sci 99: 546–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi Y, Murase K, Saito M, Imamura M, Oh K 2010. Mechanisms of estrogen receptor-α upregulation in breast cancers. Med Mol Morphol 43: 193–196 [DOI] [PubMed] [Google Scholar]
- Mo R, Freer AM, Zinyk DL, Crackower MA, Michaud J, Heng HH, Chik KW, Shi XM, Tsui LC, Cheng SH, et al. 1997. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development 124: 113–123 [DOI] [PubMed] [Google Scholar]
- Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, Nishimoto N, Kajita T, Taga T, Yoshizaki K, et al. 1997. Structure and function of a new STAT-induced STAT inhibitor. Nature 387: 924–929 [DOI] [PubMed] [Google Scholar]
- Naya FJ, Black BL, Wu H, Bassel-Duby R, Richardson JA, Hill JA, Olson EN 2002. Mitochondrial deficiency and cardiac sudden death in mice lacking the MEF2A transcription factor. Nat Med 8: 1303–1309 [DOI] [PubMed] [Google Scholar]
- Newburger DE, Bulyk ML 2009. UniPROBE: An online database of protein binding microarray data on protein–DNA interactions. Nucleic Acids Res 37: D77–D82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovcharenko I, Loots GG, Nobrega MA, Hardison RC, Miller W, Stubbs L 2005. Evolution and functional classification of vertebrate gene deserts. Genome Res 15: 137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlakian A, Tuil D, Hamard G, Tavernier G, Hentzen D, Concordet JP, Paulin D, Li Z, Daegelen D 2004. Targeted inactivation of serum response factor in the developing heart results in myocardial defects and embryonic lethality. Mol Cell Biol 24: 5281–5289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennisi E 2012. Beyond TALENs. Science 338: 1411. [DOI] [PubMed] [Google Scholar]
- Phillips JE, Corces VG 2009. CTCF: Master weaver of the genome. Cell 137: 1194–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlasek CA, Clemens JQ, Bushman W 1999. Hoxa-13 gene mutation results in abnormal seminal vesicle and prostate development. J Urol 161: 1655–1661 [PubMed] [Google Scholar]
- Pownall ME, Gustafsson MK, Emerson CP Jr 2002. Myogenic regulatory factors and the specification of muscle progenitors in vertebrate embryos. Annu Rev Cell Dev Biol 18: 747–783 [DOI] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Prescott S, Brugmann SA, Swigut T, Wysocka J 2012. Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell 11: 633–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan J, Li H, Chen Z, Coghlan A, Coin LJ, Guo Y, Heriche JK, Hu Y, Kristiansen K, Li R, et al. 2008. TreeFam: 2008 Update. Nucleic Acids Res 36: D735–D740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabourin LA, Rudnicki MA 2000. The molecular regulation of myogenesis. Clin Genet 57: 16–25 [DOI] [PubMed] [Google Scholar]
- Schmidlin H, Diehl SA, Blom B 2009. New insights into the regulation of human B-cell differentiation. Trends Immunol 30: 277–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, Kutter C, Watt S, Martinez-Jimenez CP, Mackay S, et al. 2010. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328: 1036–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenherr CJ, Paquette AJ, Anderson DJ 1996. Identification of potential target genes for the neuron-restrictive silencer factor. Proc Natl Acad Sci 93: 9881–9886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Adler AS, Field Y, Chang HY, Segal E 2008. Systematic functional characterization of cis-regulatory motifs in human core promoters. Genome Res 18: 477–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szolkiewicz M, Chmielewski M, Nogalska A, Stelmanska E, Swierczynski J, Rutkowski B 2007. The potential role of sterol regulatory element binding protein transcription factors in renal injury. J Ren Nutr 17: 62–65 [DOI] [PubMed] [Google Scholar]
- Takata R, Akamatsu S, Kubo M, Takahashi A, Hosono N, Kawaguchi T, Tsunoda T, Inazawa J, Kamatani N, Ogawa O, et al. 2010. Genome-wide association study identifies five new susceptibility loci for prostate cancer in the Japanese population. Nat Genet 42: 751–754 [DOI] [PubMed] [Google Scholar]
- Valouev A, Johnson DS, Sundquist A, Medina C, Anton E, Batzoglou S, Myers RM, Sidow A 2008. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat Methods 5: 829–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Rubin EM, Pennacchio LA 2009. Genomic views of distant-acting enhancers. Nature 461: 199–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562 [DOI] [PubMed] [Google Scholar]
- Xie X, Rigor P, Baldi P 2009. MotifMap: A human genome-wide map of candidate regulatory motif sites. Bioinformatics 25: 167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Sekine Y, Kashima K, Kubota A, Sato N, Aoki N, Matsuda T 2002. The nuclear isoform of protein-tyrosine phosphatase TC-PTP regulates interleukin-6-mediated signaling pathway through STAT3 dephosphorylation. Biochem Biophys Res Commun 297: 811–817 [DOI] [PubMed] [Google Scholar]
- Yu S, Zhao DM, Jothi R, Xue HH 2010. Critical requirement of GABPα for normal T cell development. J Biol Chem 285: 10179–10188 [DOI] [PMC free article] [PubMed] [Google Scholar]