

Abstract
Objective
In addition to its effects on cholesterol levels, apoE3 has lipid-independent effects that contribute to cardiovascular protection; one of these effects is the ability to inhibit cell cycling in VSMCs. The goal of this study was to identify and characterize cell cycle-regulatory mechanisms responsible for the anti-mitogenic effect of apoE.
Methods and results
Primary VSMCs were stimulated with serum in the absence or presence of apoE3. apoE3 upregulated expression of the cdk inhibitor, p27kip1, in primary VSMCs, and this effect required Cox2 and activation of PGI2-IP signaling. The microRNA family, miR221/222 has recently been identified as a post-translational regulator of p27, and apoE3 inhibited miR221/222 expression in a Cox2- and PGI2/IP-dependent manner. Moreover, reconstituted miR222 expression was sufficient to override the effects of apoE on p27 expression and S phase entry. The ability to repress expression of miR221/222 is shared by apoE3-containing HDL but is absent from apoA-1, LDL and apoE-depleted HDL. All three apoE isoforms regulate miR221/222, and the effect is independent of the C-terminal lipid-binding domain. miR221/222 levels are increased in the aortae of apoE3-null mice and reduced when apoE3 expression is reconstituted by adeno-associated virus infection. Thus, regulation of miR221/222 by apoE3 occurs in vivo as well as in vitro.
Conclusions: A
poE inhibits VSMC proliferation by regulating p27 through miR221/222. Control of cell cycle-regulatory microRNAs adds a new dimension to the spectrum of cardiovascular protective effects afforded by apoE and apoE-HDL.
Keywords: ApoE3, HDL, PGI2, p27, miR221/222, VSMC proliferation
1. Introduction
Apolipoprotein E (apoE), a component of high density and triglyceride-rich lipoproteins, regulates lipid homeostasis and plays an important role in preventing atherosclerotic disease [1,2]. ApoE3 is composed of a C-terminal, 10-kDa domain that is required for lipid-binding and an N-terminal, 22-kDa that binds to the LDL receptor [1,3]. Other reported properties that contribute to the anti-atherogenic behavior of HDL include anti-inflammation, anti-oxidation, anti-thrombosis and vasodilation. ApoE may also protect against cardiovascular disease by inhibiting vascular smooth muscle cell (VSMC) proliferation [4–8]. Transgenic expression of apoE inhibits, while deletion of apoE increases, VSMC proliferation after vascular injury in vivo [9].
ApoE is a polymorphic protein with three major isoforms, apoE2, apoE3 and apoE4. ApoE3 is the most common and is considered to be the parent form of the protein [1,3]. The polymorphism in apoE occurs at residues 112 and 158; the apoE4 isoform contains R at both positions while the apoE3 and apoE2 isoforms contain C/R and C/C, respectively, at these sites. The C/R interchange at position 112 that distinguishes apoE3 and apoE4 has little effect on LDLR binding activity whereas the C/R substitution at position 158 dramatically lowers the binding of apoE2 to the LDL receptor (LDLR). This is the primary defect in type III hyper-lipoproteinemia. Besides being a risk factor for atherosclerosis, apoE4 polymorphism is a major genetic risk factor for Alzheimer’s disease [1,10].
The mechanism by which apoE controls VSMC proliferation is not well understood. Others have reported that its anti-mitogenic effect is associated with a partial reduction in the expression of cyclin D1 mRNA [4]. We have not seen a strong effect of apoE on cyclin D1 [6], but did find that apoE3 increases Cox2 expression and prostacyclin (PGI2) production in primary VSMCs [6,11]. Furthermore, we showed that these effects led to a PGI2- and cyclin E/cdk2-dependent inhibition of VSMC cycling [6]. Cyclin-cdk2 complexes are typically inhibited by the binding of cip/kip family cyclin-dependent kinase (cdk) inhibitors (p21cip1, p27kip1, and p57kip2). Of these cdks, p27 has been closely linked to regulation of VSMC cycling, especially after vascular injury [12,13]. Deletion of p27 also accelerates atherosclerosis in apoE-null mice [14]. Regulators of p27 therefore have the potential to strongly influence neointima formation in atherosclerosis and during the response to injury. In this report, we show that apoE and apoE-containing HDL strongly inhibit the expression of miR221/222, a microRNA family that regulates p27 levels post-transcriptionally. Additionally, we show that the effect of apoE on miR221/222 leads to an upregulation of p27, and that the change p27 expression is sufficient to explain the anti-mitogenic effect of apoE in VSMCs.
2. Methods
2.1. Cell culture
Early passage explant cultures of mouse vascular smooth muscle cells (VSMCs) were isolated from 8 to 10 week old male C57BL/6 mouse (Jackson Labs) or IP-null mice on the C57BL/6 background ([15]; kindly provided by Garret FitzGerald, University of Pennsylvania). Explant culture VSMCs were isolated from aortae (aortic arch plus the descending thoracic aorta) of these mice as described [6] and maintained in growth medium (1:1 Dulbecco’s modified Eagle’s Medium (DME)/Ham’s F-12 supplemented with 2 mM L-glutamine and 10% FBS). The FBS was not depleted of bovine apoE before use. Cells were used between passages 2–5. For cell cycle experiments, 60–90% confluent monolayers of wild-type, IP−/−, or p27−/− VSMCs were grown in 60-mm (for RT-qPCR, S phase assays, or transfections) or 100-mm (for RT-qPCR and western blotting) culture dishes. The cells were G0-synchronized by incubation in serum-free DME containing 1 mg/ml heat-inactivated, fatty acid-free BSA (DME-BSA) for 48 h before stimulation with fresh growth medium in the absence or presence of 200 nM cicaprost (kindly provided by Bayer Schering Pharma AG), 50 μg/ml lipoprotein, 60 ± 5 μg/ml apolipoprotein, 1 μM nimesulide (a Cox-2 inhibitor) or 1 mM SC560 (a Cox-1 inhibitor). Recombinant human apoA-I, apoE2, apoE3, apoE4, the 22 kD N-terminal fragment (amino acids 1–191) of apoE3, and the 10 kD C-terminal fragment (amino acids 222–299) of apoE3 were expressed in E. coli and purified as described [16,17]. ApoE3 and its N- and C-terminal domains were tested at equivalent molarities (2 μM), as were the three apoE isoforms. Samples were dialyzed against PBS immediately before use. When S phase entry was measured, BrdU or EdU was added at the time of serum stimulation and remained in the cultures throughout the experimental incubation. LDL, total HDL, and apoE-depleted HDL were purified similarly to published procedures [18,19].
2.2. Transfections
We transfected near confluent VMSCs in 60-mm dishes containing coverslips with 3 μg of either an expression plasmid for microRNA-222 (Origene) or pCDNA (control) using 25 μl Lipofectamine 2000. After 4 h, the transfected cells were allowed to recover overnight in regular growth medium. The cells were then starved for 48 h in DME/BSA and directly stimulated with fresh growth medium containing 10% FBS and apoE3.
2.3. Quantitative real-time reverse transcriptase-PCR (qPCR)
To measure steady-state levels of miR-221 and miR-222, total RNA was isolated from cells or isolated aortae with TRIZOL and reverse transcribed using 15–40 ng of RNA in a 10-μl reaction with TaqMan MicroRNA reverse transcription kit (Applied Biosystems). An aliquot (20%) of the reaction was used for qPCR using TaqMan universal master mix, Mature MicroRNA assay ID #524 (miR221), #2276 (miR222), and #1232 (snoRNA202) (Applied Biosystems). To measure Cox-2 mRNA levels, ~50 ng of RNA was reverse transcribed in a 20-μl reaction, and the cDNA was subjected to qPCR using TaqMan gene expression assays Mm00478374_m1 (Applied Biosystems) respectively. RT-qPCR results were calculated using the standard curve or ddCt methods using 18S and SnoRNA202 as the reference for mRNAs and microRNAs, respectively.
2.4. Immunoblotting and immunofluorescence microscopy
Cells for immunoblotting were collected and lysed as described [20]. Equal amounts of protein (15–25 μg) were resolved on reducing SDS mini-gels and immunoblotted using antibodies specific for p27 (BD Biosciences Pharmingen), GAPDH (sc-25778, Santa Cruz Biotechnology) or actin (sc-8432, Santa Cruz Biotechnology). The resolved proteins were detected using ECL (Amersham). S phase incorporation assays were performed similarly to that described [21] using BrdU or EdU. Images were captured using a Nikon Eclipse 80i microscope, 20×/0.45 PL Plan Fluor objective, Hamamatsu C4742-95 digital camera and camera controller. Images were analyzed using Image-Pro Plus software, and the number of BrdU-positive and Dapi-positive nuclei was manually counted.
2.5. In vivo experimentation
The aortic arch and thoracic aorta were isolated from euthanized 11-week male wild-type and apoE-null mice on the C57BL/6J background. The isolated aortae were cleaned and stored in RNA-later (Qiagen) until isolation of total RNA using RNeasy (Qiagen). For the in vivo adeno-associated virus reconstitution experiments, an AAV8-TBG-hapoE3 vector was prepared by the Vector Core of the Penn Gene Therapy Program. Male apoE3-deficient mice (9-week old) were injected with AAV8-TBG-hapoE3 via the tail vein at a dose of 1 × 1012 genome copies. Mice were sacrificed at 11-weeks, and total RNA was prepared as described above. See Kitajima et al. [22] for methodological details of AAV vector construction, tail vein injection, cholesterol levels, and apoE3 expression.
3. Results
3.1. ApoE3 regulates p27 through the Cox2-PGI2 pathway in VSMCs
We previously reported that the anti-mitogenic effect of apoE3 requires the induction of Cox2 and production of PGI2 [6]. More recently, we showed that PGI2 inhibits VSMC cycling in a p27-dependent manner [23]. We therefore investigated the importance of p27 to the anti-mitogenic effects of apoE. Consistent with our previous studies [23], p27 levels were downregulated in response to serum-stimulation in wild-type VSMCs, and treatment with the PGI2 mimetic, cicaprost, prevented this decrease in p27 (Fig. 1A; top panels). ApoE3 also antagonized the serum-induced downregulation of p27 in wild-type VSMCs (Fig. 1A; top panels). This apoE effect was blocked by the Cox2 inhibitor, nimesulide, but not by the Cox1 inhibitor, SC560 (Fig. 1A; top panels). The level of p27 expression was similar in VSMCs treated with cicaprost, apoE, or the combination of cicaprost and apoE (Fig. 1B) suggesting that these two agents might be acting through a single pathway. Indeed, neither cicaprost nor apoE3 affected p27 expression in serum-stimulated VSMCs null for the PGI2 receptor, IP (Fig. 1A; bottom panels). Collectively, these data indicate that the increased expression of p27 seen in response to apoE can be explained by the induction of Cox2, synthesis of PGI2 and activation of PGI2/IP signaling. Moreover, the relationship between apoE3 and p27 is causal for VSMC cycling because apoE3 as well as cicaprost strongly inhibited S phase entry in wild-type VSMCs but not in p27-null VSMCs (Fig. 1C).
Fig. 1.

Anti-mitogenic effect of ApoE3 linked to p27 and the Cox2-PGI2-IP pathway in VSMCs. (A) Serum-starved early passage VSMCs from wild type and IP-null mice were stimulated with 10% FBS in the absence (control; C) or presence of cicaprost (Cica), apoE3, apoE3 with nimesulide (nim; Cox-2 inhibitor), or apoE3 with SC560 (SC; Cox-1 inhibitor). VSMCs were collected after 24 h, lysed, and immunoblotted for p27 and actin (loading control). The vertical white bar in the blot of IP-null VSMCs indicates where extraneous lanes were removed. Results from 3 to 6 independent experiments were quantified using Image J and combined to show mean ± SD. (B) VSMCs were prepared as in A but treated with cicaprost and apoE3, alone and in combination. Lysates were analyzed by immunoblotting for p27 and GAPDH (loading control). Results from 3 independent experiments were quantified using Image J and combined to show mean ± SD. (C) Serum-starved VSMCs from wild-type and p27-null mice were stimulated with 10% FBS in the absence or presence of cicaprost or apoE3 for 48 h; coverslips were analyzed for BrdU incorporation by immunofluorescence microscopy. Results are plotted as mean ± SD, n = 4. For all panels, *p < 0.05. **p < 0.01. ***p < 0.001 as determined by 2-tailed t-test.
p27 levels are commonly regulated post-transcriptionally. Initial studies linked post-transcriptional regulation of p27 to ubiquitin-mediated proteolysis by the E3 ligase, SCFSkp2; this complex targets p27 phosphorylated at T187 [24,25]. However, subsequent work indicated that SCFSkp2-mediated degradation of T187A-phosphorylated p27 is essential in S phase, but not G1 phase [26,27]. Since apoE3 blocks entry into S phase (Fig. 1D), we reasoned that it might regulate p27 independently of Skp2. Several recent studies have identified the microRNA family, miR221/222 as a novel post-transcriptional regulator of p27 [28,29]. Indeed, we observed that apoE3 inhibited the expression of miR221/222 in serum-stimulated VSMCs (Fig. 2A; bottom panel). The dose-dependency of this effect was similar to that seen for Cox2 mRNA (Fig. 2A; top panel). Moreover, the regulation of miR221/222 by apoE3 was blocked by Cox2 inhibition with nimesulide (Fig. 2B) and was dependent on IP (Fig. 2C). Thus, miR221/222 is a target of the apoE3-Cox2-PGI2/IP pathway. Moreover, when we forced expression of miR222, we eliminated the inhibitory effect of apoE3 on p27 (Fig. 2D) and a large part of its inhibitory effect on S phase entry (Fig. 2E; compare lanes 2 and 4). We did not overexpress miR221 in these experiments because miR221/222 are interchangeable in overexpression paradigms [30]. These data demonstrate a causal relationship between apoE3, miR221/222, p27, and VSMC cycling.
Fig. 2.

ApoE3 regulates miR-221/222 in VSMCs through the Cox2-PGI2-IP pathway. (A) Serum-starved mouse VSMCs were incubated with 10% FBS (control; C) and 0, 15, 30 or 60 μg/ml apoE3 for 24 h. (B) Serum-starved mouse VSMCs were incubated with 10% FBS, 10% FBS with apoE3, or 10% FBS, apoE and either nimesulide (Cox2 inhibitor) or SC560 (Cox1 inhibitor). (C) Serum-starved VSMCs from wild-type or IP-null mice were incubated with 10% FBS in the absence (control; C) or presence of apoE3 for 24 h. For A–C, total RNA was isolated, and transcript levels were determined by RT-qPCR. Results show mean ± SD, n = 3. (D) Early passage VSMCs were transiently transfected with an expression plasmid for miRNA-222 or pCDNA (control). The cells were serum starved and stimulated with 10% FBS for 24 h in the absence or presence of apoE3 before being collected and analyzed by western blotting for p27 and GAPDH (loading control). (E) The experiment in D was repeated, but the VSMCs were incubated for 48 h on coverslips. The cells were analyzed for BrdU incorporation by immunofluorescence microscopy. Results in E are expressed as mean ± SD, n = 3. For all bar graphs, significance was determined by 2-tailed t-test as defined in the legend to Fig. 1.
3.2. Regulation of miR221/222 by apoE3-containing HDL, apoE isoforms and isolated apoE domains
ApoE3 circulates in a lipid-free form and as a component of all lipoproteins other than LDL [1,3]. We therefore compared HDL and LDL for their abilities to regulate miR221/222 in mouse VSMCs. Like apoE3, HDL efficiently suppressed the expression of both micro-RNAs whereas LDL was without effect (Fig. 3A). We prepared HDL that was depleted apoE [31] and then compared the effects of HDL and apoE-depleted HDL on miR221/222. Depletion of apoE from HDL eliminated its ability to regulate miR221/222 (Fig. 3B). Additionally, suppression of miR221/222 was not detected in response to recombinant apoA-1, a major apolipoprotein in HDL (Fig. 3A). Collectively, these data indicate that regulation of miR221/222 is a selective property of apoE3 and apoE3-containing HDL rather than a general property of lipoproteins and apolipoproteins.
Fig. 3.

ApoE and HDL inhibit miR221/222 in VSMCs. (A) The experiment in Fig. 2B included serum-starved VSMCs from wild-type mice which were incubated with 10% FBS for 24 h in the absence (control; C) or presence of apoA-I, apoE3, total HDL or LDL and analyzed by RT-qPCR. (B) Serum-starved VSMCs were incubated with 10% FBS in the absence (control, C) or presence of total or apoE-depleted HDL for 24 h. For both panels, total RNA was isolated, and miR221/222 levels were determined by RT-qPCR. Results show mean ± SD, n = 3. Significance was determined by 2-tailed t-test as defined in the legend to Fig. 1.
We have previously reported that the anti-mitogenic effect of apoE3 is contained within the isolated N-terminal domain and not the C-terminal lipid-binding domains [6]. Consistent with these results, the inhibitory effect of apoE3 on miR221/222 was selectively detected with the purified N-terminal domain (Fig. 4A). Despite the single amino acid change in position 112 of their respective N-terminal domains, all three apoE isoforms inhibited the expression of miR221/222 (Fig. 4B) and S phase entry (Fig. 4C). Similar activities of the apoE isoforms have also been reported for the regulation of Cox2 mRNA [11]. Collectively, these isoform results further support the connection between apoE, Cox2 and miR221/222 and distinguish this apoE effect from the isoform-dependent LDLR binding associated with reverse cholesterol transport.
Fig. 4.

The N-terminal domain of apoE3 and its isoforms inhibit miR221/miR222 in VSMCs. (A) Serum-starved VSMCs were incubated with 10% FBS for 24 h in the absence (control; C) or presence of 2 μM ApoE3, or the N- and C-terminal fragments of ApoE3. (B) Serum-starved VSMCs were incubated with 10% FBS for 24 h in the absence or presence of 2 μM apoE2, apoE3 or apoE4. Total RNA was isolated, and miR221/222 levels were determined by RT-qPCR. (C) The experiment in B was repeated, but the VSMCs were incubated for 48 h on coverslips. The cells were analyzed for EdU incorporation by immunofluorescence microscopy. For all panels, results are expressed as mean ± SD, n = 3. Significance was determined by 2-tailed t-test as defined in the legend to Fig. 1.
3.3. Regulation of miR221/222 by apoE in vivo
To determine if the repressive effects of apoE on miR221/222 could be detected in vivo, we isolated RNA from the aortae of 11-week old wild-type and apoE-null mice and determined the relative levels of miR221/222. The results show a strong upregulation of both microRNAs in apoE-null vessels as compared to wild-type (Fig. 5A). In addition, we injected 9-week old apoE-null mice with an adeno-associated virus (AAV) encoding liver-specifically expressed human apoE3 and asked if miR221/222 would be reduced in the isolated aortae. Indeed, we found that AAV-apoE3 strongly repressed the expression levels of both microRNAs (Fig. 5B). Thus, regulation of miR221/222 by apoE is reversible in vivo. Moreover, the effect of AAV-apoE3 was detectable within 2 weeks, indicating that upregulation of miR221/222 in apoE-null aortae is not a long-term adaptation to the absence of apoE. These findings extend our earlier work showing reduced cholesterol, hyperplasia, and atherosclerotic lesions [22] as well as increased Cox2 gene expression [6] in apoE-null mice infected with AAV-apoE3.
Fig. 5.
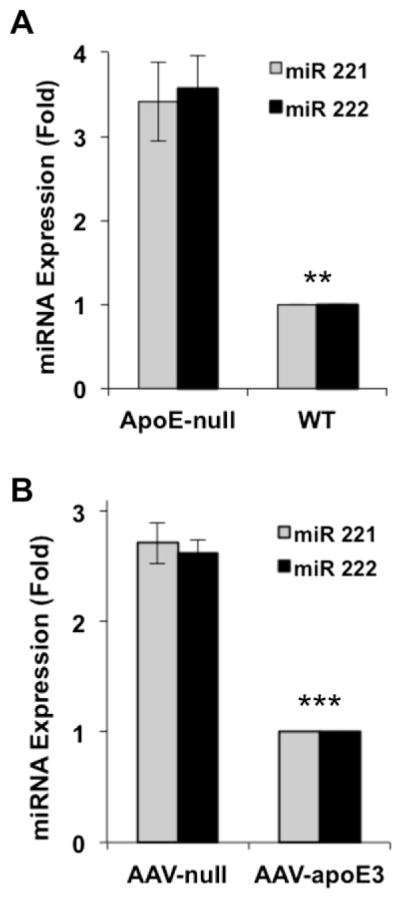
ApoE inhibits miR221/222 in vivo. (A) Total RNA was prepared from freshly isolated aortae of 11-week old male wild-type or apoE-null mice. miR221/222 levels were determined by RT-qPCR. n = 3. (B) Male 9-wk old apoE-null mice were given equal MOIs of null AAV or AAV-apoE3. Aortae from the mice of each condition were harvested after two weeks and analyzed by RT-qPCR for miR221/222, n = 4. Error bars show mean ± SEM, significance was determined by 2-tailed t-test as defined in the legend to Fig. 1.
4. Discussion
Our results show that apoE3 inhibits the p27-dependent proliferation of VSMCs by antagonizing the mitogen-dependent induction of miR221/222. Many studies have shown that increased expression of p27 inhibits cyclin E-cdk2 activity which, in turn, leads to reduced phosphorylation of the retinoblastoma protein (Rb) and release of E2F transcription factors. Although our work has focused on regulation of miR221/222, the link between miR221/222 and Rb phosphorylation, and the fact that Skp2 is an E2F-dependent gene [32,33], raises the possibility that apoE3 might also regulate Skp2 expression and Skp2-dependent p27 degradation as a secondary consequence of Rb phosphorylation and E2F release.
ApoE is present as ~6% of the total HDL protein, and about two-thirds of apoE in plasma of normolipidemic subjects is present in the HDL fraction of lipoproteins [19,34]. Our results show that physiologically relevant concentrations of HDL repress miR221/222 levels in primary VSMCs. LDL, which lacks apoE, fails to regulate cell cycling [6] or these microRNAs. All three isoforms of apoE, but not apoA-I, the major apolipoprotein in HDL, regulate miR221/222. These data indicate that vascular smooth muscle cell cycle regulation is specific to apoE and apoE-containing HDL. It will be interesting to determine the exact roles of the HDL particle and apoE lipidation on suppression of miR221/222.
Others have reported that apoE inhibits PDGF-mediated S-phase entry by inducing iNOS [5] as well as heparan sulfate and perlecan [7]. These antimitogenic effects involve the C-terminal heparin-binding domain of apoE, which binds to heparan sulfate proteoglycan [8]. These antimitogenic actions of apoE are isoform-selective [35]. However, in our studies the antimitogenic effect of apoE, as well as its inhibitory effect on miR221/222, is restricted to the N-terminal receptor–binding domain ([6] and Fig. 4A) and does not exhibit isoform selectivity. Although the basis for these different results is not well understood, it may be due to the fact that we stimulate cell cycling with an optimal concentration of serum whereas others used minimal FBS and stimulated cycling with PDGF [5,8]. The mitogenic activities in serum extend well beyond PDGF, so serum may affect apoE-sensitive signaling pathways that are not regulated by PDGF. Additionally, the serum we used was not depleted of endogenous apoE. While the large differences in signals we observed using FBS with and without exogenous apoE indicates that the endogenous bovine apoE is not making a significant contribution to the total apoE pool in our experiments, we cannot exclude the possibility that the absence [4,5,8] or presence ([6] and this report) of endogenous apoE may affect cell-cycle regulatory signaling pathways. Finally, some of the work by others used the A7r5 smooth muscle cell line and smooth muscle cells isolated by collagenase digestion whereas we used mouse smooth muscle cells prepared by explant culture. VSMCs are highly plastic, and their response to apoE may be affected by their method of preparation and state of differentiation.
The etiology of atherosclerosis and the response to vascular injury are complex, but p27 and VSMC proliferation are thought to contribute to both pathologies, particularly the response to injury. In fact, p27 is a major target of the anti-restenotic drug, rapamycin [12,13]. ApoE3 and the Cox2-PGI2-IP pathway limit VSMC proliferation by regulating p27 through miR221/222, and these results fit well with the increased disease seen upon deletion or down-regulation of apoE, IP, and p27 in mice ([9,15] and this report). Interestingly, miR221/222 also inhibits c-Kit expression in VSMCs and endothelial cells [36] and reduces eNOS levels in endothelial cells [37]. Although apoE does not regulate Cox2, nor does PGI2 regulate p27, in endothelial cells [6,23], the potent effects of the apoE-Cox2-PGI2 pathway on miR221/222 expression in VSMCs may lead to changes in c-kit that contribute to the overall systemic response to apoE and apoE-containing HDL. In fact, the miR221/222 family has more than 200 potential mRNA targets as determined by TargetScan. Thus, regulation of miR221/222 by apoE may have wide-spread effects on VSMC behavior and function.
Acknowledgments
Sources of funding
This work was supported by NIH grant HL62250, HL56083 and the Gene Therapy Resource Program of the National Heart, Lung and Blood Institute.
We thank Elizabeth Hawthorne and Tina Xu for generating primary mouse VSMCs, Garret FitzGerald for IP-null mice, and Bayer Schering Pharma AG for cicaprost.
Abbreviations
- Apo
apolipoprotein
- HDL
high-density lipoprotein
- LDL
low-density lipoprotein
- VSMC
vascular smooth muscle cell
- cdk
cyclin-dependent kinase
- qPCR
quantitative PCR
Footnotes
Disclosures
None.
References
- 1.Mahley RW, Rall SC., Jr Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–37. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 2.Ribalta J, Vallve JC, Girona J, et al. Apolipoprotein and apolipoprotein receptor genes, blood lipids and disease. Curr Opin Clin Nutr Metab Care. 2003;6:177–87. doi: 10.1097/00075197-200303000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Weisgraber KH. Apolipoprotein E: structure-function relationships. Adv Protein Chem. 1994;45:249–302. doi: 10.1016/s0065-3233(08)60642-7. [DOI] [PubMed] [Google Scholar]
- 4.Ishigami M, Swertfeger DK, Granholm NA, et al. Apolipoprotein E inhibits platelet-derived growth factor-induced vascular smooth muscle cell migration and proliferation by suppressing signal transduction and preventing cell entry to G1 phase. J Biol Chem. 1998;273:20156–61. doi: 10.1074/jbc.273.32.20156. [DOI] [PubMed] [Google Scholar]
- 5.Ishigami M, Swertfeger DK, Hui MS, et al. Apolipoprotein E inhibition of vascular smooth muscle cell proliferation but not the inhibition of migration is mediated through activation of inducible nitric oxide synthase. Arterioscler Thromb Vasc Biol. 2000;20:1020–6. doi: 10.1161/01.atv.20.4.1020. [DOI] [PubMed] [Google Scholar]
- 6.Kothapalli D, Fuki I, Ali K, et al. Antimitogenic effects of HDL and APOE mediated by Cox-2-dependent IP activation. J Clin Invest. 2004;113:609–18. doi: 10.1172/JCI19097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paka L, Goldberg IJ, Obunike JC, et al. Perlecan mediates the antiproliferative effect of apolipoprotein E on smooth muscle cells. An underlying mechanism for the modulation of smooth muscle cell growth? J Biol Chem. 1999;274:36403–8. doi: 10.1074/jbc.274.51.36403. [DOI] [PubMed] [Google Scholar]
- 8.Swertfeger DK, Hui DY. Apolipoprotein E receptor binding versus heparan sulfate proteoglycan binding in its regulation of smooth muscle cell migration and proliferation. J Biol Chem. 2001;276:25043–8. doi: 10.1074/jbc.M102357200. [DOI] [PubMed] [Google Scholar]
- 9.Zhu B, Kuhel DG, Witte DP, et al. Apolipoprotein E inhibits neointimal hyperplasia after arterial injury in mice. Am J Pathol. 2000;157:1839–48. doi: 10.1016/S0002-9440(10)64823-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weisgraber KH, Mahley RW. Human apolipoprotein E: the Alzheimer’s disease connection. FASEB J. 1996;10:1485–94. doi: 10.1096/fasebj.10.13.8940294. [DOI] [PubMed] [Google Scholar]
- 11.Ali K, Lund-Katz S, Lawson J, et al. Structure-function properties of the apoE-dependent COX-2 pathway in vascular smooth muscle cells. Atherosclerosis. 2008;196:201–9. doi: 10.1016/j.atherosclerosis.2007.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marks AR. Sirolimus for the prevention of in-stent restenosis in a coronary artery. N Engl J Med. 2003;349:1307–9. doi: 10.1056/NEJMp038141. [DOI] [PubMed] [Google Scholar]
- 13.Marx SO, Marks AR. Bench to bedside: the development of rapamycin and its application to stent restenosis. Circulation. 2001;104:852–5. doi: 10.1161/01.cir.104.8.852. [DOI] [PubMed] [Google Scholar]
- 14.Diez-Juan A, Andres V. The growth suppressor p27(Kip1) protects against diet-induced atherosclerosis. FASEB J. 2001;15:1989–95. doi: 10.1096/fj.01-0130com. [DOI] [PubMed] [Google Scholar]
- 15.Cheng Y, Austin SC, Rocca B, et al. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science. 2002;296:539–41. doi: 10.1126/science.1068711. [DOI] [PubMed] [Google Scholar]
- 16.Morrow JA, Arnold KS, Weisgraber KH. Functional characterization of apolipoprotein E isoforms overexpressed in Escherichia coli. Protein Expr Purif. 1999;16:224–30. doi: 10.1006/prep.1999.1069. [DOI] [PubMed] [Google Scholar]
- 17.Lund-Katz S, Zaiou M, Wehrli S, et al. Effects of lipid interaction on the lysine microenvironments in apolipoprotein E. J Biol Chem. 2000;275:34459–64. doi: 10.1074/jbc.M005265200. [DOI] [PubMed] [Google Scholar]
- 18.Hatch FT. Practical methods for plasma lipoprotein analysis. Adv Lipid Res. 1968;6:1–68. [PubMed] [Google Scholar]
- 19.Weisgraber KH, Mahley RW. Subfractionation of human high density lipoproteins by heparin-sepharose affinity chromatography. J Lipid Res. 1980;21:316–25. [PubMed] [Google Scholar]
- 20.Welsh CF, Roovers K, Villanueva J, et al. Timing of cyclin D1 expression within G1 phase is controlled by Rho. Nat Cell Biol. 2001;3:950–7. doi: 10.1038/ncb1101-950. [DOI] [PubMed] [Google Scholar]
- 21.Kothapalli D, Zhao L, Hawthorne EA, et al. Hyaluronan and CD44 antagonize mitogen-dependent cyclin D1 expression in mesenchymal cells. J Cell Biol. 2007;176:535–44. doi: 10.1083/jcb.200611058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitajima K, Marchadier DH, Miller GC, et al. Complete prevention of atherosclerosis in apoE-deficient mice by hepatic human apoE gene transfer with adeno-associated virus serotypes 7 and 8. Arterioscler Thromb Vasc Biol. 2006;26:1852–7. doi: 10.1161/01.ATV.0000231520.26490.54. [DOI] [PubMed] [Google Scholar]
- 23.Castagnino P, Kothapalli D, Hawthorne EA, et al. Cell-type- and cell-cycle-specific anti-mitogenesis by cicaprost. Prostaglandins Other Lipid Mediat. 2010;93:20–4. doi: 10.1016/j.prostaglandins.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carrano AC, Eytan E, Hershko A, et al. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–9. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 25.DeSalle LM, Pagano M. Regulation of the G1 to S transition by the ubiquitin pathway. FEBS Lett. 2001;490:179–89. doi: 10.1016/s0014-5793(01)02121-4. [DOI] [PubMed] [Google Scholar]
- 26.Malek NP, Sundberg H, McGrew S, et al. A mouse knock-in model exposes sequential proteolytic pathways that regulate p27Kip1 in G1 and S phase. Nature. 2001;413:323–7. doi: 10.1038/35095083. [DOI] [PubMed] [Google Scholar]
- 27.Kamura T, Hara T, Matsumoto M, et al. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27Kip1 at G1 phase. Nat Cell Biol. 2004;6:1229–35. doi: 10.1038/ncb1194. [DOI] [PubMed] [Google Scholar]
- 28.Davis BN, Hilyard AC, Nguyen PH, et al. Induction of microRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. J Biol Chem. 2009;284:3728–38. doi: 10.1074/jbc.M808788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.le Sage C, Nagel R, Egan DA, et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. Embo J. 2007;26:3699–708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu X, Cheng Y, Zhang S, et al. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2009;104:476–87. doi: 10.1161/CIRCRESAHA.108.185363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kothapalli D, Liu S-L, Bae Y-H, et al. Cardiovascular protection by apoE and apoE-HDL linked to suppression of ECM gene expression and arterial stiffening. Cell reports. 2012 doi: 10.1016/j.celrep.2012.09.018. http://dx.doi.org/10.1016/j.celrep.2012.09.018. [DOI] [PMC free article] [PubMed]
- 32.Yung Y, Walker JL, Roberts JM, et al. A Skp2 autoinduction loop and restriction point control. J Cell Biol. 2007;178:741–7. doi: 10.1083/jcb.200703034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang L, Wang C. F-box protein Skp2: a novel transcriptional target of E2F. Oncogene. 2006;25:2615–27. doi: 10.1038/sj.onc.1209286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cohn JS, Tremblay M, Amiot M, et al. Plasma concentration of apolipoprotein E in intermediate-sized remnant-like lipoproteins in normolipidemic and hyperlipidemic subjects. Arterioscler Thromb Vasc Biol. 1996;16:149–59. doi: 10.1161/01.atv.16.1.149. [DOI] [PubMed] [Google Scholar]
- 35.Zeleny M, Swertfeger DK, Weisgraber KH, et al. Distinct apolipoprotein E isoform preference for inhibition of smooth muscle cell migration and proliferation. Biochemistry. 2002;41:11820–3. doi: 10.1021/bi026202k. [DOI] [PubMed] [Google Scholar]
- 36.Liu X, Cheng Y, Yang J, et al. Cell-specific effects of miR-221/222 in vessels: molecular mechanism and therapeutic application. J Mol Cell Cardiol. 2012;52:245–55. doi: 10.1016/j.yjmcc.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rippe C, Blimline M, Magerko KA, et al. MicroRNA changes in human arterial endothelial cells with senescence: relation to apoptosis, eNOS and inflammation. Exp Gerontol. 2012;47:45–51. doi: 10.1016/j.exger.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]