

Abstract
Background
Hepatitis C virus (HCV) infections occur worldwide and either spontaneously resolve or persist and markedly increase the person’s lifetime risk of cirrhosis and hepatocellular carcinoma. Although HCV persistence occurs more often in persons of African ancestry and in persons with a genetic variant near IL28B, the genetic basis is not well understood.
Objective
To evaluate the host genetic basis for spontaneous resolution of HCV infection.
Design
Two-stage genome wide association study (GWAS).
Setting
13 international multicenter study sites.
Patients
919 individuals with serum HCV antibodies but no HCV RNA (spontaneous resolution) and 1482 individuals with serum HCV antibodies and RNA (persistence).
Measurements
Frequencies of 792,721 SNPs.
Results
Differences in allele frequencies between persons with spontaneous resolution and persistence were identified on chromosomes 19q13.13 and 6p21.32. On chromosome 19, allele frequency differences localized near IL28B and included rs12979860 (overall per-allele OR = 0.45, P = 2.17 × 10−30) and 10 additional SNPs spanning 55,000 bases. On chromosome 6, allele frequency differences localized near genes for class II human leukocyte antigens (HLA) and included rs4273729 (overall per-allele OR= 0.59, P = 1.71 × 10−16) near DQB1*03:01 and an additional 116 SNPs spanning 1,090,000 base pairs. The associations in chromosomes 19 and 6 were independent, additive, and explain an estimated 14.9% (95% CI: 8.5–22.6%) of the variation in HCV resolution in those of European-Ancestry, and 15.8% (95% CI:4.4–31.0%) in individuals of African-Ancestry. Replication of the chromosome 6 SNP, rs4272729 in an additional 746 individuals confirmed the findings (p=0.015).
Limitations
Epigenetic effects were not studied.
Conclusions
IL28B and HLA class II are independently associated with spontaneous resolution of HCV infection and SNPs marking IL28B and DQB1*03:01 may explain ~15% of spontaneous resolution of HCV infection.
Introduction
Hepatitis C virus (HCV) infection culminates in one of two distinct clinical outcomes. Approximately 60% of individuals have life-long chronic infection that produces an average of 109–12 viruses per day with the associated risks of cirrhosis and hepatocellular carcinoma, while the remainder spontaneously eliminate infection (1,2). The virus itself cannot be chiefly responsible for these dichotomous outcomes because they occurred even when there was accidental infection with the same HCV inoculum(3). Likewise, persons of African ancestry are less likely to have spontaneous resolution than persons of European or Asian backgrounds infected with the same virus genotype, strongly suggesting there is a host genetic basis(4). The most consistently replicated genetic association has been with variants near the gene for interleukin 28B (IL28B) also known as lambda interferon 3(5,6). However, prior studies have either focused on one particular SNP(5) or had limited sample sizes from individual outcome groups and thus have restricted ability to find additional susceptibility alleles for spontaneous resolution of HCV infection. To investigate comprehensively the host genetic basis for spontaneous control of HCV infection, a multicenter, collaborative two-stage genome-wide association study was conducted.
A genome wide association study (GWAS) tests common variation across the human genome for association with an outcome and utilizes 100 thousand to 5 million single nucleotide polymorphisms (SNPs). Unlike the more traditional candidate gene studies that evaluate biologically plausible genes that may be related to the disease outcome, GWAS does not have an a priori hypotheses on which gene may be involved and evaluates common SNPs across the genome. A complete GWAS study relies on large sample sizes and replication studies to confirm the initial results and identification of important genetic regions or genes, but on their own they do not identify causal alleles. For a variety of diseases and traits, GWAS has been fruitful in identifying novel genes that may play a role in disease pathogenesis (7) Using this GWAS approach, we evaluate 2401 individuals from 13 distinct study groups (Appendix Table 1) for genetic associations with spontaneous resolution of HCV.
Table 1.
Genome-wide association study (GWAS) results and meta-analysis for HCV spontaneous clearance and persistence a) Original GWAS results and meta-analysis b) Replication association results and replication meta-analysis. Ancestry determined by self report in replication cohort which did not undergo GWAS only specific allele testing. OR is the per allele odds ratio. An OR <1 means the SNP is associated with persistence, and OR >1 means the SNP is associated with clearance. MAF is the minor allele frequency. Chromosome 19 SNPs were not included in this replication because the findings in IL28b have previously been reported. All replication SNPs can be seen in Appendix Table 6.
|
a.
| ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Chr | Position | European Ancestry (N=1,581) | African Ancestry (N=447) | Mixed/Other Ancestry (N=373) | Meta-Analysis | ||||||||
| MAF | OR | P | MAF | OR | P | MAF | OR | P | OR | P | ||||
| rs12979860 | 19 | 39738787 | 0.31(T) | 0.45 | 1.90E-18 | 0.45 (C) | 1.841 | 1.26E-05 | 0.34 (T) | 0.29 | 6.81E-11 | 0.45 | 2.17E-30 | |
| rs12980275 | 19 | 39731783 | 0.31 (G) | 0.46 | 3.45E-18 | 0.50 (G) | 0.58 | 8.43E-05 | 0.32 (G) | 0.31 | 2.82E-10 | 0.46 | 1.32E-28 | |
| rs11881222 | 19 | 39734923 | 0.29 (G) | 0.46 | 2.83E-17 | 0.30 (G) | 0.63 | 2.54E-03 | 0.28 (G) | 0.29 | 5.41E-10 | 0.46 | 1.07E-25 | |
| rs8099917 | 19 | 39743165 | 0.19 (C) | 0.40 | 6.30E-17 | 0.06 (C) | 0.70 | 2.71E-01 | 0.19 (C) | 0.29 | 7.49E-08 | 0.40 | 1.90E-22 | |
| rs4273729 | 6 | 32678597 | 0.39 (C) | 0.61 | 3.42E-10 | 0.42 (C) | 0.55 | 3.37E-05 | 0.34 (C) | 0.56 | 5.76E-04 | 0.59 | 1.71E-16 | |
| rs2647051 | 6 | 32670897 | 0.36 (A) | 0.60 | 2.12E-10 | 0.34 (A) | 0.56 | 1.57E-04 | 0.26 (A) | 0.54 | 7.37E-04 | 0.59 | 5.35E-16 | |
| rs9275224 | 6 | 32659878 | 0.47 (A) | 0.63 | 4.82E-10 | 0.46 (A) | 0.72 | 1.71E-02 | 0.39 (A) | 0.57 | 4.24E-04 | 0.64 | 9.46E-14 | |
| rs2395522 | 6 | 32664722 | 0.47 (T) | 0.63 | 5.59E-10 | 0.46 (T) | 0.73 | 2.01E-02 | 0.39 (T) | 0.57 | 3.63E-04 | 0.64 | 1.22E-13 | |
| rs9469220 | 6 | 32658310 | 0.46 (G) | 0.63 | 9.35E-10 | 0.42 (G) | 0.58 | 1.60E-04 | 0.43 (G) | 0.73 | 3.75E-02 | 0.63 | 1.30E-13 | |
|
b.
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Chr | Position | European Ancestry (N=292) | African Ancestry (N=284) | Egyptian Ancestry (N=169) | Meta-analysis | I2 | I2 P | |||||||
| MAF | OR | P | MAF | OR | P | MAF | OR | P | OR | P | |||||
| rs4273729 | 6 | 32678597 | 0.40 (C) | 0.85 | 0.337 | 0.44(C) | 0.61 | 0.006 | 0.39 (C) | 0.92 | 0.693 | 0.77 | 0.015 | 23.2 | 0.27 |
| rs2647051 | 6 | 32670897 | 0.38 (T) | 0.89 | 0.501 | 0.35 (T) | 0.58 | 0.007 | 0.37 (T) | 0.87 | 0.524 | 0.77 | 0.022 | 29.5 | 0.24 |
| rs9275224 | 6 | 32659878 | 0.48 (A) | 0.88 | 0.464 | 0.46 (A) | 0.59 | 0.005 | 0.49 (G) | 1.27 | 0.260 | 0.75 | 0.007 | 24.6 | 0.27 |
| rs2395522 | 6 | 32664722 | 0.48 (T) | 0.88 | 0.464 | 0.47 (T) | 0.61 | 0.007 | 0.48 (A) | 1.39 | 0.112 | 0.74 | 0.004 | 5.79 | 0.35 |
| rs9469220 | 6 | 32658310 | 0.48 (G) | 0.75 | 0.111 | 0.43 (G) | 0.71 | 0.071 | 0.50 (G) | 0.82 | 0.338 | 0.75 | 0.011 | 0 | 0.88 |
In African Americans the C alleles in the minor allele; the allele frequency for the T allele is 0.54 and the per-allele OR for the T allele is 0.54. I2 is a meta-analysis heterogeneity factor and I2P is the associated p-value
Methods
Samples
A total of 2401 individuals were selected from 13 distinct study groups. The study sites include the AIDS Link to the Intravenous Experience (ALIVE)(8), Baltimore Before and After Acute Study of Hepatitis (BBAASH)(9), Boston Area HCV Study Transmission, Immunity, Outcomes Network (BAHSTION)(10), Cramp (11), Hemophilia Growth and Development Cohort (HGDS)(12), Mangia(13), Multicenter Hemophilia Cohort Studies (MHCS I and II)(14), Correlates of Resolved Versus Low-Level Viremic Hepatitis C Infection in Blood Donors (REVELL Study) (15), The Swan Project(16), Toulouse cohort(17), Women’s Interagency HIV Study (WIHS)(18), and United Kingdom Drug Use cohort(19). Study sites were selected because they had well established HCV outcomes, available DNA and permission for genetic testing. Case definitions were determined by each study and are detailed in the Appendix. No HCV treatments were reported prior to the determination of persistence/spontaneous resolution. The following cohorts also were involved in a prior study in which rs12979860 near IL28B was reported to be associated with HCV recovery (ALIVE, n=281, MHCS, n=305, HGDS, n=106, REVELL, n=85, and United Kingdom Drug Use cohort, n=180)(5). Each individual study obtained consent for genetic testing as approved by the governing Institutional Review Board and provided DNA without identifiers to Johns Hopkins School of Medicine where DNA samples were prepared for testing, a process approved by the Johns Hopkins School of Medicine Institutional Review Board.
Genome-wide association genotyping
The Center for Inherited Disease Research performed the genotyping for the GWAS using the Illumina Human Omni-Quad array. There were 1,000,559 SNPs released with genotyped and intensity data. A series of standard quality control measures were employed (see Appendix) for both samples and markers including deviations from Hardy-Weinberg equilibrium, percent missingness, cryptic relatedness and determination of ancestry using principal components analysis (PCA). Q-Q plots and the inflation factor (λ) were evaluated for confounding by population stratification (Appendix Table 2, Appendix Figures 1 and 2)
Statistical Analysis
Using principal components and a random subset of 21,710 SNPs across the genome, we genetically determined ethnicity across the studies. Principal components analysis is used to summarize the background genetic variation of populations into a few variables that represent ethnic origin. Three distinct ethnic groups emerged across the 13 studies and participants from each study may have contributed to each of the different ethnic groups: European ancestry, African ancestry and Mixed/Other ancestry (Appendix Figures 3 and 4). Minor allele frequencies were tabulated for each ethnic group (Appendix Table 3). Ethnicity-specific groups were tested for association using an additive logistic regression model, adjusting for the first two principal components and HIV status. An additive genetic model represents genotypes as 0, 1,2 copies of the minor allele. These results were then meta-analyzed in a fixed effects inverse variance model. All covariate-stratified analyses were conducted similarly with each ethnicity being stratified by the variable, and then the three ethnic groups being meta-analyzed using the same fixed effects inverse variance method in META. To assess significance, we evaluated the p-value from the meta-analysis using an accepted GWAS threshold of 5 × 10−8. Heterogeneity was evaluated using the I2 metric and the associated p-value which can range from 0–100% and shows the extent of heterogeneity between studies.
The log10 p-values were plotted for all SNPs across the genome in a Manhattan plot, which graphically depicts all of the p-values in a GWAS and the “peaks” or “skyscrapers” are the regions of probable importance (Figure 1). The p-value a threshold of 5 × 10−8 is depicted in the figures and SNPs at or below this p-value were considered statistically significant. This p-value threshold of 5 × 10−8 is based upon a Bonferroni correction for 1 million SNPs and is consistent with a modified Bonferroni correction for the entire genome and simulations to control for the potential Type 1 error (20,21)). SNPs with p-values in the range of 10−6– 10−8 suggest a possible association.
Figure 1.
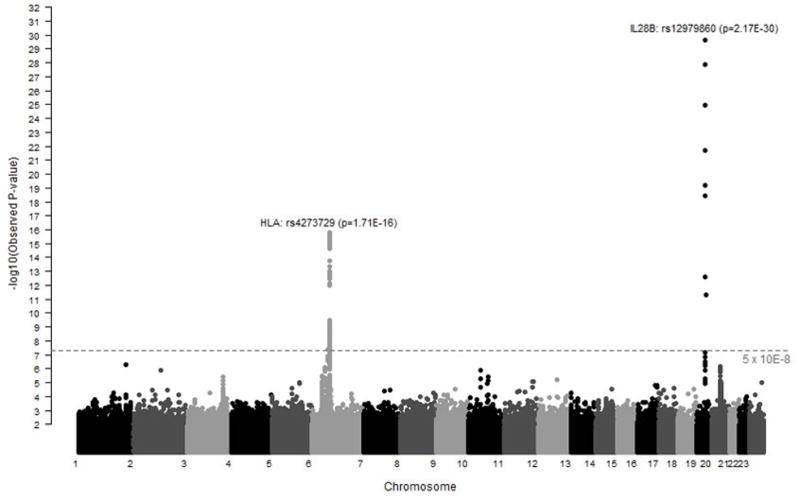
Manhattan Plot summarizing the genome-wide association results in 919 individuals with spontaneous resolution of HCV infection and 1,482 individuals with chronic HCV infection. Each point corresponds to a p-value from a test of association for a single SNP. The −log10 p values are plotted by location of the individual SNP across the genome. The dotted grey line represents an accepted level of genome wide significance, p=5 × 10−8. SNPs in the MHC and Il28b region on chromosomes 6 and 19, respectively exceed this threshold.
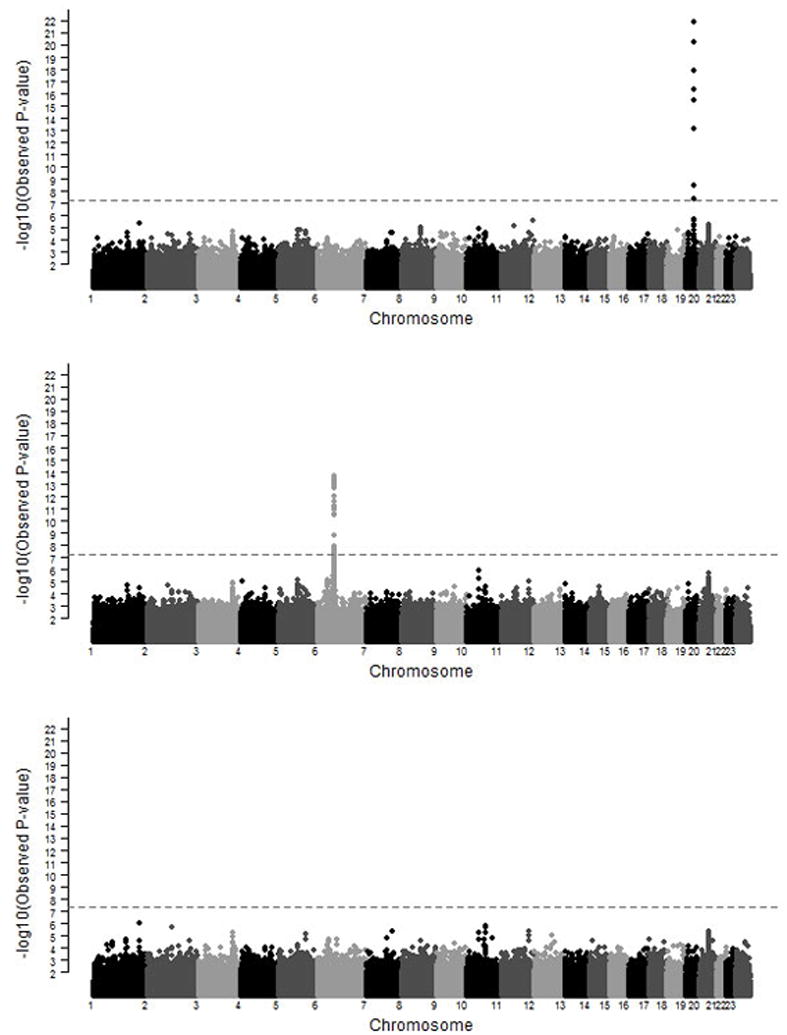
We also performed a conditional analysis to determine if a single SNP “explained” a significant genetic region. We included the SNP in the model (for some regions, more than one SNP was included consecutively to explain the significance of a region) and then re-evaluated the plots (Figure 2).
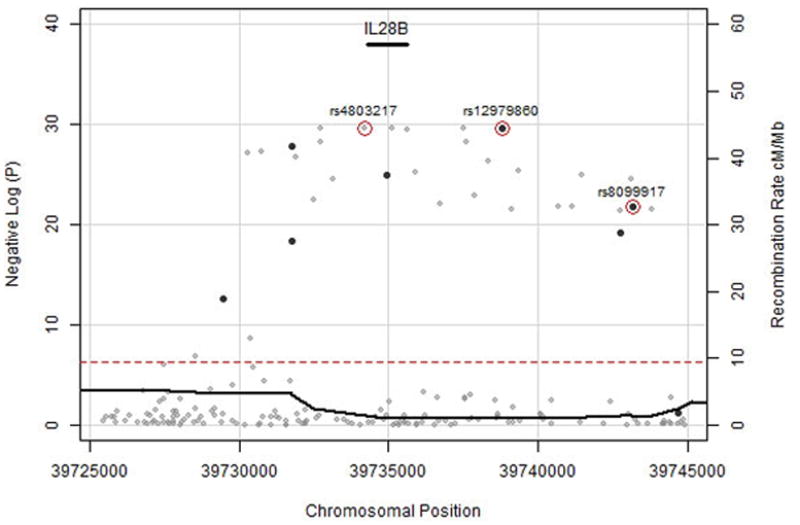
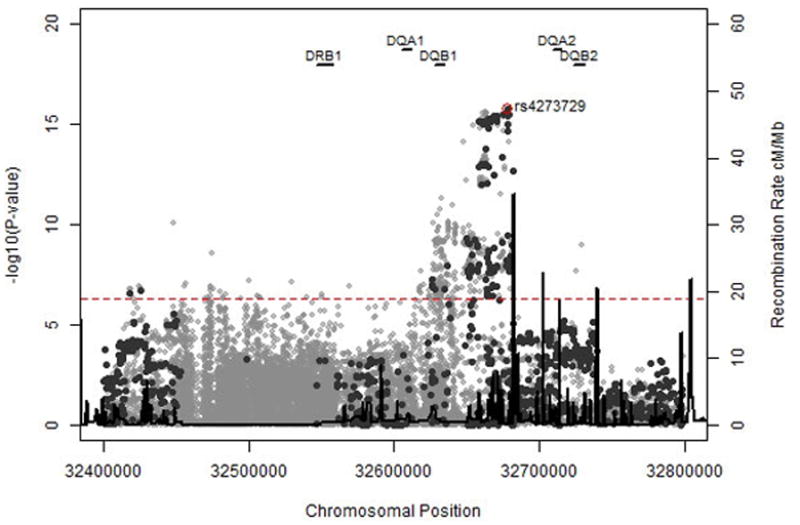
Figure 2. Genotyped and Imputed SNPs for HLA Class II (A) and IL28B (B) Regions.
Genotyped SNPs are in dark grey, 1000 genomes imputed SNPs are in light grey. The dotted red line is p=5 × 10−8.
The percent variance explained was estimated using the R program by So et al. (22), and a 10–40% prevalence of clearance. Statistical analyses were performed in PLINK v1.07 (23) or R and the meta analysis was completed in META v1.40)(24).
We also repeated a logistic regression model of spontaneous clearance in all available members of the unmatched Baltimore ALIVE cohort (4). The details of the model were previously reported, but the independent association of factors associated with spontaneous clearance (P<0.10) were considered by using logistic regression including self-assigned race, age (classified as above or below the median), HIV status (CD4+ lymphocyte counts stratified by convention as ≥500/mm3; 200–499/mm3, and <200/mm3), and hepatitis B status (determined by presence or absence of HBsAg)(4). The rs12979860 allele was genotyped in the ALIVE cohort and the model repeated.
Imputation
Imputation, which determines probable genotypes for SNPs that were not on the array, was conducted for chromosomes 6 and 19. The reference panel consisted of 1,092 individuals representing four continental populations (Africa, Americas, Asia and Europe) as part of the 1000 Genomes Project. The goal of imputation is to increase the resolution of genetic markers in these regions in an attempt to narrow down the possible causal locations by finding stronger signals(25). This is done by evaluating the underlying linkage disequilibrium in these regions in the reference population with known genotypes and then comparing this to the populations being studied. The imputed genotypes were analyzed by PCA-determined ethnicity controlling for the first two principal components and HIV status, and then meta-analyzed in the same way as the original analysis.
Replication
To confirm the novel findings from the GWAS we performed a replication study using convenience samples from three cohorts (Table 1): Multicenter AIDS Cohort Study (MACS), ALIVE participants not included in the original GWAS and an Egyptian cohort. The Egyptian samples were included because Egypt has the highest prevalence of HCV infection in the world due to accidental transmission during efforts to prevent schistosomiasis(26).
For this replication, we identified SNPs from the GWAS that either reached genome wide significance p<5 × 10−8 or were correlated with one of these SNPs or were located in the nearest gene. Since genome wide markers were not available for these replicate samples, we used self-reported ethnicity to categorize an individual: Black, White or North African/Egyptian. Genotyping was performed using either commercial or custom TaqMan® SNP Genotyping Assays from Applied Biosystems (Life Technologies, Carlsbad, CA), following the manufacturer’s standard protocol for an 40X Assay mix. The Roche LightCycler 480 Real-Time System (Roche Applied Science, Indianapolis, IN) was used to amplify the DNA and measure the fluorescence from each reaction to determine the genotype using Roche’s software. Test of associations were assessed in Plink (PLINK v1.07)(23) using an additive model for each ethnic group, and then meta-analyzed using META [v1.4.0](24). We used the conventional threshold for replication (p=0.01) which represents the nominal p-value and also performed a joint statistical analysis in which we expect the overall p value to be lower (27) (Appendix Table 6). We did not genotype chromosome 19 SNPs since findings in IL28b have been previously reported and confirmed.
HLA Classical Allele Analysis
To determine if a specific HLA class II allele explained the genome wide significant association on chromosome 6 in the MHC region, we tested classical HLA alleles for association with HCV spontaneous resolution. The class II HLA genotyping was performed using high-resolution (4-digit) SBT (sequence based typing) protocols recommended by the 13th International Histocompatibility Workshop. HLA classical allele analysis is presented in Appendix Tables 4 and 5. Only alleles with at least 10% frequency were included in the analysis. P-values were ascertained using logistic regression, adjusting for the first two ethnicity-specific principal components and HIV status. Counts were ascertained for HLA-DQB1*03:01 alleles versus rs4273729, the most significant SNP from the association analysis in the original GWAS for HLA.
Funding
This project was funded in whole or in part with federal funds from the office of AIDS Research through the Center for Inherited Diseases at Johns Hopkins University, the National Institutes of Drug Abuse R01013324 (DT); the National Institutes of the Frederick National Laboratory for Cancer Research, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This Research was supported in part by the Intramural Research Program of the NIH, Frederick National Lab, Center for Cancer Research.
Results
There were 919 individuals with spontaneous resolution of HCV infection identified by detection in blood of HCV specific antibodies but no HCV RNA (Appendix Table 1) and 1,482 individuals with chronic HCV infection identified by detection in blood of HCV specific antibodies and HCV RNA. After quality control measures were exercised there was information on 792,721 SNPs. Marked differences in allele frequency between persons with HCV clearance and persistence were evident on chromosomes 19q13.13 and 6p21.32 (Figure 1). These associations were consistent when both adjusted and stratified by HIV status, sex, and ancestry (Appendix Figures 5–8).
On chromosome 19, significant differences in allele frequency were detected for 11 SNPs representing 55,000 base pairs (bp) spanning IL28B, as previously described(5,6,28). The most significant association was with rs12979860, located ~3kb upstream of the IL28B start codon which has an allele frequency of 20–100% (C allele) in worldwide populations (5). Overall, the favorable homozygous CC genotype was detected in 523 (56.9%) HCV spontaneous resolvers and 470 (38.9%) HCV persistent individuals compared to CT in 297 (32.3%) HCV spontaneous resolvers and 739 (61.1%) HCV persisters and TT 84 (9.1%)HCV spontaneous resolvers and 257 (21.3%) HCV persisters (per-allele ORmeta = 0.45, P = 2.17 × 10−30). Since this finding included rs12979860 that was previously reported to be associated with spontaneous clearance, no confirmatory testing was performed.
This finding was consistent in substrata classified by genetic ancestry, gender, HIV-infection, and mode of HCV infection. The linkage disequilibrium block marked by rs12979860 chiefly explained the association, as the other 7 SNPs were no longer significantly associated with clearance in analyses conditioned on rs12979860 (P > 5 × 10−8, Appendix Figure 9). In contrast, rs12979860 remained strongly associated with HCV clearance in analyses conditioned on the other SNPs, even rs8099917, which was associated with spontaneous clearance in a previous Swiss study (Appendix Figure 10–11–14)(29). Imputation to 1000 genomes sequence data revealed another strongly associated SNP, rs4803217 (odds ratiometa = 0.44, p = 2.12 x10 −30), located in the 3′ untranslated region of IL28B (Figure 2). However, the association of rs4803217 could not be assessed separately from rs12979860 because of strong pairwise linkage disequilibrium in persons of both European (CEU, r2 =0.91) and African (YRI, r2 =0.86) ancestry. We also typed the rs12979860 allele on all available members of the Baltimore, Maryland ALIVE cohort in whom the greater HCV persistence in persons of African ancestry was originally discovered (4). In the same, fully-adjusted model of spontaneous resolution that was previously reported, by adding the results of rs12979860 testing, the fully-adjusted odds of persistence in persons of African compared to Caucasian ancestry was reduced from 4.53 (95% CI 2.16, 9.52) to 2.75 (95% CI 1.24, 6.12).
On chromosome 6, differences in allele frequencies between HCV spontaneous clearance and persistence were detected in 117 SNPs spanning 1,090,000 base pairs and encompassing genes for human leukocyte antigen (HLA). The strongest associations were detected for SNPs in the HLA class II region, such as the C allele at rs4273729 (Table 1, Figure 2). Overall, the homozygous GG genotype was detected in 429 (46.7%) HCV spontaneous resolvers and 495 (40.9%) HCV persisters compared to GC in 403 (43.9%) HCV spontaneous clearers and 697 (57.7%) HCV persisters and CC 84 (9.1%) HCV spontaneous clearers and 287 (23.7%) HCV persisters (per-allele ORmeta= 0.59, P = 1.71x 10−16). The finding was consistent in substrata classified by genetic ancestry, gender, HIV-infection, and mode of HCV infection. Using conditional analyses, a single SNP (rs4273729) explained the significant findings in this region (p>5.0 x10−8, Appendix Figure 12). Imputation to 1000 genomes refined the locus to the DQA1-DQB2 HLA region, although no imputed SNP was more significant than rs4273729. Additional analyses incorporating study site in the model confirmed the findings (Appendix Table 9). The significance of this region was confirmed in a replication dataset comprising 745 individuals from 3 cohorts including 284 individuals with spontaneous clearance and 461 individuals with chronic infection (Table 1b, rs4273729 odds ratio replication = 0.77, p=0.015 and OR meta+rep= 0.63, p= 5.2 × 10−17).
Although strong, long-distance linkage disequilibrium made it impossible to pin point a single casual allele, classical HLA allelic typing available on 823 individuals from this study demonstrated that DQB1*03:01 contributes significantly to the association (OR=1.93, p=1.84 × 10−5 European ancestry, OR =1.82, p=0.016 African ancestry, Appendix Table 4–5). Since DQB1*03:01 exists in haplotypes with multiple DR alleles, this finding may explain why other studies have reported DQB1*03:01 and/or diverse additional DR associations (13,17,30–32). Interestingly, in similar GWAS, host control of other chronic viral infections has been mapped to different major histocompatibility (MHC) genes such as HLA-B*57 for HIV, HLA-DP for HBV, and MHC class I polypeptide-related sequence B for dengue, underscoring differences in the roles of HLA loci in the host control of chronic viral infections (33–35).
The associations in chromosomes 19 and 6 with HCV clearance were independent and additive. Conditioning on the chromosome 19 SNP, rs12979860, did not alter the chromosome 6 associations, and conditioning on chromosome 6 did not alter chromosome 19 associations (Figure 3). There was a stepwise increase in the percent spontaneous resolution with the presence of each protective allele, indicating an additive effect of the protective variants (Figure 4). In a population with prevalence of spontaneous clearance of 30%, an overall estimate of 15% of the variance in spontaneous HCV resolution is explained by SNPs at chromosome 19 (8.4African, 10.1European%, rs12979860) and chromosome 6 (7.4African, 4.8European%, rs4273729). For individuals of European ancestry, the total variance explained is 14.9% (95% CI 8.5–22.6%) and for individuals of African ancestry the total variance explained is 15.8% (95% CI 4.4–31%) (Appendix Figure 13).
Figure 3. Conditional Analyses of HLA Class II and IL28B Regions.
The first plot shows the genome-wide association results for HCV clearance and persistence conditioned upon rs4273729 in HLA Class II region. The second is conditioned upon rs12979860 in the IL28B region. The third plot is conditioned on both SNPs. The dotted grey line represents p= 5 × 10−8.
Figure 4. Additive Relationship for HCV clearance for HLA Class II and IL28B SNPs.

Among 919 individuals with spontaneous clearance and 1482 with viral persistence, the % with viral clearance are shown for (A) rs12979860 in the IL28B region by ethnicity; (B) rs4273729 in the HLA Class II region by ethnicity; and (C) both rs4273729 and rs12979860 by ethnicity. As a case control study the relative differences in % are more informative than absolute values, which vary by the ratio of clearance to persistence in the study population.
Discussion
The results of this investigation provide incontrovertible evidence that IL28B and HLA class II are independently associated with spontaneous resolution of HCV infection and SNPs marking IL28B and DQB1*03:01 may explain ~15% of spontaneous resolution of HCV infection.
The mechanisms through which DNA sequences near IL28B and DQB affect spontaneous clearance from HCV infection are not known. Interestingly, the same alleles near IL28B (but not DQB) are also associated with resolution of HCV infection following treatment with peginterferon and ribavirin (6,28,36). This observation suggests interesting similarities and differences in the pathogenesis of spontaneous clearance and interferon alfa-based treatment of HCV infection.
Spontaneous resolution of HCV infection has been linked to preservation of proliferative CD4+ lymphocyte responses(37), and we show data here that among class II molecules, DQB1*03:01 appears to be expressed at higher levels on the surface of peripheral blood cells compared to all other DQB alleles (Appendix Figure 17). However, the mechanisms through which DNA sequence differences in class II contribute to spontaneous resolution of HCV infection have not yet been established.
There are potential clinical implications to this research. Genetic testing can be used to predict the likelihood of spontaneous resolution of HCV infection. Guidelines for treatment of acute HCV infection recommend that interferon alpha based treatment be given if infection is not spontaneously resolved before the infection transitions to the chronic phase (38). This recommendation is based on the observation that those with longer HCV infection (more than 12 months) are less likely to respond to treatment than those infected for shorter periods of time. However, the precise timing of when treatment responses begin to decline is unknown and unnecessary treatment can cause significant adverse effects. IL28B testing, which is commercially available, could provide a basis for prioritizing persons with T alleles at rs12979860 for early treatment since those who have CC genotypes have a high rate of spontaneous (and treatment related) resolution(28). Likewise, testing for rs4273729 (or DQB1*03:01) might provide a basis for prioritizing early treatment based on risk of spontaneous resolution, despite not altering the expectation for treatment response.
In this investigation we were able to explain ~15% of the variance of spontaneous HCV resolution with 2 SNPs. This compares favorably to other complex traits including Type 1 diabetes (45 variants) explaining ~11% of the variance, Type 2 diabetes (25 variants) explaining ~12% of the variance, and Crohn’s disease (32 variants) explaining ~7% of the variance (22). Explanation of a significant proportion of the phenotypic variance with 2 SNPs may have been enhanced by adjusting for other potential contributing factors such as HIV infection and race, as well as the unambiguous classification of the phenotype by RNA testing. Even more of the variance in the outcome might have been explained if we could have adjusted for the infecting virus. Unfortunately, viral genotype information in those who spontaneously resolve is rarely available since HCV infection is seldom recognized when it first occurs(2).
These data may provide insights into why individuals of African descent are less likely to spontaneously clear HCV infection. The T allele at rs12979860 that is associated with viral persistence is the ancestral allele (5). We have no insights into why there are T allele frequency differences in human populations. Specifically, HCV infection itself should not provide a large competitive advantage for a human allele since it does not commonly affect human fecundity. In contrast, the virus has vast capability to evolve its own sequence to optimize its own fecundity in a particular host genetic background (39,40). Thus, if HCV originally adapted to an African genetic background, where a higher prevalence of this ancestral allele exists, then a higher rate of spontaneous resolution might be anticipated when infection occurred in a new genetic background.
This hypothesis could not be confirmed in our discovery DNA panel because some cases and controls were matched on race. However, the rs12979860 allele was genotyped on all available members of the Baltimore, Maryland ALIVE cohort in whom the greater HCV persistence in persons of African ancestry was originally discovered (4). By adding the results of rs12979860 testing, the odds of persistence in persons of African compared to Caucasian descent was reduced from 4.53 to 2.75 suggesting that the lower frequency of favorable IL28B alleles may explain some, but not all, of the lower likelihood of spontaneous resolution in persons of African ancestry.
In summary, this investigation demonstrates that that IL28B and HLA class II are independently and unequivocally associated with spontaneous resolution of HCV infection and SNPs marking IL28B and DQB1*03:01 may explain ~15% of spontaneous resolution. Additional research is needed to ascertain the mechanisms through which these DNA sequences affect the outcome of HCV infection.
Acknowledgments
This project was funded in whole or in part with federal funds from the office of AIDS Research through the Center for Inherited Diseases at Johns Hopkins University, the National Institutes of Drug Abuse R01013324 (DT); the National Institutes of the Frederick National Laboratory for Cancer Research, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This Research was supported in part by the Intramural Research Program of the NIH, Frederick National Lab, Center for Cancer Research.
The study protocol is not available. Primary data are available through the central NIH GWAS data repositoryat the National Center for Biotechnology Information (NCBI), National Library of Medicine (http://grants.nih.gov/grants/guide/notice-files/NOT-OD-07-088.html).
Footnotes
Author contributions follow: PD, CT, GW, JG, MC and DT lead the writing team; PD and GW performed the primary analysis in conjunction with DT, MC, RL, JG, and CT. HLA imputations and testing were done by MC and XG. HLA II expression by MC and RA. DNA and key data for discovery panel was contributed by JG, AM, AK, GL, RC, MP, GK, AC, SK, LA, MC, SD, BE, LT, MB, GA, and HR and for the confirmatory panel by MAH, SM, GK, and CT. All the authors reviewed and approved the final manuscript.
HCV GWAS Paper Authors & Affiliations
1. David L. Thomas, Johns Hopkins University, School of Medicine, Division of Infectious Diseases
2. Georg M. Lauer, MD, PhD, Department of Medicine, Massachusetts General Hospital, Harvard Medical School, Warren 1019A, 55 Fruit Street, Boston MA 02114
3. Raymond T. Chung, Department of Medicine, Massachusetts General Hospital, Harvard Medical School, Warren 1019A, 55 Fruit Street, Boston MA 02114
4. Arthur Y. Kim, Department of Medicine, Massachusetts General Hospital, Harvard Medical School, Warren 1019A, 55 Fruit Street, Boston MA 02114
5. Laurent Alric, CHU Purpan, UMR 152 –IRD, Toulouse III University, France
6. Graeme Alexander MA, MD, FRCP, Division of Gastroenetrology & Hepatology, University Department of Medicine, Cambridge University Hospitals NHS Foundation Trust, Addenbrooke’s Hospital, Hill’s Road, Cambridge, CB2 0QQ, United Kingdom.
7. Salim Khakoo, Faculty of Medicine, University of Southampton, Mailpoint 811, Level E South Academic Block, Southampton General Hospital, Tremona Road, Southampton, SO16 6YD
8. Leslie H. Tobler, Viral Reference Laboratory and Repository Core, Blood Systems Research Institute, San Francisco, CA
9. Michael P. Busch, Viral Reference Laboratory and Repository Core, Blood Systems Research Institute, San Francisco, CA
10. Mohamed Abdel-Hamid, Department of Microbiology and Immunology, Faculty of Medicine, Minia University and Director, Viral Hepatitis Research Lab (VHRL) National Hepatology and Tropical Disease Research Institute (NH-TMRI)
11. James J. Goedert, Infections and Immunoepidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, Rockville, Maryland 20852
12. Alessandra Mangia, Liver Unit, IRCCS “Casa Sollievo della Sofferenza”, 71013 San Giovanni Rotondo, ITALY
13. Mary Carrington, Cancer and Inflammation Program, Laboratory of Experimental Immunology, SAIC-Frederick, Inc., Frederick National Laboratory for Cancer Research, Frederick, MD 21702 and Ragon Institute of MGH, MIT and Harvard, Boston, MA 02114
14. Richard Apps, Cancer and Inflammation Program, Laboratory of Experimental Immunology, SAIC-Frederick, Inc., Frederick National Laboratory for Cancer Research, Frederick, MD 21702 and Ragon Institute of MGH, MIT and Harvard, Boston, MA 02114
15. Xiaojiang Gao, Cancer and Inflammation Program, Laboratory of Experimental Immunology, SAIC-Frederick, Inc., Frederick National Laboratory for Cancer Research, Frederick, MD 21702 and Ragon Institute of MGH, MIT and Harvard, Boston, MA 02114\
16. Sharyne M. Donfield, Ph.D., Senior Research Scientist, Principal Investigator, Rho, Inc., 6330 Quadrangle Drive, Chapel Hill, NC 27517
17. Marion Peters, MD, Chief of Hepatology Research, UCSF, 513 Parnassus Ave S357
18. Matthew E. Cramp MD FRCP, South West Liver Unit, Plymouth, PL6 8DH, United Kingdom
19. Brian R. Edlin, MD, Professor of Medicine, SUNY Downstate College of Medicine, Brooklyn, NY, and Adjunct Associate Professor of Medicine and Public Health, Center for the Study of Hepatitis C, Weill Medical College of Cornell University, New York, NY
20. Hugo R. Rosen, MD, FACP, Waterman Professor of Medicine and Immunology Head, Division of Gastroenterology & Hepatology, Department of medicine, University of Colorado
21. Priya Duggal, PhD, Johns Hopkins University, Bloomberg School of Public Health
22. Chloe Thio, Johns Hopkins University, School of Medicine, Division of Infectious Diseases
23. Genevieve L. Wojcik, MHS, Johns Hopkins University, Bloomberg School of Public Health
24. Rachel Latanich, Johns Hopkins University, School of Medicine, Division of Infectious Diseases
25. Gregory Kirk, Johns Hopkins University, Bloomberg School of Public Health, 26. Shruti Mehta, Johns Hopkins University, Bloomberg School of Public Health, 27. Andrea Cox, Johns Hopkins University, School of Medicine, Division of Infectious Diseases
Reference List
- 1.Neumann AU, Lam NP, Dahari H, Gretch DR, Wiley TE, Layden TJ, et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science. 1998;282:103–7. doi: 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
- 2.Villano SA, Vlahov D, Nelson KE, Cohn S, Thomas DL. Persistence of viremia and the importance of long-term follow-up after acute hepatitis C infection. Hepatology. 1999;29:908–14. doi: 10.1002/hep.510290311. [DOI] [PubMed] [Google Scholar]
- 3.Kenny-Walsh E. Clinical outcomes after hepatitis C infection from contaminated anti-D immune globulin. Irish Hepatology Research Group. New England Journal of Medicine. 1999;340:1228–33. doi: 10.1056/NEJM199904223401602. [DOI] [PubMed] [Google Scholar]
- 4.Thomas DL, Astemborski J, Rai RM, Anania FA, Schaeffer M, Galai N, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA. 2000;284:450–456. doi: 10.1001/jama.284.4.450. [DOI] [PubMed] [Google Scholar]
- 5.Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O’Huigin C, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rauch A, Kutalik Z, Descombes P, Cai T, di IJ, Mueller T, et al. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology. 2010;138:1338–45. doi: 10.1053/j.gastro.2009.12.056. [DOI] [PubMed] [Google Scholar]
- 7.Hindorff LA, MacArthur J, Wise A, Junkins HA, Hall PN, Klemm AK, Manolio TA European Bioinformatics Institute. A Catalog of Published Genome-Wide Association Studies. Available at: www.genome.gov/gwastudies. Accessed [date of access]
- 8.Vlahov D, Munoz A, Anthony JC, Cohn S, Celentano DD, Nelson KE. Association of drug injection patterns with antibody to human immunodeficiency virus type 1 among intravenous drug users in Baltimore, Maryland. American Journal of Epidemiology. 1990;132:847–56. doi: 10.1093/oxfordjournals.aje.a115727. [DOI] [PubMed] [Google Scholar]
- 9.Cox AL, Netski DM, Mosbruger T, Sherman SG, Strathdee S, Ompad D, et al. Prospective evaluation of community-acquired acute-phase hepatitis C virus infection. Clin Infect Dis. 2005;40:951–58. doi: 10.1086/428578. [DOI] [PubMed] [Google Scholar]
- 10.Kim AY, Kuntzen T, Timm J, Nolan BE, Baca MA, Reyor LL, et al. Spontaneous control of HCV is associated with expression of HLA-B 57 and preservation of targeted epitopes. Gastroenterology. 2011;140:686–96. doi: 10.1053/j.gastro.2010.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cramp ME, Carucci P, Underhill J, Naoumov N, Williams R, Donaldson PT. Association of class II genotype and spontaneous clearance of hepatitis C viraemia. J Hepatol. 1998;29:207–13. doi: 10.1016/s0168-8278(98)80005-6. [DOI] [PubMed] [Google Scholar]
- 12.Hilgartner MW, Donfield SM, Willoughby A, Contant CF, Jr, Evatt BL, Gomperts ED, et al. Hemophilia growth and development study. Design, methods, and entry data. Am J Pediatr Hematol Oncol. 1993;15:208–18. doi: 10.1097/00043426-199305000-00009. [DOI] [PubMed] [Google Scholar]
- 13.Mangia A, Gentile R, Cascavilla I, Margaglione M, Villani MR, Stella F, et al. HLA class II favors clearance of HCV infection and progression of the chronic liver damage. J Hepatol. 1999;30:984–89. doi: 10.1016/s0168-8278(99)80250-5. [DOI] [PubMed] [Google Scholar]
- 14.Goedert JJ, Chen BE, Preiss L, Aledort LM, Rosenberg PS. Reconstruction of the hepatitis C virus epidemic in the US hemophilia population, 1940–1990. Am J Epidemiol. 2007;165:1443–53. doi: 10.1093/aje/kwm030. [DOI] [PubMed] [Google Scholar]
- 15.Tobler LH, Bahrami SH, Kaidarova Z, Pitina L, Winkelman VK, Vanderpool SK, et al. A case-control study of factors associated with resolution of hepatitis C viremia in former blood donors (CME) Transfusion. 2010;50:1513–23. doi: 10.1111/j.1537-2995.2010.02634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edlin BR, Shu MA, Winkelstein E, Des Jarlais DC, Busch MP, Rehermann B, et al. More rare birds, and the occasional swan. Gastroenterology. 2009;136:2412–14. doi: 10.1053/j.gastro.2009.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alric L, Fort M, Izopet J, Vinel JP, Charlet JP, Selves J, et al. Genes of the major histocompatibility complex class II influence the outcome of hepatitis C virus infection. Gastroenterology. 1997;113:1675–81. doi: 10.1053/gast.1997.v113.pm9352872. [DOI] [PubMed] [Google Scholar]
- 18.Kuniholm MH, Gao X, Xue X, Kovacs A, Marti D, Thio CL, et al. The relation of HLA genotype to hepatitis C viral load and markers of liver fibrosis in HIV-infected and HIV-uninfected women. J Infect Dis. 2011;203:1807–14. doi: 10.1093/infdis/jir192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khakoo SI, Thio CL, Martin MP, Brooks C, Gao X, Astemborski J, et al. HLA and NK Cell Inhibitory Receptor Genes in Resolving Hepatitis C Virus Infection. Science. 2004;305:872–74. doi: 10.1126/science.1097670. [DOI] [PubMed] [Google Scholar]
- 20.Dudbridge F, Gusnanto A. Estimation of significance thresholds for genomewide association scans. Genet Epidemiol. 2008;32:227–34. doi: 10.1002/gepi.20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duggal P, Gillanders EM, Holmes TN, Bailey-Wilson JE. Establishing an adjusted p-value threshold to control the family-wide type 1 error in genome wide association studies. BMC Genomics. 2008;9:516. doi: 10.1186/1471-2164-9-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.So HC, Li M, Sham PC. Uncovering the total heritability explained by all true susceptibility variants in a genome-wide association study. Genet Epidemiol. 2011;35:447–56. doi: 10.1002/gepi.20593. [DOI] [PubMed] [Google Scholar]
- 23.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.META: The genetics software suite. The Universtity of Oxford; Oxford, UK: [Google Scholar]
- 25.Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 26.Frank C, Mohamed MK, Strickland GT, Lavanchy D, Arthur RR, Magder LS, et al. The role of parenteral antischistosomal therapy in the spread of hepatitis C virus in Egypt. Lancet. 2000;355:887–91. doi: 10.1016/s0140-6736(99)06527-7. [DOI] [PubMed] [Google Scholar]
- 27.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet. 2006;38:209–13. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- 28.Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 29.Rauch A, Kutalik Z, Descombes P, Cai T, di IJ, Mueller T, et al. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology. 2010;138:1338–45. doi: 10.1053/j.gastro.2009.12.056. [DOI] [PubMed] [Google Scholar]
- 30.Fanning LJ, Levis J, Kenny-Walsh E, Whelton M, O’sullivan K, Shanahan F. HLA class II genes determine the natural variance of hepatitis C viral load. Hepatology. 2001;33:224–30. doi: 10.1053/jhep.2001.20642. [DOI] [PubMed] [Google Scholar]
- 31.Kuniholm MH, Kovacs A, Gao X, Xue X, Marti D, Thio CL, et al. Specific human leukocyte antigen class I and II alleles associated with hepatitis C virus viremia. Hepatology. 2010;51:1514–22. doi: 10.1002/hep.23515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thio CL, Thomas DL, Goedert JJ, Vlahov D, Nelson KE, Hilgartner MW, et al. Racial differences in HLA class II associations with hepatitis C virus outcomes. J Infect Dis. 2001;184:16–21. doi: 10.1086/321005. [DOI] [PubMed] [Google Scholar]
- 33.Fellay J, Ge D, Shianna KV, Colombo S, Ledergerber B, Cirulli ET, et al. Common genetic variation and the control of HIV-1 in humans. PLoS Genet. 2009;5:e1000791. doi: 10.1371/journal.pgen.1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamatani Y, Wattanapokayakit S, Ochi H, Kawaguchi T, Takahashi A, Hosono N, et al. A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nat Genet. 2009;41:591–95. doi: 10.1038/ng.348. [DOI] [PubMed] [Google Scholar]
- 35.Khor CC, Chau TN, Pang J, Davila S, Long HT, Ong RT, et al. Genome-wide association study identifies susceptibility loci for dengue shock syndrome at MICB and PLCE1. Nat Genet. 2011;43:1139–41. doi: 10.1038/ng.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rhodes SL, Erlich H, Im KA, Wang J, Li J, Bugawan T, et al. Associations between the human MHC and sustained virologic response in the treatment of chronic hepatitis C virus infection. Genes Immun. 2008;9:328–33. doi: 10.1038/gene.2008.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schulze zur WJ, Ciuffreda D, Lewis-Ximenez L, Kasprowicz V, Nolan BE, Streeck H, et al. Broadly directed virus-specific CD4+ T cell responses are primed during acute hepatitis C infection, but rapidly disappear from human blood with viral persistence. J Exp Med. 2012;209:61–75. doi: 10.1084/jem.20100388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghany MG, Strader DB, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49:1335–74. doi: 10.1002/hep.22759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ray SC, Fanning L, Wang XH, Netski DM, Kenny-Walsh E, Thomas DL. Divergent and convergent evolution after a common-source outbreak of hepatitis C virus. J Exp Med. 2005;201:1753–59. doi: 10.1084/jem.20050122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Timm J, Lauer GM, Kavanagh DG, Sheridan I, Kim AY, Lucas M, et al. CD8 epitope escape and reversion in acute HCV infection. J Exp Med. 2004;200:1593–604. doi: 10.1084/jem.20041006. [DOI] [PMC free article] [PubMed] [Google Scholar]