

Abstract
Activation of the MAPK/ERK signaling cascade in the suprachiasmatic nucleus (SCN) is a key event that couples light to circadian clock entrainment. However, we do not fully understand the mechanisms that shape the properties of MAPK/ERK signaling in the SCN, and how these mechanisms may influence overt circadian rhythms. Here we show that Raf kinase inhibitor protein (RKIP) controls the kinetics of light-induced MAPK/ERK activity in the SCN and photic entrainment of behavioral rhythms. Light triggers robust phosphorylation of RKIP in the murine SCN and dissociation of RKIP and c-Raf. Overexpression of a nonphosphorylatable form of RKIP in the SCN of transgenic mice blocks light-induced ERK1/2 activation in the SCN and severely dampens light-induced phase delays in behavioral rhythms. Conversely, in RKIP knock-out (RKIP−/−) mice, light-induced ERK1/2 activity in the SCN is prolonged in the early and late subjective night, resulting in augmentation of the phase-delaying and -advancing effects of light. Reentrainment to an advancing light cycle was also accelerated in RKIP−/− mice. In relation to the molecular clockwork, genetic deletion of RKIP potentiated light-evoked PER1 and PER2 protein expression in the SCN in the early night. Additionally, RKIP−/− mice displayed enhanced transcriptional activation of mPeriod1 and the immediate early gene c-Fos in the SCN in response to a phase-delaying light pulse. Collectively, our data reveal an important role of RKIP in the regulation of MAPK/ERK signaling in the SCN and photic entrainment of the SCN clock.
Introduction
In mammals, the ability to anticipate and adapt to environmental changes resides in the suprachiasmatic nucleus (SCN), a bilateral structure of the ventral hypothalamus with intrinsic pacemaker activity. The SCN circadian clock not only drives daily oscillations in physiology and behavior with near 24 h periodicity, but can be reset by various external time cues such as light. Photic information is relayed from the retina to the SCN via the retinohypothalamic tract, which releases the excitatory neurotransmitter glutamate and neuropeptide pituitary adenylate cyclase-activating peptide (PACAP) (Hannibal, 2002). Receptor-mediated postsynaptic activation of SCN neurons then triggers a series of intracellular signaling events that ultimately resets (entrains) the molecular clock.
Various signal transduction pathways have been implicated in photic entrainment of the SCN. Foremost among these is the p44/p42 MAPK/ERK cascade, which involves sequential activation of the kinases Raf, MEK and ERK. Light, but also glutamate and PACAP, triggers robust activation of this pathway in a phase-restricted manner (Obrietan et al., 1998; Butcher et al., 2005). MAPK/ERK activation is a prerequisite for light-induced phase shifts in locomotor activity rhythms as well as transcriptional activation of immediate early genes (IEGs) and clock genes (Butcher et al., 2002; Dziema et al., 2003; Coogan and Piggins, 2003). More recent evidence points to an additional role for members of the protein kinase C (PKC) family in photic entrainment (Jakubcakova et al., 2007; Lee et al., 2007).
Further advances in our understanding of the entrainment process require the identification of novel mechanisms that regulate these signaling cascades in the SCN in response to light. Along this line, Raf kinase inhibitor protein (RKIP) represents one avenue for regulation of light-evoked MAPK/ERK activity as well as light-dependent coupling of the MAPK/ERK and PKC pathways. Previously identified as a novel suppressor of cancer metastasis, RKIP is a widely expressed protein that negatively regulates MAPK/ERK signaling by binding to Raf and blocking Raf-mediated phosphorylation of MEK (Yeung et al., 1999). Phosphorylation of RKIP at Ser-153 (in humans) by PKC disrupts the RKIP-Raf association, thereby inactivating RKIP's function as a Raf inhibitor (Corbit et al., 2003). Collectively, these data raise the possibility that RKIP plays a critical role in photic entrainment by modulating the degree of MAPK/ERK signaling in the SCN in response to light.
Here we used RKIP transgenic and knock-out mice to probe the function of RKIP in light-induced clock entrainment. Our data indicate that light triggers the phosphorylation of RKIP and its dissociation from c-Raf in the SCN. Transgenic mice expressing a nonphosphorylatable form of RKIP did not exhibit light-induced ERK1/2 activation in the SCN or phase delays in behavioral rhythms. Conversely, RKIP−/− mice displayed prolonged ERK1/2 activation in the SCN in response to light and enhanced light-induced clock resetting. Furthermore, the absence of RKIP potentiated light-induced expression of PER1 and PER2, and enhanced transcriptional activation of the mPeriod1 and c-Fos genes in the early subjective night. Together, our data identify RKIP as a novel player in photic entrainment of the SCN clock.
Materials and Methods
Antibody production.
A polyclonal antibody that specifically recognizes the Thr-153-phosphorylated form of murine RKIP was generated by GenScript USA Inc. Rabbits were immunized with the KLH-conjugated phosphopeptide cDNRGKFKVE{pT}FRKK (representing aa 144–157 of the murine RKIP protein phosphorylated at Thr-153). Antisera were purified by 2-step column chromatography using resin conjugated to the nonphosphorylated form of the immunizing peptide, and subsequently passing the flow-through on a second column with resin conjugated to the phospho-peptide. Specificity of the phospho-RKIP antisera was determined by ELISA. Previously, the analogous epitope was used to successfully generate antibodies specific for the Ser-153-phosphorylated form of human RKIP (Corbit et al., 2003).
Animals.
Mice in which the rkip gene was disrupted (RKIP−/−) using a gene trap approach (Theroux et al., 2007) were backcrossed for a minimum of 8 generations onto a C57BL/6J background (The Jackson Laboratory) before analysis, and compared with wild-type (RKIP+/+) littermate controls. Complete absence of RKIP expression in the brains of RKIP−/− mice was confirmed by Western blotting (data not shown). mPeriod1::VENUS transgenic mice (Cheng et al., 2009), backcrossed for a minimum of 10 generations onto C57BL/6J, were bred onto a RKIP−/− or RKIP+/+ background. To generate RKIP(T153V) transgenic mice, the point mutation threonine 153 → valine was introduced into the I.M.A.G.E. clone encoding the cDNA of murine RKIP (ATCC) using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). Full-length RKIP(T153V) was subcloned into the expression vector pIRES2-eGFP (Clontech), and the RKIP(T153V)-IRES2-eGFP cassette was subsequently inserted into the tetracycline-inducible expression vector pTRE-TIGHT (Clontech). Using the construct TRE-TIGHT-RKIP(T153V)-IRES-eGFP, three independent transgenic mouse lines were established, which showed similar patterns and levels of eGFP expression in founder screens. Data are presented from double-transgenic mice generated from intercrosses between founder line #3793 and CaMKIIα-tetracycline transactivator (tTA) mice (Mayford et al., 1996). All animal handling and experimental procedures were approved by the Animal Welfare Committees of the University of Ottawa and the University of Toronto in accordance with institutional guidelines.
Behavioral analyses.
Male mice, aged 6–8 weeks at the start of the experiment, were housed in cages equipped with running wheels placed into a light-tight ventilated circadian activity chamber with computer-controlled light schedules (Phenome Technologies Inc.). Light intensity at cage level was ∼80 lux. Mice had ad libitum access to food and water throughout the experiment. Mice were initially entrained to a 12 h-light:12 h-dark (LD) cycle and then released in complete darkness (DD) to measure initial free-running period. After ∼14 d in DD, mice received a brief light pulse (30 lux intensity) for 15 min at circadian time (CT) 15 or CT 22, and returned to DD for an additional 2 weeks. A separate cohort of mice was initially entrained to a 12:12 LD cycle for at least 2 weeks, and then subjected to an abrupt 8 h advance in the phase of the LD cycle. Behavioral analyses were performed with the ClockLab software (Actimetrics). Period and circadian amplitude were measured with a χ2 periodogram. Light-induced phase shifts were measured using the ClockLab software by fitting regression lines through the daily activity onsets 10 d before and after the light pulse, and measuring the displacement. Phase shifts to an 8 h advance in the LD cycle were determined by measuring the difference between the actual onset of daily locomotor activity and the projected activity onset from the previous LD schedule. Data were analyzed with two-tailed Student's t tests or ANOVA followed by Student–Neuman–Keuls tests with α set at 0.05.
Cell culture, transfection and luciferase assay.
Neuro2A cells (ATCC) were grown on 24-well plates in DMEM (Invitrogen Canada Inc.) containing 5% fetal bovine serum (WISENT Inc.) and penicillin-streptomycin (Invitrogen Canada Inc.) at 37°C/5% CO2 until they reached 80% confluence. Cells were then transfected using Lipofectamine 2000 (Invitrogen Canada Inc.) with the following constructs: 500 ng of pcDNA3.1+Hygro, 500 ng of pcDNA3.1-RKIP(T153V), 100 ng of E1B-luciferase (Chung and Brautigan, 1999), 200 ng of Gal4-Elk1 (Chung and Brautigan, 1999), and 25 ng of pRL-TK (Promega Corporation). Cells were treated 24 h after transfection with 100 nm 12-O-tetradecanoylphorbol-13-acetate (TPA; Sigma-Aldrich), and cell lysates were prepared 6 h after stimulation. Luciferase activity was measured using the Dual Glo Luciferase Assay System (Promega Corporation). The experiment was repeated three times with similar results. Data were analyzed with ANOVA followed by Student–Neuman–Keuls tests with α set at 0.05.
Light treatment and tissue harvest.
Mice were stably entrained to a fixed LD cycle and subsequently released into constant dark conditions for 2 consecutive days. For light pulse experiments, mice received a 15 min light pulse of a specific intensity at the indicated CT, and were killed at the designated times. Mice were killed by cervical dislocation under dim red light, and eyes were covered with black electrical tape. Brains were rapidly dissected and cut into 600 μm thick coronal sections containing the SCN in cooled oxygenated media using an oscillating tissue slicer (Electron Microscopy Sciences). For coimmunoprecipitation (co-IP) or Western blotting, the tissue was immediately frozen on dry ice, and the SCN was isolated using a razor blade and stored in −80°C until use. For immunohistochemistry or immunofluorescence labeling, tissues were fixed in 4% paraformaldehyde in PBS for 6–8 h at room temperature, cryoprotected in 30% sucrose in PBS at 4°C, and cut into thin (40 μm) sections using a freezing microtome (Leica Microsystems Inc.).
co-IP.
co-IP was performed using the Pierce Co-Immunoprecipitation Kit (Fisher Scientific Ltd.) according to manufacturer's instructions. SCN tissues from 10 mice were pooled for each condition. Resins were conjugated with rabbit anti-RKIP antibody (Cell Signaling Technology Ltd.) or mouse anti-c-Raf antibody (BD Biosciences).
Western blotting.
SCN tissues from 3 mice were pooled for each condition and homogenized in ice-cold RIPA lysis buffer supplemented with Protease Inhibitor Cocktail. Proteins were quantified by the Bradford method using the Coomassie Plus Protein Assay Reagent (Fisher Scientific Ltd.), and 30 μg samples were resolved by SDS-PAGE. Samples were blotted onto PVDF membranes (Immobilon-P; Fisher Scientific Ltd.) by wet transfer. The following primary antibodies were used: rabbit anti-phospho-RKIP (1:1000; GenScript USA Inc.); rabbit anti-RKIP (1:20,000; Cell Signaling Technology Ltd.); and mouse anti-c-Raf (1:4000; BD Biosciences). Pierce ImmunoPure HRP-conjugated goat anti-rabbit or anti-mouse IgG(H+L) secondary antibodies (1:250,000; Fisher Scientific Ltd.) were also used. Signals were detected by chemiluminescence using the SuperSignal West Femto Maximum Sensitivity Substrate (Fisher Scientific Ltd.).
Immunofluorescence and immunohistochemistry.
For immunofluorescence (IF), sections were washed in PBST (PBS with 0.1% Triton X-100), blocked (1 h, room temperature) in 10% horse serum/PBST, and incubated (overnight, 4°C) in goat anti-GFP (1:4000; Eusera) or rabbit anti-phospho-p44/42 MAPK (Thr202/Tyr204) (1:2000; Cell Signaling Technology Ltd.). The following day, sections were washed five times in PBST and incubated (2 h, room temperature) with Alexa Fluor 488- and/or Alexa Fluor 594-conjugated secondary antibodies (1:1000; Invitrogen Canada Inc.) against the IgG domains of the primary antibodies. Sections were washed five times in PBST and mounted on slides with Dako Fluorescence Mounting Medium. For immunohistochemistry (IHC), sections were washed in PBST, treated with 0.3% H2O2 in PBS for 20 min, washed again in PBST, blocked (1 h, room temperature) in 10% horse serum/PBST, and incubated (overnight, 4°C) in primary antibodies. The following primary antibodies were used: rabbit anti-RKIP (1:5000; Cell Signaling Technology Ltd.); rabbit anti-phospho-p44/42 MAPK (Thr202/Tyr204) (1:6000; Cell Signaling Technology Ltd.); rabbit anti-PER1 (1:4000; gift from S. Reppert, University of Massachusetts Medical School, Worcester, MA); rabbit anti-PER2 (1:6000; gift from D. Weaver, University of Massachusetts Medical School, Worcester, MA); and rabbit anti-c-Fos (1:50,000; EMD Biosciences). Sections were washed five times in PBST, and incubated (2 h, room temperature) with biotinylated anti-rabbit IgG(H+L) secondary antibodies (1:300, Vector Laboratories Inc.). Immunodetection was accomplished using the Vectastain Elite ABC Kit (Vector Laboratories Inc.) and Peroxidase DAB Substrate Kit (Vector Laboratories Inc.) according to the manufacturer's instructions.
Image acquisition and analysis.
IHC images were acquired using a 20× objective with a Zeiss Axiovert Observer Z1 epifluorescent/light microscope equipped with an AxioCam MRm cooled-color camera. IF images were captured using a Zeiss 510 laser scanning confocal microscope with the argon (488 nm) and helium/neon (546 nm) lasers. Quantitation was performed using the ImageJ software (http://rsbweb.nih.gov/ij). Areas of interest were marked by placing a digital circle (for the ventral SCN), a digital crescent (for the dorsal SCN) or a digital oval (for the SCN or the adjacent lateral hypothalamus) over the image. For p-ERK1/2 quantitation, each unilateral SCN was measured individually by the sum of all grayscale values from 0 to 255 divided by the total number of pixels within the region of interest. Background values were obtained from nonimmunoreactive tissue in the lateral hypothalamus and subtracted from SCN mean gray values. For PER1, PER2 and c-FOS cell counting, images were assigned a minimal threshold using a grayscale threshold value between 0–100 and 0–120. Summation of all particles within the area of interest yielded a total area of immunoreactive cells within the SCN. Only particle areas of >50 pixels were included in the calculation of immunoreactive cells (50 pixels was experimentally determined to be above nonspecific DAB staining). Total area was divided by an average cell size of 120 pixels to yield a cell count estimate. For analysis of VENUS expression, a digital oval was placed in the central SCN and the mean intensity values were determined. A digital oval was placed in the lateral hypothalamus and the mean value was subtracted from the adjacent SCN to generate a normalized SCN intensity value. For all experiments, two central SCN sections per animal were used for analysis. Data were analyzed with ANOVA followed by Student–Neuman–Keuls tests with α set at 0.05.
Results
Light triggers RKIP phosphorylation and RKIP-Raf dissociation in the murine SCN
Previous in vitro studies have shown that diverse stimuli can trigger the phosphorylation of human RKIP at Ser-153, leading to disruption of the RKIP-Raf complex and activation of the MAPK/ERK cascade (Corbit et al., 2003). Our initial experiments showed that RKIP protein is expressed in the murine SCN (Fig. 1A), consistent with recent reports of rkip gene expression using a β-geo reporter (Theroux et al., 2007). To test the physiological relevance of the RKIP-Raf association in the SCN, we used co-IP to examine this interaction in the SCN of mice that had received a 15 min light pulse (LP) in the early subjective night, at CT 15. Compared with control mice that had not received a light pulse (DD) before killing at the same circadian time, SCN samples from light-pulsed mice exhibited lower levels of c-Raf co-immunoprecipitating with RKIP, and vice versa (Fig. 1B). Immunoblotting for the immunoprecipitated protein confirmed equal sample loading, whereas absence of detectable signal when DD samples were immunoprecipitated with rabbit IgG ruled out the possibility of nonspecific binding.
Figure 1.

Light induces the phosphorylation of RKIP and its dissociation from c-Raf in the SCN. A, Representative micrographs of total RKIP expression in the murine SCN at ZT 8. Boxed region (left) is shown in high magnification on the right. B, Light-responsive interaction between RKIP and c-Raf in the SCN as determined by co-IP. Mice received a 15 min light pulse (LP: 400 lux intensity) at CT 15 and were killed 5 min later. Control mice were killed at the same circadian time without receiving a light pulse (DD). SCN protein extracts were immunoprecipitated (IP) with antibodies against total RKIP (top) or total c-Raf (bottom) and immunoblotted (IB) with both antibodies. IP with rabbit IgG was used as a negative control. A brief light pulse in the early subjective night reduces the binding interaction of RKIP and c-Raf. C, Light-induced phosphorylation of RKIP as determined by Western blot analysis. Mice received a 15 min light pulse (LP: 400 lux intensity) at CT 15 and were killed 5 min later. Control mice were killed at the same circadian time without receiving a light pulse (DD). Blots were probed with antibodies against the Thr-153-phosphorylated form of RKIP (p-RKIP) (top), total RKIP (middle), and total c-Raf (bottom). Light triggers rapid and robust phosphorylation of RKIP at Thr-153 in the SCN.
Next, we tested whether light modulates the phosphorylation status of RKIP in the SCN. Using antibodies that specifically recognize the Thr-153-phosphorylated form of murine RKIP, we compared the expression of phospho-RKIP (p-RKIP) in the SCN of mice 5 min after receiving a brief (15 min) light pulse (LP) at CT 15 against that of dark control mice (DD). Western blot analysis revealed a robust increase in p-RKIP levels in the SCN of light-pulsed mice relative to dark controls (Fig. 1C). There was no effect of light on levels of total RKIP or c-Raf (Fig. 1C). Together, our data indicate that light triggers the phosphorylation of RKIP in the murine SCN and subsequent dissociation of the RKIP-Raf complex.
Phosphorylation of RKIP and photic entrainment
We further explored the physiological importance of RKIP phosphorylation in the context of circadian clock entrainment. Mutation of Ser-153 of human RKIP to the nonphosphorylatable valine residue has been shown to block PKC-dependent activation of MAPK/ERK signaling in vitro (Corbit et al., 2003). Along similar lines, we generated a transgenic mouse strain that expresses a dominant-negative mutant form of RKIP bearing the point mutation threonine 153 → valine (T153V), in conjunction with eGFP as the transgene reporter, under the control of a tetracycline-responsive promoter (Fig. 2A). These mice were then bred with a second transgenic strain—the CaMKIIα-tTA mice—that expresses the tetracycline transactivator under the control of the CaMKIIα promoter, to generate RKIP(T153V)-IRES-GFP::CaMKIIα-tTA double-transgenic mice (hereafter referred to as RKIP[T153V]::tTA). In the absence of the tetracycline analog doxycycline, double-transgenic mice coexpress RKIP(T153V) and eGFP in CaMKIIα-expressing cells of the mouse forebrain, including the SCN (Fig. 2C, data not shown; Mayford et al., 1996; Cheng et al., 2006).
Figure 2.
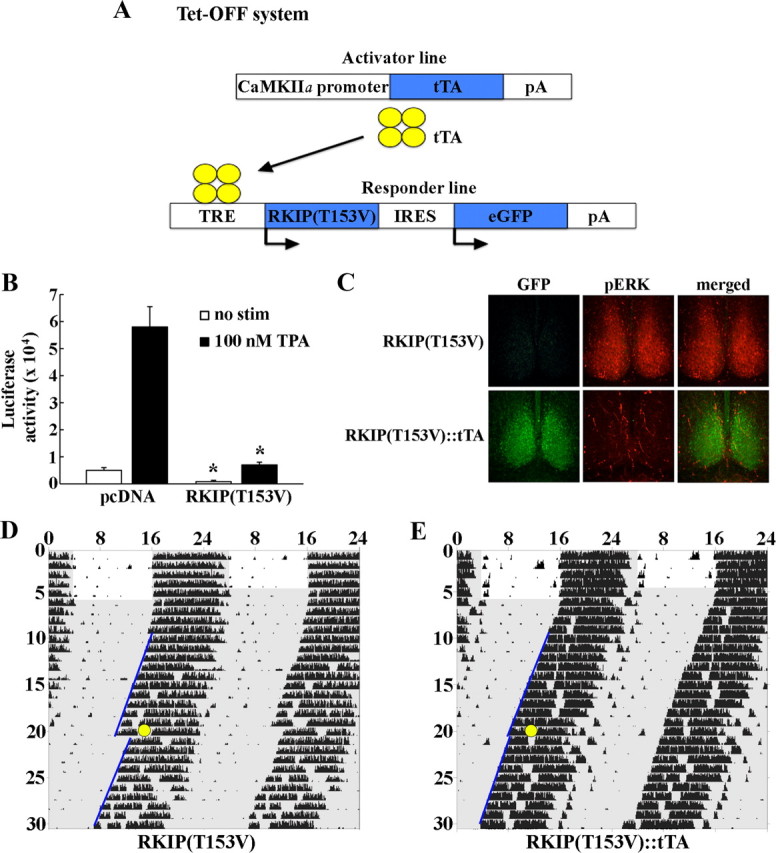
Phosphorylation of RKIP at Thr-153 mediates stimulus-dependent MAPK/ERK activation and light-induced phase shifts in circadian locomotor rhythms. A, Schematic diagram of the tetracycline-inducible system used to drive expression of dominant-negative RKIP in the murine forebrain. A single transgenic construct carrying two genes, dominant-negative RKIP harboring the T153V mutation and eGFP, in a polycistronic unit under the control of the tetracycline-responsive (TRE) promoter was generated. RKIP(T153V)-IRES-eGFP transgenic mice (hereafter referred to as RKIP[T153] single transgenics) were bred with the CaMKIIα-tTA mice. In the absence of the tetracycline analog doxycycline, double-transgenic mice coexpress RKIP(T153V) and eGFP in CaMKIIα-expressing cells. B, Neuro2A cells were transfected with an E1B-luciferase reporter gene construct and the Gal4-Elk1 expression vector, in combination with the pcDNA3.1-RKIP(T153V) expression construct or pcDNA empty vector control. Twenty-four hours post-transfection, cultures were treated with 100 nm TPA and assayed 6 h later. Basal (no stimulus) and TPA-induced Gal4-Elk1 activation were markedly attenuated in cells overexpressing dominant-negative RKIP compared with pcDNA controls. Data are presented as mean ± SEM of quadruplicate determinations. *p < 0.05 vs pcDNA control. C, Overexpression of dominant-negative RKIP in the SCN blocks light-induced ERK1/2 activation. Single-transgenic RKIP(T153V) (top) and double-transgenic RKIP(T153V)::tTA (bottom) mice received a 15 min light pulse (30 lux light intensity) at CT 15 and were killed 15 min later. SCN tissues were probed with antibodies against GFP (green) or phospho-p44/p42 MAP kinase (Thr202/Tyr204) (red). Light triggers robust phosphorylation of ERK1/2 in the SCN of single-transgenic mice, but not in double-transgenic mice expressing a nonphosphorylatable form of RKIP. D, E, Overexpression of dominant-negative RKIP in the SCN attenuates light-induced phase delays in circadian locomotor rhythms. Representative double-plotted activity profiles of single-transgenic RKIP(T153V) (D) and double-transgenic RKIP(T153V)::tTA (E) mice that were stably entrained in a fixed 12:12 LD cycle before release into constant darkness (DD). After 2 weeks in DD, mice received a 15 min light pulse (30 lux intensity: yellow circle) at CT 15 and returned to DD for an additional 2 weeks. Periods of darkness are shaded in gray. Activity onsets are indicated by blue lines. y-axis indicates the nth day of study. x-axis indicates time in hours. Double-transgenic RKIP(T153V)::tTA mice showed a statistically significant reduction in light-induced phase delays compared with single-transgenic controls.
We first validated the functionality of the T153V mutation using a GAL4-Elk1/E1B-driven luciferase reporter assay in cultured Neuro2A cells to monitor activation of the MAPK/ERK pathway (Chung and Brautigan, 1999; Cheng et al., 2006). Cotransfection of a RKIP(T153V) expression vector reduced Elk1 transactivation in response to the phorbol ester TPA, a PKC activator, by 88% relative to pcDNA-transfected controls (Fig. 2B). Basal (no stimulus) Elk1 activity was also reduced in RKIP(T153V)-transfected samples by 84% compared with pcDNA controls (Fig. 2B).
Next, we examined the effects of expression of the RKIP(T153V) mutant in the SCN on light-evoked ERK1/2 activity. Consistent with previous findings in wild-type mice, a brief (15 min, 30 lux intensity) light pulse at CT 15 triggered a robust increase in levels of phospho-active ERK1/2 in the SCN of RKIP(T153V) single-transgenic mice, which do not express the transgene (Fig. 2C, top). In contrast, expression of RKIP(T153V) in the SCN of RKIP(T153V)::tTA double-transgenic mice (under doxycycline-free conditions) abrogated light-induced p-ERK1/2 expression (Fig. 2C, bottom). Most importantly, uncoupling of light from ERK1/2 activation in the SCN by RKIP(T153V) expression coincides with disruption in light-induced phase delays of circadian locomotor rhythms. Double-transgenic mice expressing RKIP(T153V) in the SCN exhibited markedly reduced phase delays in response to a 15 min (30 lux intensity) light pulse at CT 15, relative to single-transgenic littermate controls [mean ± SEM phase shift (minutes) of single-transgenic (n = 8) vs double-transgenic (n = 12) mice: −98.4 ± 8.5 vs −20.8 ± 5.4, p < 0.05] (Fig. 2D,E). Collectively, these findings indicate that phosphorylation of RKIP at Thr-153 is a critical step in the activation of the MAPK/ERK pathway in the SCN and entrainment of the circadian clock in response to a phase-delaying light pulse.
RKIP modulates light-induced SCN clock entrainment
To determine whether RKIP is essential for proper regulation of photic entrainment, we first examined the phase-delaying and -advancing effects of a brief (15 min, 30 lux) light pulse at CT 15 and CT 22, respectively, in mice deficient for the rkip gene (RKIP−/−). This mouse strain was previously generated by Theroux et al. (2007) using mouse embryonic stem cells carrying a gene trap in intron 1 of the rkip gene, resulting in a null mutation by Western blot analysis (data not shown and Theroux et al., 2007). Mice were stably entrained and then dark adapted for at least 2 weeks before administration of the light pulse. Under these conditions, RKIP−/− mice exhibited an ∼38% increase in the magnitude of phase delays compared with wild-type (RKIP+/+) littermate controls following photic stimulation at CT 15 (Fig. 3A,B,E). In addition, whereas a 30 lux light pulse administered at CT 22 produced relatively small phase advances in RKIP+/+ mice (9.2 ± 3.4 min), the same light treatment evoked a more pronounced phase-advancing response in RKIP−/− mice (28.2 ± 7.4 min, p < 0.05) (Fig. 3C–E). The data suggest that both the phase-delaying and -advancing effects of acute photic stimulation are negatively modulated by RKIP.
Figure 3.
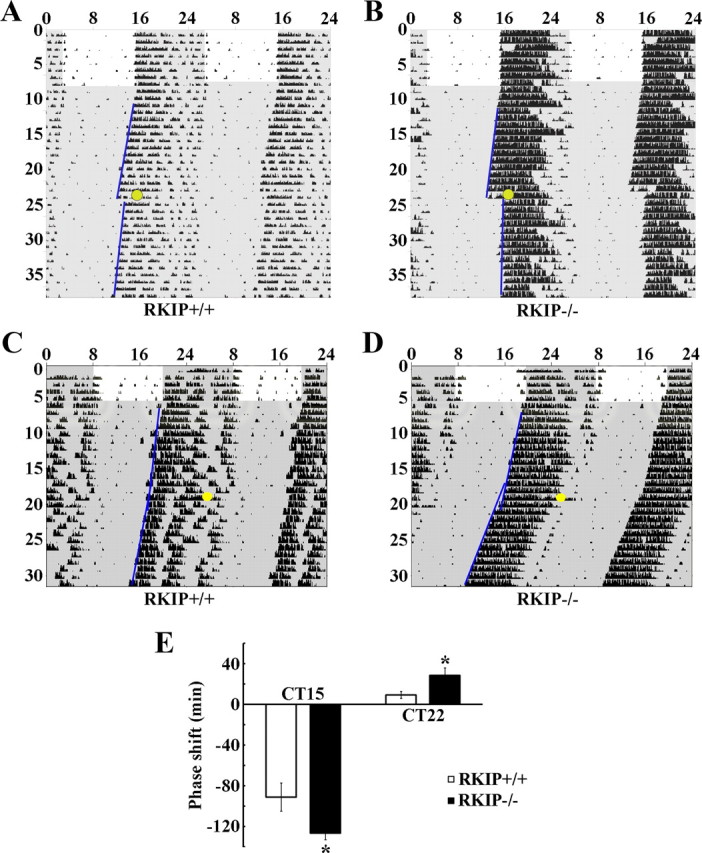
Absence of RKIP potentiates light-induced phase delays and phase advances in circadian locomotor rhythms. A–D, Representative double-plotted activity profiles of RKIP+/+ (A, C) and RKIP−/− (B, D) mice. Mice were stably entrained to a fixed LD cycle before release into DD. After 2 weeks in DD, mice received a 15 min light pulse (30 lux intensity: yellow circle) at either CT 15 (A, B) or CT 22 (C, D) and returned to DD for an additional 2 weeks. Periods of darkness are shaded in gray. Activity onsets are indicated by blue lines. y-axis indicates the nth day of study. x-axis indicates time in hours. E, Quantification of light-induced phase shifts. Values represent mean ± SEM (minutes) phase shift. Negative values indicate phase delays, whereas positive values indicate phase advances. n = 9–13 per group. *p < 0.05 vs RKIP+/+ controls.
Many studies have documented the relatively small phase advances exhibited by C57BL/6 mice in response to an acute light pulse in the late night (Daan and Pittendrigh, 1976; Schwartz and Zimmerman, 1990; Pendergast et al., 2010). The maximal phase advance is ∼0.5 h in response to a CT 22 light pulse that is of a much stronger intensity (100–200 lux) than the one used in our experiment (Fig. 3C). Because small changes in circadian phase are arguably challenging to quantify, we sought additional evidence to confirm that RKIP is indeed involved in the phase-advancing effects of light. To this end, we used reentrainment to an abrupt shift in the LD cycle as a second behavioral paradigm. Briefly, RKIP+/+ and RKIP−/− mice were stably entrained to a fixed LD cycle for at least 2 weeks before an abrupt 8 h phase advance of the LD cycle. For each animal, the rate of reentrainment to the new LD cycle was assessed by measuring the cumulative phase shift on each day after the change in the LD cycle. Our data show that RKIP−/− mice reentrained at a much faster rate than RKIP+/+ mice (Fig. 4A–C). On average, RKIP+/+ and RKIP−/− mice were fully reentrained to the new LD cycle after 11.7 ± 0.6 and 7.7 ± 0.7 d, respectively (p < 0.05). Collectively, these results demonstrate that RKIP is a critical negative modulator of the phase-advancing and phase-delaying effects of light.
Figure 4.

Absence of RKIP accelerates the rate of reentrainment to an 8 h advanced LD cycle. A, B, Representative double-plotted activity profiles of RKIP+/+ (A) and RKIP−/− (B) mice that were stably entrained to an initial fixed LD cycle for at least 2 weeks. Subsequently, the LD cycle was advanced by 8 h and mice were maintained under the new light cycle until they were stably entrained for a minimum of 10 d. Periods of darkness are shaded in gray. y-axis indicates the nth day of study. x-axis indicates time in hours. C, Quantification of the rate of reentrainment to an 8 h advanced LD cycle. Values represent mean ± SEM (hours) cumulative phase shift on each day after the change in the LD cycle. Positive values indicate a phase advance. n = 7 per group. *p < 0.05 vs RKIP+/+ controls.
As an aside, other parameters of circadian locomotor behavior were also examined but no differences were found between RKIP+/+ and RKIP−/− mice. Free-running period in DD (RKIP+/+ vs RKIP−/−: 23.59 ± 0.09 h vs 23.64 ± 0.07 h, p > 0.05) as well as circadian amplitude of wheel-running activity (RKIP+/+ vs RKIP−/−: 865 ± 88 vs 1080 ± 108, p > 0.05) were comparable between the two genotypes. Thus far, our behavioral data suggest that the role of RKIP in the SCN is confined to regulation of photic inputs and any potential effect that it may have on core clock mechanisms is not detectable at the level of locomotor activity rhythms.
RKIP modulates the duration of light-evoked MAPK/ERK activity in the SCN
Previous studies demonstrating the regulatory effects of RKIP on the Raf/MEK/ERK cascade (Yeung et al., 1999; Corbit et al., 2003), coupled with the prominent role of the MAPK/ERK pathway in photic entrainment of the SCN circadian clock (Obrietan et al., 1998; Butcher et al., 2002, 2005; Dziema et al., 2003), led us to consider the possibility that photic entrainment is enhanced in RKIP−/− mice as a consequence of a potentiation in light-evoked MAPK/ERK signaling in the SCN. To address this, we examined the effects of a 15 min light pulse (LP: 80 lux) at CT 15 on the kinetics of p-ERK1/2 expression in the SCN of RKIP+/+ and RKIP−/− mice (Fig. 5A). Mice were killed at various times (from 15 to 90 min) after the start of the light pulse, with time = 0 corresponding to dark controls. In both genotypes, levels of p-ERK1/2 in the SCN reached a maximum at the 15 min time point, immediately after cessation of the LP (Fig. 5A,B). There was no statistical difference in p-ERK1/2 expression in the SCN of RKIP+/+ and RKIP−/− mice at this time point. However, whereas p-ERK1/2 levels rapidly decayed in the SCN of RKIP+/+ mice from 30 min onwards, p-ERK1/2 expression remained significantly elevated in the SCN of RKIP−/− mice relative to wild-type controls at these later time points (Fig. 5A,B). A similar trend was also observed when we examined the kinetics of p-ERK1/2 expression in the SCN of RKIP+/+ and RKIP−/− mice following a light pulse (15 min, 30 lux) administered at CT 22 (Fig. 5C,D). Relative to their wild-type counterparts, RKIP−/− mice exhibited significantly higher levels of p-ERK1/2 in the SCN at the 45 and 60 min time point following initiation of the light pulse (Fig. 5C,D).
Figure 5.

Absence of RKIP prolongs light-induced MAPK/ERK signaling in the SCN but does not alter the gating of photic inputs. A, C, Representative micrographs of p-ERK1/2 expression in the SCN of (top row) RKIP+/+ and (bottom row) RKIP−/− mice at various times (minutes) after a 15 min light pulse is administered at CT 15 (80 lux intensity) (A) or CT 22 (30 lux intensity) (C). Time = 0 represents dark control, whereas samples corresponding to time = 15′ were harvested immediately following termination of the light pulse. B, D, Quantification of light-induced p-ERK1/2 levels in the SCN of RKIP+/+ (white bars) and RKIP−/− (black bars) mice as a function of time (minutes) after the start of a light pulse administered at CT 15 (B) or CT 22 (D). Time = 0 represents dark control. Values are presented as mean ± SEM relative grayscale intensity (in arbitrary units) normalized to background staining. n = 4 mice per group. *p < 0.05 vs RKIP+/+ controls. E, Light-induced ERK1/2 activation in the SCN of RKIP+/+ (top) and RKIP−/− (bottom) mice following a 15 min light pulse (LP: 80 lux) delivered at various circadian times. Single light pulses were given at CT 6, 10, 14 and 22, and mice were killed immediately after cessation of the light pulse. Control mice were killed at the same circadian time without receiving a prior light pulse (DD). F, Quantification of p-ERK1/2 expression in the SCN of light-pulsed (white bars) and dark control (black bars) mice of both genotypes. Values are presented as mean ± SEM relative grayscale intensity (in arbitrary units) normalized to background staining. n = 4 mice per group. *p < 0.05 vs dark control mice of the same genotype.
In another experiment, we explored the role of RKIP in phase-restricted activation of MAPK/ERK signaling in the SCN in response to light. RKIP+/+ and RKIP−/− mice received a 15 min light pulse (LP: 80 lux) in the middle (CT 6) and late (CT 10) subjective day, as well as early (CT 14) and late (CT 22) subjective night (Fig. 5E). Mice were killed immediately after cessation of the LP to assess p-ERK1/2 expression. As expected, compared with dark controls, a robust increase in p-ERK1/2 levels in the SCN of RKIP+/+ mice was observed in response to nighttime, but not daytime, light exposure (Fig. 5E,F). Importantly, this temporal restriction in light-induced p-ERK1/2 expression was also observed in the SCN of RKIP−/− mice (Fig. 5E,F). Together, the data reveal that RKIP is essential in negatively modulating the duration of light-evoked MAPK/ERK activity in the SCN but it does not influence the temporal gating of photically induced MAPK/ERK activation.
Absence of RKIP alters rhythmic p-ERK1/2 expression in the SCN
Previous studies have noted circadian rhythms in p-ERK1/2 expression in the SCN (Obrietan et al., 1998), although the underlying cause or the physiological relevance of this rhythmicity remains a mystery. To determine whether RKIP affects p-ERK1/2 rhythms in the SCN, RKIP+/+ and RKIP−/− mice were dark adapted for 2 consecutive days and then killed at 4 h intervals across a full 24 h cycle. Levels of p-ERK1/2 in the SCN of both RKIP+/+ and RKIP−/− mice were robustly rhythmic (Fig. 6A). In RKIP+/+ mice, the dorsomedial (shell) region of the SCN exhibited strong and rhythmic p-ERK1/2 expression that peaked at CT 10 (Fig. 6A,B). Although the amplitude of p-ERK1/2 rhythms in the shell SCN of RKIP−/− mice was unaltered relative to wild-type controls, peak expression was advanced by ∼4 h to CT 6 (Fig. 6A,B). In addition, within the ventrolateral (core) region of the SCN of RKIP+/+ mice, there was a cluster of cells that expressed high levels of p-ERK1/2 in the early night (CT 14) (Fig. 6A). p-ERK1/2 expression in the core SCN, and particularly in this cluster of cells in the center of the region, of RKIP−/− mice was significantly greater compared with wild-type controls at CT 14 (Fig. 6A,C).
Figure 6.
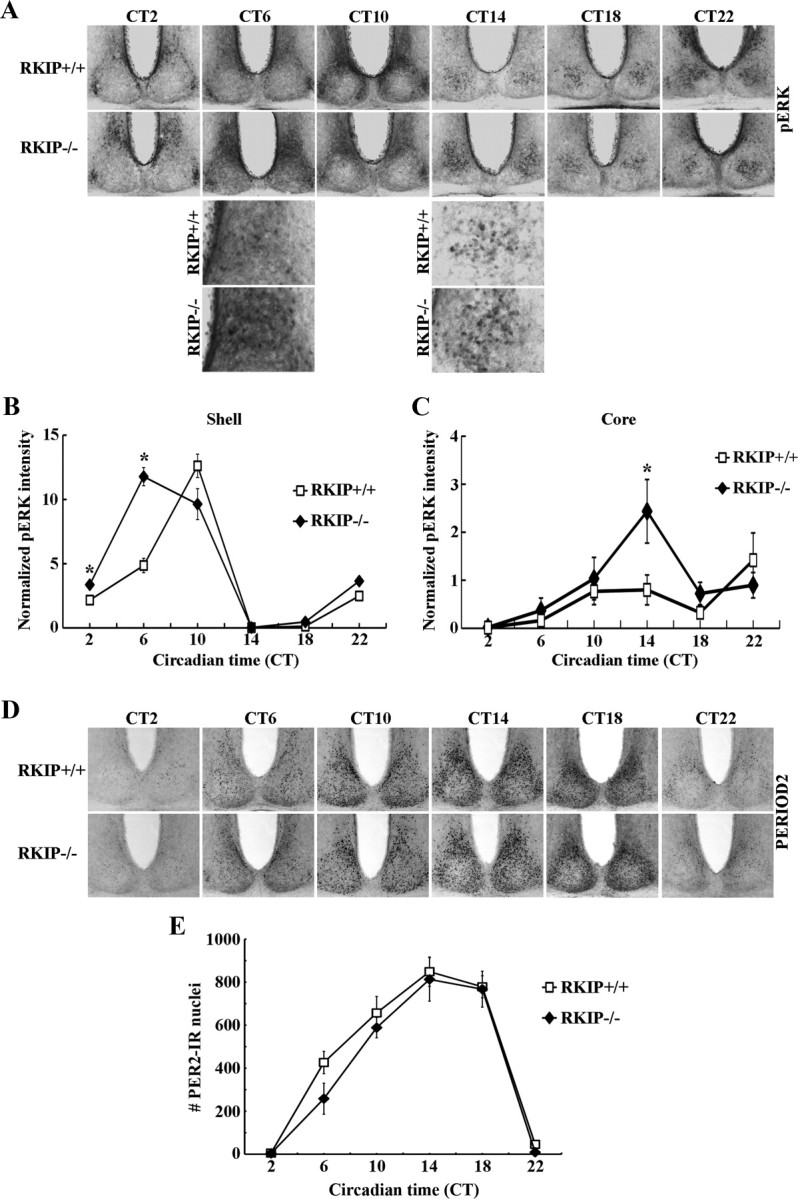
Absence of RKIP alters the circadian expression profile of p-ERK1/2 in the SCN but does not perturb PER2 rhythms. A, D, Representative micrographs of rhythmic p-ERK1/2 (A) or PER2 (D) expression in the SCN of RKIP+/+ and RKIP−/− mice. Mice were stably entrained to a fixed LD cycle and then released into DD for 2 consecutive days before tissue harvest at the designated circadian times. For (A), higher-magnification images (third and fourth rows) are given to highlight the increase in p-ERK1/2 expression at CT 6 in the shell SCN of RKIP−/− mice, and at CT 14 in the core SCN of RKIP−/− mice, relative to wild-type controls. B, C, Quantification of p-ERK1/2 expression in the shell (B) and core (C) SCN of RKIP+/+ and RKIP−/− mice. Values are presented as mean ± SEM relative grayscale intensity (in arbitrary units) normalized to background staining. n = 4 mice per group. *p < 0.05 vs RKIP+/+ controls. E, Quantification of PER2 expression in the SCN of RKIP+/+ and RKIP−/− mice. Values represent mean ± SEM number of immunoreactive nuclei in the bilateral SCN. n = 4 mice per group.
A previous study suggested that ERK1/2 rhythms in the SCN are important for sustaining robust rhythms in clock gene expression (Akashi et al., 2008). To examine whether the alterations in p-ERK1/2 rhythms in the SCN of RKIP−/− mice had an effect on the molecular clock machinery, we examined PER2 protein rhythms in the SCN of these animals (Fig. 6D,E). The data revealed no effect of RKIP ablation on either the amplitude or phase of PER2 rhythms (Fig. 6D,E). PER1 rhythms were similarly unaltered in the SCN of RKIP−/− mice compared with wild-type controls (data not shown). Thus, while our data show that RKIP has a role in shaping rhythmic MAPK activation in the SCN, we have yet to understand the potential impact of this on core clock mechanisms.
RKIP modulates light-induced PERIOD expression in the SCN
Various studies have shown that induction of Period1 and Period2 gene expression in the SCN accompanies and is essential for photic resetting of the circadian clock (Shigeyoshi et al., 1997; Takumi et al., 1998). Based on these findings, we examined whether altered inducibility of PER1 and PER2 protein levels in the SCN in response to a phase-delaying light pulse underlies the enhanced behavioral resetting in RKIP−/− mice. RKIP+/+ and RKIP−/− mice received a 15 min light pulse (LP: 80 lux) at CT 15, and were returned to darkness and killed 4 h later. Control mice were killed at the same circadian time without receiving a light pulse (DD). Consistent with previous results (Yan and Silver, 2004), we observed a significant increase in the number of PER1-IR nuclei in the ventrolateral SCN (Fig. 7A,B), as well as in the number of PER2-IR nuclei in the dorsomedial SCN (Fig. 7C,D), of both RKIP+/+ and RKIP−/− mice relative to their respective dark controls. However, light induction of both PER1 and PER2 was significantly potentiated in the SCN of RKIP−/− mice by ∼37% and ∼41%, respectively, compared with wild-type controls (Fig. 7A–D). There was no effect of genotype on the basal (CT 19) expression of PER1 and PER2 in the SCN (Fig. 7A–D). Thus, the data indicate that RKIP negatively modulates photic induction of PER1 and PER2 within the SCN in the early night.
Figure 7.

Absence of RKIP potentiates light-induced expression of PER1 and PER2 in the SCN. A, C, Representative micrographs of PER1 (A) and PER2 (C) expression in the SCN of RKIP+/+ and RKIP−/− mice 4 h after a 15 min light pulse (LP: 80 lux intensity) at CT 15. Control mice were killed at the same circadian time, CT 19, without receiving a light pulse (DD). B, D, Quantification of PER1 expression in the ventrolateral SCN (B) and PER2 expression in the dorsomedial SCN (C) of light-pulsed (white bars) and dark control (black bars) mice of both genotypes. Values represent mean ± SEM number of immunoreactive nuclei in the designated subregion of the unilateral SCN. n = 4 mice per group. *p < 0.05.
RKIP and light-evoked transcriptional activation in the SCN
Transcriptional activation has long been considered as a pivotal event in clock entrainment. Within this context, studies have shown that the MAPK/ERK pathway couples light to CREB-dependent transcription and expression of IEGs in the SCN (Butcher et al., 2002; Dziema et al., 2003). Additionally, RKIP expression has been shown to modulate AP-1-dependent transcription (Yeung et al., 1999). To determine whether RKIP regulates photic resetting in the early night by affecting light-evoked de novo transcription, we examined the expression of two light-inducible and CREB-regulated genes, c-Fos (an IEG) and mPeriod1, in the SCN of RKIP+/+ and RKIP−/− mice in response to light. For c-Fos expression, mice received a 15 min light pulse (LP: 80 lux) at CT 15 and were killed at 1, 2 and 4 h post-LP. In RKIP+/+ mice, c-FOS protein levels in the SCN peaked at 1 h post-LP and declined thereafter (Fig. 8A,B). In contrast, c-FOS expression at 1 and 2 h post-LP was significantly augmented in the SCN of RKIP−/− mice by ∼46% and ∼36%, respectively, relative to wild-type controls (Fig. 8A,B). To examine Period1 gene expression, we used mPer1::VENUS transgenic mice, which express the VENUS protein under the control of the mPeriod1 gene promoter, bred onto an RKIP+/+ or RKIP−/− background (Cheng et al., 2009). These mice received a 15 min light pulse (LP: 80 lux) at CT 15 and were killed 4 h later. Compared with dark controls, there was a significant increase in VENUS expression in the SCN of both RKIP+/+ and RKIP−/− mice following a light pulse (Fig. 8C,D). However, light-induced VENUS expression in the SCN was markedly elevated by ∼53% in the absence of RKIP (Fig. 8C,D). There was no difference in basal (CT 19) VENUS expression in the SCN of RKIP+/+ and RKIP−/− mice (Fig. 8C,D). Collectively, the data reveal that RKIP negatively modulates light-evoked transcriptional activation of the c-Fos and mPeriod1 genes in the SCN in the early night.
Figure 8.

Absence of RKIP results in enhanced transcriptional activation of the c-Fos and mPeriod1 genes in the SCN in response to a light pulse. A, Representative micrographs of c-FOS protein expression in the SCN of (top) RKIP+/+ and (bottom) RKIP−/− mice at various times (hours) after a 15 min light pulse (80 lux intensity) at CT 15. B, Quantification of c-FOS expression in the SCN of RKIP+/+ (white bars) and RKIP−/− (black bars) mice at the designated times after a 15 min light pulse at CT 15. Values represent mean ± SEM number of immunoreactive nuclei in the unilateral SCN. n = 4 mice per group. *p < 0.05 vs RKIP+/+ controls. C, Representative micrographs of VENUS expression in the SCN of (top) RKIP+/+ and (bottom) RKIP−/− mice carrying the mPER1::VENUS transgene, 4 h after a 15 min light pulse (LP: 80 lux) at CT 15. Control mice were killed at the same circadian time without receiving a light pulse (DD). Tissues were probed with anti-GFP antibody that recognizes VENUS. D, Quantification of VENUS expression in the SCN of light-pulsed (white bars) and dark control (black bars) mice of both genotypes. Values represent mean ± SEM fluorescence intensity (in arbitrary units) normalized to background staining. n = 4 mice per group. *p < 0.05 vs RKIP+/+ controls.
Discussion
Previous studies have highlighted the role of the MAPK/ERK and PKC signaling cascades in the coupling of light to entrainment of the SCN circadian clock (Butcher et al., 2002; Dziema et al., 2003; Jakubcakova et al., 2007). In this study, we examined the potential involvement of RKIP, a PKC target and a potent modulator of MAPK/ERK signaling, in the entrainment process. Our results show that RKIP is not only light-responsive but also negatively modulates photic responsiveness of the SCN. Overexpression of a nonphosphorylatable form of RKIP in the SCN of transgenic mice severely dampened light-induced phase delays and ERK1/2 activation in the SCN. Conversely, the absence of RKIP expression facilitated the phase-delaying as well as the phase-advancing effects of light at the behavioral level. Acute photic stimulation in both the early and late subjective night resulted in prolonged ERK1/2 activation in the SCN of RKIP−/− mice. Furthermore, in the early night, sustained ERK1/2 signaling within the RKIP−/− SCN in response to light was accompanied by enhanced PER1 and PER2 induction and more robust transcriptional activation of the c-Fos and mPeriod1 genes. Collectively, our data identify an essential role of RKIP in modulating the responses of the SCN clock to environmental light.
RKIP was originally identified in a yeast 2-hybrid screen for Raf1 interaction partners (Yeung et al., 1999). Subsequent studies showed that RKIP inhibits the phosphorylation of MEK by Raf1 through sequestration of Raf1 and MEK and interference with the formation of a kinase-substrate complex (Yeung et al., 2000). RKIP is not a substrate for Raf1 or MEK, but is phosphorylated by PKC at Ser/Thr-153, an event that triggers the dissociation of RKIP from Raf1 (Corbit et al., 2003). Overexpression of RKIP interferes with ERK activation and AP-1-dependent transcription, whereas downregulation of endogenous RKIP expression elicits the opposite effects (Yeung et al., 1999). Given the importance of MAPK/ERK signaling in the entrainment process, we hypothesized that RKIP may play a vital role in modulating the responsiveness of the SCN to photic stimuli.
Here we found that a brief light pulse in the early subjective night triggers rapid phosphorylation of RKIP at Thr-153 within the murine SCN and reduces the abundance of the RKIP-c-Raf complex. This is consistent with previous findings indicating that activation of the upstream kinase, PKC, within the SCN is induced by nocturnal light exposure (Lee et al., 2007). Biochemical studies have shown that RKIP is a target of the classical (α, β and γ) and atypical (ζ, ι and λ) but not novel (δ, ε, η and θ) isoforms of PKC (Corbit et al., 2003). Along these lines, light selectively triggers the activation of the α, ζ and λ isoforms, but reduces the phosphoactivation of the δ isoform (Lee et al., 2007). Although it remains to be determined which isoform(s) of PKC is/are responsible for mediating light-induced phosphorylation of RKIP, emerging evidence points to a role of a PKC-mediated pathway in photic entrainment. In particular, PKCα-deficient mice exhibit deficits in light-induced phase delays (Jakubcakova et al., 2007).
In vitro studies have shown that stimulus-dependent activation of the MAPK/ERK pathway can be antagonized by overexpression of a mutant form of RKIP that cannot be phosphorylated at Ser-/Thr-153 (Corbit et al., 2003). Within the context of photic entrainment, we demonstrate that both light-induced ERK1/2 activation in the SCN and phase delays of behavioral rhythms are abrogated in transgenic mice that express mutant RKIP(T153V) in the SCN. The dual effects of RKIP(T153V) on ERK1/2 activation and behavioral resetting in response to light mirror previous experiments showing that pharmacological inhibition of the MAPK/ERK pathway uncouples light to clock entrainment (Butcher et al., 2002). Our data place RKIP as an upstream regulator of ERK activity in the SCN, and further raise the possibility that RKIP functions to couple PKC and MAPK/ERK signaling events in the SCN in response to light.
To address the physiological relevance of RKIP in photic entrainment, we examined the behavior of mice in which the RKIP gene was genetically ablated. RKIP−/− mice exhibited enhanced responsiveness to the phase-delaying and -advancing effects of light in the early and late subjective night, respectively, and a faster rate of reentrainment to an abrupt 8 h phase advance in the light cycle. (It should be noted that the faster rate of LD reentrainment may be based on incremental phase advances, which we predict to be larger in the knock-outs based on the results of the CT 22 light pulse experiments, but it may also involve other mechanisms within and outside the SCN that modulate the speed of resynchronization to the new phase of the LD cycle). On the other hand, the period of the free-running rhythm was not significantly altered by the absence of RKIP. Consistent with the lack of an overt effect on the period (or amplitude) of behavioral rhythms, we observed no effect of RKIP ablation on either the amplitude or phase of PER1 and PER2 rhythms in the SCN, despite the fact that p-ERK1/2 rhythms were altered in the SCN of RKIP−/− mice. The data suggest that while RKIP may not be essential for the regulation of the inherent pacemaker activity of the SCN, it serves as a key negative modulator of the responsiveness of the SCN to the entraining effects of light.
The role of the MAPK/ERK pathway in photic entrainment has been extensively documented. Light-induced MAPK/ERK activation in the SCN not only mediates behavioral resetting, but through a number of downstream effectors actuates changes in gene transcription, mRNA translation, and mRNA/protein stability. Early work showed that light-evoked MAPK activity in the SCN is coupled to transcriptional activation of CREB, and results in induced transcription of mPeriod1 and immediate early genes including c-Fos, JunB, and egr-1 (Travnickova-Bendova et al., 2002; Dziema et al., 2003). Recent studies revealed that the MAPK/ERK pathway mediates mTOR-dependent control of mRNA translation in the SCN (Cao et al., 2010), as well as microRNA-132-dependent control of chromatin remodeling and translation in the SCN (Cheng et al., 2007; Alvarez-Saavedra et al., 2011). Additionally, insights into the mechanisms that shape MAPK/ERK signaling within the SCN have come from functional studies of: MAP kinase phosphatase 1 (MKP1), which dephosphorylates p-ERK1/2; pleckstrin homology domain leucine-rich repeat protein phosphatase 1 (PHLPP1), a K-Ras inhibitor; and Dexras1, a time-of-day-specific regulator of MAPK/ERK signaling in the SCN (Cheng et al., 2004, 2006; Doi et al., 2007; Masubuchi et al., 2010). The current work provides evidence for a new layer of regulation of the MAPK/ERK pathway within the SCN, one in which signaling is terminated at the level of MEK activation by the Raf inhibitor RKIP.
Our data reveal that genetic ablation of RKIP in mice prolongs the duration, but not maximal intensity, of light-induced ERK1/2 activation in the SCN. Absence of RKIP expression did not affect phase-restricted ERK1/2 activation in the SCN in response to light. These results are consistent with mathematical modeling studies that predict that loss of RKIP would result in sustained ERK1/2 signaling but not necessarily a change in peak activation (Thalhauser and Komarova, 2009). Our data showing phase-restricted ERK1/2 activation in the SCN of RKIP−/− mice are also in line with previous studies indicating that loss of RKIP does not by itself trigger ERK1/2 signaling in the absence of a context-appropriate stimulus (Trakul et al., 2005). Until now, the functional consequence of altered kinetics of ERK1/2 activity, rather than the simple presence or absence of ERK1/2 signaling, in the SCN following photic stimulation has not been studied. Given that the duration of ERK1/2 signaling is associated with distinct cellular responses in other biological systems (Murphy et al., 2002), it is possible that prolonged ERK1/2 signaling in the SCN results in differential activation of diverse downstream effector pathways that modulate the resetting response to light.
In line with the larger phase delays and prolonged ERK1/2 activation, RKIP−/− mice exhibited greater expression of light-evoked PER1 and PER2 in the SCN in the early night. The anatomical distribution of PER1 and PER2 induction is consistent with previous reports (Yan and Silver, 2004; Cheng et al., 2009). Possible mechanisms underlying this increase in steady-state PER protein levels include changes in gene transcription, mRNA translation, or protein stability. Indeed, there is ample evidence to indicate that transcriptional activation plays a key role in coupling light to clock entrainment, and that RKIP can modulate transcription of AP-1 target genes (Yeung et al., 1999). Along these lines, our data reveal that light-evoked transcriptional activation of the c-Fos and mPeriod1 genes in the SCN is augmented in the absence of RKIP.
One unresolved issue is the physiological significance of altered p-ERK1/2 rhythms in the SCN of RKIP−/− mice. U0126-mediated pharmacological inhibition of ERK1/2 signaling has been shown to dampen rhythms of clock gene expression in the SCN (Akashi et al., 2008), yet the alterations in p-ERK1/2 rhythms arising from RKIP ablation did not cause a change in PER1 or PER2 oscillations, as one might arguably expect. On the other hand, changes in light-activated p-ERK1/2 expression in the SCN of RKIP−/− mice were indeed accompanied by changes in the inducibility of PER1 and PER2. These data suggest that RKIP functions in a context-dependent manner, and that its role may differ in different cell types within the SCN.
In conclusion, the collective findings uncover a novel role of the Raf inhibitor RKIP in photic entrainment of the circadian clock. By shaping the duration of MAPK/ERK signaling in the SCN in response to light, RKIP modulates the resetting capacity of the SCN clock. Additional studies should examine how RKIP influences the ability of ERK1/2 to couple to diverse downstream effector pathways and how these discrete signaling events contribute to clock entrainment.
Footnotes
This work was supported by operating grants to H.-Y.M.C. from the Canadian Institute of Health Research and the National Sciences and Engineering Research Council of Canada, and an infrastructure grant from the Canadian Foundation for Innovation. We thank Dr. Jan Klysik for providing us the RKIP−/− mice.
The authors declare no competing financial interests.
References
- Akashi M, Hayasaka N, Yamazaki S, Node K. Mitogen-activated protein kinase is a functional component of the autonomous circadian system in the suprachiasmatic nucleus. J Neurosci. 2008;28:4619–4623. doi: 10.1523/JNEUROSCI.3410-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Saavedra M, Antoun G, Yanagiya A, Oliva-Hernandez R, Cornejo-Palma D, Perez-Iratxeta C, Sonenberg N, Cheng HY. miRNA-132 orchestrates chromatin remodeling and translational control of the circadian clock. Hum Mol Genet. 2011;20:731–751. doi: 10.1093/hmg/ddq519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher GQ, Doner J, Dziema H, Collamore M, Burgoon PW, Obrietan K. The p42/44 mitogen-activated protein kinase pathway couples photic input to circadian clock entrainment. J Biol Chem. 2002;277:29519–29525. doi: 10.1074/jbc.M203301200. [DOI] [PubMed] [Google Scholar]
- Butcher GQ, Lee B, Cheng HY, Obrietan K. Light stimulates MSK1 activation in the suprachiasmatic nucleus via a PACAP-ERK/MAP kinase-dependent mechanism. J Neurosci. 2005;25:5305–5313. doi: 10.1523/JNEUROSCI.4361-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Li A, Cho HY, Lee B, Obrietan K. Mammalian target of rapamycin signaling modulates photic entrainment of the suprachiasmatic circadian clock. J Neurosci. 2010;30:6302–6314. doi: 10.1523/JNEUROSCI.5482-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HY, Obrietan K, Cain SW, Lee BY, Agostino PV, Joza NA, Harrington ME, Ralph MR, Penninger JM. Dexras1 potentiates photic and suppresses nonphotic responses of the circadian clock. Neuron. 2004;43:715–728. doi: 10.1016/j.neuron.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Cheng HY, Dziema H, Papp J, Mathur DP, Koletar M, Ralph MR, Penninger JM, Obrietan K. The molecular gatekeeper Dexras1 sculpts the photic responsiveness of the mammalian circadian clock. J Neurosci. 2006;26:12984–12995. doi: 10.1523/JNEUROSCI.4253-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HY, Papp JW, Varlamova O, Dziema H, Russell B, Curfman JP, Nakazawa T, Shimizu K, Okamura H, Impey S, Obrietan K. microRNA modulation of circadian-clock period and entrainment. Neuron. 2007;54:813–829. doi: 10.1016/j.neuron.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HY, Alvarez-Saavedra M, Dziema H, Choi YS, Li A, Obrietan K. Segregation of expression of mPeriod gene homologs in neurons and glia: possible divergent roles of mPeriod1 and mPeriod2 in the brain. Hum Mol Genet. 2009;18:3110–3124. doi: 10.1093/hmg/ddp252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Brautigan DL. Protein phosphatase 2A suppresses MAP kinase signalling and ectopic protein expression. Cell Signal. 1999;11:575–580. doi: 10.1016/s0898-6568(99)00033-9. [DOI] [PubMed] [Google Scholar]
- Coogan AN, Piggins HD. Circadian and photic regulation of phosphorylation of ERK1/2 and Elk-1 in the suprachiasmatic nuclei of the Syrian hamster. J Neurosci. 2003;23:3085–3093. doi: 10.1523/JNEUROSCI.23-07-03085.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit KC, Trakul N, Eves EM, Diaz B, Marshall M, Rosner MR. Activation of Raf-1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J Biol Chem. 2003;278:13061–13068. doi: 10.1074/jbc.M210015200. [DOI] [PubMed] [Google Scholar]
- Daan S, Pittendrigh CS. A functional analysis of circadian pacemakers in nocturnal rodents. II. The variability of phase response curves. J Comp Physiol. 1976;106:253–266. [Google Scholar]
- Doi M, Cho S, Yujnovsky I, Hirayama J, Cermakian N, Cato AC, Sassone-Corsi P. Light-inducible and clock-controlled expression of MAP kinase phosphatase 1 in mouse central pacemaker neurons. J Biol Rhythms. 2007;22:127–139. doi: 10.1177/0748730406298332. [DOI] [PubMed] [Google Scholar]
- Dziema H, Oatis B, Butcher GQ, Yates R, Hoyt KR, Obrietan K. The ERK/MAP kinase pathway couples light to immediate-early gene expression in the suprachiasmatic nucleus. Eur J Neurosci. 2003;17:1617–1627. doi: 10.1046/j.1460-9568.2003.02592.x. [DOI] [PubMed] [Google Scholar]
- Hannibal J. Neurotransmitters of the retino-hypothalamic tract. Cell Tissue Res. 2002;309:73–88. doi: 10.1007/s00441-002-0574-3. [DOI] [PubMed] [Google Scholar]
- Jakubcakova V, Oster H, Tamanini F, Cadenas C, Leitges M, van der Horst GT, Eichele G. Light entrainment of the mammalian circadian clock by a PRKCA-dependent posttranslational mechanism. Neuron. 2007;54:831–843. doi: 10.1016/j.neuron.2007.04.031. [DOI] [PubMed] [Google Scholar]
- Lee B, Almad A, Butcher GQ, Obrietan K. Protein kinase C modulates the phase-delaying effects of light in the mammalian circadian clock. Eur J Neurosci. 2007;26:451–462. doi: 10.1111/j.1460-9568.2007.05664.x. [DOI] [PubMed] [Google Scholar]
- Masubuchi S, Gao T, O'Neill A, Eckel-Mahan K, Newton AC, Sassone-Corsi P. Protein phosphatase PHLPP1 controls the light-induced resetting of the circadian clock. Proc Natl Acad Sci U S A. 2010;107:1642–1647. doi: 10.1073/pnas.0910292107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol. 2002;4:556–564. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Impey S, Storm DR. Light and circadian rhythmicity regulate MAP kinase activation in the suprachiasmatic nuclei. Nat Neurosci. 1998;1:693–700. doi: 10.1038/3695. [DOI] [PubMed] [Google Scholar]
- Pendergast JS, Friday RC, Yamazaki S. Photic entrainment of period mutant mice is predicted from their phase response curves. J Neurosci. 2010;30:12179–12184. doi: 10.1523/JNEUROSCI.2607-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz WJ, Zimmerman P. Circadian timekeeping in BALB/c and C57BL/6 inbred mouse strains. J Neurosci. 1990;10:3685–3694. doi: 10.1523/JNEUROSCI.10-11-03685.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeyoshi Y, Taguchi K, Yamamoto S, Takekida S, Yan L, Tei H, Moriya T, Shibata S, Loros JJ, Dunlap JC, Okamura H. Light-induced resetting of a mammalian circadian clock is associated with rapid induction of the mPer1 transcript. Cell. 1997;91:1043–1053. doi: 10.1016/s0092-8674(00)80494-8. [DOI] [PubMed] [Google Scholar]
- Takumi T, Matsubara C, Shigeyoshi Y, Taguchi K, Yagita K, Maebayashi Y, Sakakida Y, Okumura K, Takashima N, Okamura H. A new mammalian period gene predominantly expressed in the suprachiasmatic nucleus. Genes Cells. 1998;3:167–176. doi: 10.1046/j.1365-2443.1998.00178.x. [DOI] [PubMed] [Google Scholar]
- Thalhauser CJ, Komarova NL. Specificity and robustness of the mammalian MAPK-IEG network. Biophys J. 2009;96:3471–3482. doi: 10.1016/j.bpj.2008.10.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theroux S, Pereira M, Casten KS, Burwell RD, Yeung KC, Sedivy JM, Klysik J. Raf kinase inhibitory protein knockout mice: expression in the brain and olfaction deficit. Brain Res Bull. 2007;71:559–567. doi: 10.1016/j.brainresbull.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakul N, Menard RE, Schade GR, Qian Z, Rosner MR. Raf kinase inhibitory protein regulates Raf-1 but not B-Raf kinase activation. J Biol Chem. 2005;280:24931–24940. doi: 10.1074/jbc.M413929200. [DOI] [PubMed] [Google Scholar]
- Travnickova-Bendova Z, Cermakian N, Reppert SM, Sassone-Corsi P. Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMAL1 activity. Proc Natl Acad Sci U S A. 2002;99:7728–7733. doi: 10.1073/pnas.102075599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Silver R. Resetting the brain clock: time course and localization of mPER1 and mPER2 protein expression in suprachiasmatic nuclei during phase shifts. Eur J Neurosci. 2004;19:1105–1109. doi: 10.1111/j.1460-9568.2004.03189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, Sedivy JM, Kolch W. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–177. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- Yeung K, Janosch P, McFerran B, Rose DW, Mischak H, Sedivy JM, Kolch W. Mechanism of suppression of the Raf/MEK/extracellular signal-regulated kinase pathway by the raf kinase inhibitor protein. Mol Cell Biol. 2000;20:3079–3085. doi: 10.1128/mcb.20.9.3079-3085.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]