

Abstract
Both IL-23- and IL-1-mediated signaling pathways play important roles in Th17 cell differentiation, cytokine production, and autoimmune diseases. The IL-1 receptor associated kinase 4 (IRAK4) is critical for IL-1/TLR signaling. We show here that inactivation of IRAK4 kinase in mice (IRAK4 KI) results in significant resistance to experimental autoimmune encephalomyelitis (EAE) due to a reduction in infiltrating inflammatory cells into the CNS and reduced antigen-specific CD4+ T cell-mediated IL-17 production. Adoptive transfer of MOG35-55-specific IRAK4 KI Th17 cells failed to induce EAE in either wild-type or IRAK4 KI recipient mice, indicating the lack of autoantigen-specific Th17 cell activities in the absence of IRAK4 kinase activity. Furthermore, the absence of IRAK4 kinase activity blocked induction of IL-23 receptor expression, STAT3 activation by IL-23, and Th17 cytokine expression in differentiated Th17 cells. Importantly, blockade of IL-1 signaling by IL-1RA inhibited Th17 differentiation and IL-23-induced cytokine expression in differentiated Th17 cells. The results of these studies demonstrate that IL-1-mediated IRAK4 kinase activity in T cells is essential for induction of IL-23 receptor expression, Th17 differentiation, and autoimmune disease.
Keywords: EAE, IRAK4, Th17 cells, IL-23 receptor
Introduction
IL-17 is a key proinflammatory cytokine that plays an important role in protective immunity against infections and also induces a variety of inflammatory effects (1, 2). The expression of IL-17 has been demonstrated to be increased in many inflammatory conditions such as multiple sclerosis, rheumatoid arthritis, lung airway infections, and psoriasis (3–6). IL-17 binds to its ubiquitously expressed receptor, IL-17R, and induces the secretion of many other proinflammatory cytokines, chemokines, and metalloproteases in multiple cell types resulting in recruitment of neutrophils and/or macrophages to the area of inflammation as well as enabling cell movement and tissue damage (7, 8). IL-17 deficiency in mice delays the onset and reduces severity of experimental autoimmune encephalomyelitis (EAE) and collagen-induced arthritis (CIA) (9). Similar to the IL-17 deficient mice, treatment with anti-IL-17 monoclonal antibody produces a significant, but modest reduction of CNS or joint inflammation and clinical disease score in mouse models of EAE and RA, respectively, indicating that IL-17 plays important roles under those inflammatory conditions (10–12).
CD4+ helper T (Th) cells are the orchestrators in the adaptive immune response. Upon activation, naïve Th cells differentiate into effector cells that have been historically classified as Th1 or Th2 lineage, based on their unique cytokine secretion and immune regulatory functions (13, 14). However, recent studies have led to the discovery of a new lineage of effector CD4+ T cells, Th17 cells, which originate under different polarizing conditions (15–17). Several cytokines including IL-6, TGF-β, and IL-21 have been shown to be required for optimal Th17 differentiation in vitro. Both, IL-6 and IL-21 bind to their respective receptors and activate STAT3, which is necessary for the induction of IL-17. In addition to STAT3, two additional related transcription factors, RORα and RORγt, have been recently shown to be selectively expressed in Th17 cells. The expression of both transcription factors is necessary and sufficient for IL-17 expression. Overexpression of RORα and/or RORγt in CD4+ T cells is sufficient to induce IL-17 production in the absence of exogenous cytokines, whereas the deficiency of either factor alone leads to partial inhibition of IL-17 production. Loss of both factors completely blocks Th17 cell differentiation and cytokine production (18). Consistent with their unique expression, the expression of RORα and RORγt is increased by TGF-β and IL-6 during Th17 differentiation and both are now considered Th17 lineage specific transcription factors (18).
In addition to TGF-β and IL-6, IL-23 is another critical cytokine involved in Th17 differentiation, cell expansion/survival and stabilization (19). The importance of IL-23 in Th17-mediated inflammation has been revealed mainly through studies in mice. Mice lacking IL-23p19 were shown to be resistant to disease induction in Th17 cell-dependent CIA, EAE, and IBD diseases models (20–22). Administration of anti-IL-23p19 monoclonal antibody (mAb) or anti-IL-12p40 mAb inhibited the production of multiple inflammatory cytokines including IL-17, IL-6, IFN-γ, IL-1β, and TNF-α, and consequently, reduced EAE development (20–22). Furthermore, adoptive transfer of myelin oligodendrocyte (MOG33-55)-specific IL-17-producing T cells but not IFN-γ-producing T cells can efficiently induce EAE in recipient mice, indicating that Th17 cells, not Th1 cells, are the pathological effector T cells in this autoimmune disease model (20). Although IL-23 seems to be more directly involved in maintaining or stabilizing Th17 cells, it has been recently shown that IL-23 along with TGF-β and inflammatory cytokines such as IL-6 and IL-1β is essential for human Th17 differentiation and cytokine modulation (23, 24). Sutton et al. recently demonstrated that IL-1 also plays a vital role in promoting antigen-specific Th17 cells in vivo in response to immunization with foreign or self-antigen and a TLR ligand (25). Upon binding to IL-1R, IL-1β triggers the binding of intracellular adaptor protein, MyD88, to the receptor and consequently leads to the recruitment of IL-1R associated kinases (IRAKs), IRAK4 and IRAK1, to the receptor complex (26). IRAK1 is then phosphorylated, ubiquitinylated and degraded resulting in activation of multiple downstream signaling pathways including JNK, p38, and NF-κB (26). IRAK4 is pivotal for IL-1R and TLR-induced signaling and deficiency of IRAK4 in mice abolishes the innate immune response (27). However, the role of IRAK4 in the adaptive immune response is still controversial. IRAK4 has been shown to be recruited to T cell lipid rafts upon T cell receptor activation and IRAK4 deficiency led to impaired T cell responses in vivo, suggesting that IRAK4 is involved in cross-talk between signaling in the innate and adaptive immune responses (28). In contrast, a more recent study with both IRAK4−/− and IRAK4 kinase-dead knock-in (KI) mice showed no defects in T cell response (29). Therefore, whether the IRAK4 kinase activity is required for its function in T cell signaling and response remains to be resolved. We and others recently described IRAK4 kinase dead “knock-in” mice in which the wild-type gene coding for IRAK4 was replaced with a mutant form in which the two conserved lysine residues in the ATP binding pocket were replaced by alanine (29, 30). Deficiency of IRAK4 kinase activity prevented the recruitment of IRAK1 to the IL-1R complex and its subsequent phosphorylation and degradation. Despite these extensive studies, the precise role of IRAK4 kinase activity in adaptive immune responses including Th17 differentiation is unknown. We report here that IRAK4 kinase dead knock-in mice (IRAK4 KI) are resistant to the induction and progression of Th17-cell driven experimental autoimmune encephalomyelitis (EAE). CD4+ T cells from IRAK4 KI mice failed to produce IL-17 upon IL-23 stimulation and adoptive transfer of autoantigen-specific IRAK4 KI Th17 cells failed to induce EAE in recipient mice. The absence of IRAK4 kinase activity also diminished IL-23 receptor induction and stabilization of the differentiated Th17 cells mediated by IL-23. In addition, IL-23 was found to be essential for the continued expression of IL-23 receptor on differentiated WT Th17 cells and their Th17 cytokine expression. In contrast, the differentiated IRAK4 KI Th17 cells failed to induce IL-23 receptor expression during differentiation and the continuous expression upon stimulation of IL-23, suggesting that IRAK4 plays important roles in regulating IL-23 receptor expression during Th17 differentiation, which is subsequently required for the continued Th17 polarization by IL-23. As a result, IL-23 failed to activate STAT3 and stimulate Th17 cytokine expression in IRAK4 KI Th17 cells. Furthermore, blockade of IL-1 signaling with recombinant IL-1RA had similar effects on Th17 cell differentiation of wild-type cells as those observed in Th17 cells derived from IRAK4 KI mice. Taken together, our results provide evidence that IRAK4, an essential component of IL-1 signaling, plays a crucial role in Th17 cell-mediated autoimmune disease by regulating IL-23 receptor expression and polarization of pathogenic Th17 cells.
Materials and Methods
Mice and reagents
IRAK4 kinase-inactive knock-in (IRAK4 KI) mice were described previously (30). Breeding colonies for both IRAK4 KI mice and their wild-type (WT) littermates were established and maintained at Taconic (Germantown, NY). All animal experimental procedures used in this study were approved by Eli Lilly and Company Animal Care and Use Committee and carried out in accordance with the guidelines of Eli Lilly and Company (Indianapolis, IN).
All antibodies for flow cytometry, anti-CD3, CD28, IL-4, IFN-γ, IL-1α, and IL-1β were purchased from BD Pharmingen (San Diego, CA). Anti-phospho-STAT3 (Y705) antibody was purchased from Cell Signaling (Danvers, MA). Naïve CD4+CD62L+ T cells were purified by AutoMacs according to the manufacturer’s protocol (Miltenyi Biotech, Auburn, CA). Recombinant cytokines including IL-6, TGF-β, IL-23, and IL-1β were purchased from R&D Systems, Inc. (Minneapolis, MN). Recombinant IL-1RA was commercially prescribed as Kineret® (Amgen Inc. Thousand Oaks, CA).
Induction and clinical evaluation of MOG35-55-induced EAE
All mice were age- and sex-matched (6- to 10-week old females) at the start of experiments, and WT littermates were used as controls. IRAK4 KI and WT mice were immunized subcutaneously (s.c.) at two sites on the back with 300 μg MOG35-55 peptide (MEVGWYRSPFSRVVHLYRNGK, Peptides International, Louisville, KY) emulsified in a total of 200 μl Complete Freunds Adjuvant (CFA; Difco, Detroit, MI) containing 500 μg M. tuberculosis H37 Ra (Difco) on days 0 and 7, supplemented with intraperotoneal (i.p.) injections of 500 ng pertussis toxin (Calbiochem, San Diego, CA) on days 0 and 2. Clinical symptoms of EAE were scored daily by two separate observers using the following scale; 0 = no symptoms, 0.5 = distal weak or spastic tail, 1 = completely limp tail, 1.5 = limp tail and hind limb weakness (feet slip through cage grill), 2.0 = unilateral partial hind limb paralysis, 2.5 = bilateral partial hind limb paralysis, 3.0 = complete bilateral hind limb paralysis, 3.5 = complete hind limb and unilateral partial forelimb paralysis, 4.0 = moribund or death. The data was recorded as the mean daily clinical score.
For Adoptive transfer of EAE, mice were immunized with MOG35-55 plus CFA via conditions that induce active EAE. Lymph nodes were collected 10 days later and single cell suspensions were prepared. Cells (6 × 106 cells/ml) were cultured in RPMI 1640 medium (supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 100 IU/ml penicillin/streptomycin and 2 × 10−5 M 2-ME) with MOG35-55 (20 μg/ml) and IL-23 (20 ng/ml). Four days after initiation of culture, cells were harvested, washed in PBS and then injected into recipient mice (2 × 107 cells per mouse) sublethally irradiated (600 rad) 4h before injection.
Histology, immunohistochemistry, and flow cytometry
MOG35-55-immunized IRAK4 KI and WT mice were euthanized at days 21–25. Spinal cords were removed, fixed overnight in IHC Zinc fixative (BD Biosciences PharMingen, San Diego, CA) solution. Sample process and immunohistochemistry staining was done as described previously (36).
All flow cytometric data were collected and analyzed with a Cytomics™ FC500 (Becton Coulter, Miami, FL) using RXP™ software (Becton Coulter). The following antibodies for staining were purchased from BD PharMingen: anti-CD45-Cy-chrome (20-F1), anti-CD4-PE (L3T4), anti-CD8a-FITC (53–6.7), isotype control Abs, (rat IgG-FITC and rat IgG-PE). CD11b-PE (M1/70) was purchased from Biolegend (San Diego, CA). F4/80-FITC (MCA497F) was purchased from AbD Serotec (Münsterstr, Germany).
Proliferation and cytokine ELISA analysis
Splenocyte cell suspensions were isolated from MOG35-55-immunized WT or IRAK4 KI mice at 21, along with naïve WT mice. Pooled splenocytes of 5 individual mice from the same group were plated in triplicate in 96-well round bottom plates at 2 × 105 cells/well in 200 μl complete RPMI 1640 medium (Invitrogen) supplemented with 5% FCS and cultured at 37°C, 5% CO2. Proliferation was measured by incorporation of [3H]-Methylthymidine (1 μCi/well, ICN Radiochemicals, Irvine, CA) during the last 8 hr of culture using a filtermate harvester (Packard Instrument Co., Downers Grove, IL) and a 1450 microbeta liquid scintillation counter (Pharmacia Biotech AB). Cytokine levels produced by cultured splenocytes following stimulation with MOG35-55 peptide were analyzed by ELISA or using luminex beads. ELISA assays for IL-17, IL-17F, or IL-22 were performed according to the manufacturer’s protocol (R&D systems Inc., Minneapolis, MN). IFN-γ, IL-2, IL-5, IL-1α and IL-1β were determined by luminex (Linco Research, Missouri).
TaqMan quantitative RT-PCR analysis, Western analysis and phospho-Stat3 ELISA detection
For RNA expression, total cellular RNA was pooled from spinal cords or Th17 T cells of WT or IRAK4 KI mice. RNA was prepared using RNeasy Mini Kit (QIAGEN Inc. Valencia, CA), DNase treated and quantified by spectrophotometric analysis. A two-step quantitative RT-PCR was performed to determine gene expression by using the relative standard curve method, with the cellular housekeeping gene, β-actin as the normalization control. cDNA was synthesized with a High Capacity cDNA Archive Kit (ABI 4322171) using Gene Amp PCR 9700 (PE Applied Biosystems). A second PCR step was performed using TaqMan Universal PCR Master Mix (ABI 4304437) and ABI Prism™ 7900HT Sequence Detection System (Applied Biosystems). IL-17 assay-on-demand TaqMan Gene Expression (ABI Mm00439619_m1), IL-17F, IL-21, IL-22, RORα, RORγt and β-actin primers and probe (PE Applied Biosystems 4352339E) were used. Threshold cycle numbers (Ct) were determined with Sequence Detector Software (version 2.1.1; PE Applied Biosystems) and transformed using a standard curve generated from serially diluted cDNA samples from WT cells. The mRNA expression amount was divided by the β-actin amount to obtain a normalized target value.
For Western blot analysis of Stat3 protein expression, naïve CD4+ T cells were differentiated into Th17 cells in vitro, which were incubated with plate-bound mAbs of anti-CD3 and anti-CD28 in the presence of soluble mAb of anti-IL-4 mAb (5 μg/ml), anti-IFNγ (5 μg/ml), IL-6 (10 ng/ml), and TGF-β (5 ng/ml) (R&D System Inc., MN) at 37 °C for 4 days. The Th17 cells were then starved in serum free medium for 4 hrs before stimulating with 10 ng/ml of recombinant IL-23 for 15 min at 37 °C. Cells were then washed and total cell lysates were prepared in cell lysis buffer (Cell Signaling, Beverly, MA). Equal amount of protein was loaded and blotted using antibodies recognizing total Stat3 or phospho-Stat3 (Y705) purchased from Cell Signaling, Beverly MA. Phosphorylation of Stat3 was quantitated in cell lysates using the PathScanPhospho-Stat3 (Y705) ELISA kit (Cell Signaling, Beverly MA).
Naïve CD4+ T cell Th1, Th2 and Th17 differentiation in vitro
Naïve CD4+ T cells were purified from both WT and IRAK4 KI mice with AutoMACS and differentiated in vitro. For Th17 differentiation: cells were incubated with plate-bound mAbs of anti-CD3 and anti-CD28 in the presence of soluble mAb of anti-IL-4 mAb (5 μg/ml), anti-IFNγ (5 μg/ml), IL-6 (10 ng/ml), and TGF-β (5 ng/ml) (R&D System Inc., MN) at 37 °C for 4 days. In some experiments, IL-1β (5 ng/ml), IL-23 (5 ng/ml) (R&D System Inc., MN), or IL-1-RA (1 μg/ml) was added as indicated. Following differentiation, cells were washed with PBS or growth medium and used as indicated. For Th1 and Th2 differentiation, naive CD4+ T cells were incubated with plate-bound mAbs of anti-CD3 and anti-CD28 in the presence of mIL-4 plus anti-IFNγ mAb (Th2 condition) or mIL-12 plus anti-IL-4 mAb (Th1 condition). Differentiated Th cells were washed and restimulated with plate-bound anti-CD3 mAb for 24 hrs and cell supernatants were used for measuring cytokine levels by ELISA (R&D System Inc. MN).
Statistical analysis
Comparison of clinical scores and cytokine production levels between the various of treatment groups were analyzed by unpaired Student’s t test. Values of p < 0.05 were considered significant.
Results
Reduced MOG35-55-induced experimental autoimmune encephalomyelitis in IRAK4 KI mice
Our previous work has shown that IRAK4 kinase activity is critical in Toll-like receptor mediated-innate immunity (30). To investigate the role of IRAK4 kinase activity in the adaptive T cell immune response to autoantigen and disease induction, we immunized WT and IRAK4 KI mice with MOG35-55 peptide to induce EAE. As shown in Fig. 1A, the disease onset in WT mice occurred at approximately day 9–11 and peaked between day 18–21 with a mean disease score of 3.2. In contrast, disease onset in IRAK4 KI mice was significantly delayed occurring on day 13–15, and peaked at day 20–22 with a mean disease score of 1.5. Disease incidence (EAE score >1.0) was also reduced in IRAK4 KI mice (42% incidence) as compared to WT mice (92% incidence). The cumulative disease index (CDI) based on each individual animal’s disease score from day 8–21 also was significantly reduced in IRAK4 KI mice (CDI = 7.9 ± 1.83) versus WT mice (CDI = 20.1 ± 1.71). These data show that IRAK4 kinase activity is critical for the full induction of MOG35-55-induced EAE in mice. To determine whether resistance to EAE induction in IRAK4 KI mice was associated with a reduction in CNS inflammation, we examined leukocyte infiltration in the pooled spinal cords by flow cytometric analysis at the peak of disease (on day 21). To quantitatively measure the inflammatory cell infiltration in the CNS, mononuclear cells isolated from the pooled spinal cords of four individual WT (average disease score ~3.0) and IRAK4 KI (average disease score ~0.8) mice were stained for cell surface antigens, and analyzed by flow cytometry (Fig. 1, B–D). As compared to WT mice, there was an approximate 4-fold reduction in total CD45+ cells (WT: 21.3%; IRAK4 KI: 5.4%) (Fig. 1B) and a 7-fold reduction of CD45hiF4/80+ macrophages (WT: 14.2%; IRAK4 KI: 2.1%) in IRAK4 KI mice (Fig. 1C). In addition, there was approximately a 3-fold decrease in infiltrating CD4+ T cells (WT: 23.6%; IRAK-4 KI: 8.3%) and a 4-fold reduction in infiltrating CD8+ T cells (WT: 11.3, IRAK4 KI: 2.8 %) in IRAK4 KI spinal cord as compared to WT (Fig. 1D). These results suggest that the resistance of IRAK4 KI mice to MOG35-55-induced EAE is primarily due to a reduction in mononuclear cell infiltration in the CNS during EAE induction.
Fig. 1. Reduced induction and progression of EAE in IRAK4 KI mice.


(A) EAE was induced following immunization of MOG35-55 (300 μg/mouse, n = 15) in CFA on days 0 and 7, as described in Methods and Materials. The severity of EAE is presented as mean disease scores in each group. The data represent one of two independent experiments. There was also higher percentage of disease incidence (EAE score >1.0) after combined from two independent experiments in WT mice (23/25, 92%) than IRAK4 KI mice (11/26, 42%). (B) Mononuclear cells isolated from the pooled spinal cords of four individuals from IRAK4 KI (disease score: 0, 0, 0.5, 2.8) or WT mice (disease score 3.5, 3.0, 3.0, 3.0) on day 21 were stained for CD45. The CD45+ cells were gated and the percentage of positive cells is indicated. (C–D) Mononuclear cells isolated from the same pooled spinal cords as in panel B were stained for anti-CD45 plus F4/80 for infiltrated macrophages (C) or with anti-CD4 antibody and anti-CD8 antibody for infiltrated T cells (D). Percentages of CD4+-, CD8+-T cells or F4/80+-macrophages were counted on gated CD45+ cells. The data represent one of two independent experiments with similar results.
Reduced IL-17 production in MOG35-55-specific T cells in IRAK4 KI mice
Previous studies using IRAK4 deficient mice showed that deficiency of IRAK4 protein completely impaired in vivo T cell immune response (28). To determine whether the reduction in EAE disease in IRAK4 KI mice was simply due to defective T cell responses to immunized antigen, we examined self-antigen-stimulated T cell proliferation and cytokine production from MOG35-55-immunized IRAK4 KI or WT mouse splenocytes on day 21 post immunization (peak of the disease) in the presence of different concentrations of MOG35-55 peptide. There was no difference in MOG35-55-induced T cell proliferation between WT and IRAK4 KI mice (Fig. 2A). Moreover, compared with WT animals, antigen-stimulated T cells from IRAK4 KI mice produced similar amounts or slight higher level of IL-2, IL-1α, IL-1β, Th1 and Th2 cytokines including IFN-γ and IL-5, respectively. In contrast, unlike the other cytokines, the production of IL-17 was significantly reduced in IRAK4 KI mice compared with WT mice (Fig. 2B). These data indicate that loss of IRAK4 kinase activity does not affect T cell proliferation or Th1 and Th2 cytokine production in response to self-antigen (MOG35-55 peptide) immunization. Instead, antigen-induced IL-17 expression is specifically regulated by IRAK4 kinase activity.
Figure 2. Impaired IL-17 production in MOG35-55-primed lymphocytes from IRAK4 KI mice.

Splenocytes were isolated from five each of IRAK4 KI and WT mice on days 21 following immunization with MOG35-55 peptide in CFA. The MOG35-55-primed cells were then cultured in the presence of the indicated concentration of MOG35-55. (A). T cell proliferation was determined using tritium-labeled thymidine incorporation assay. The results were plotted as mean c.p.m. ± SEM from triplicates. The data represent one of two independent experiments. (B) The MOG35-55-stimulated splenocyte culture supernatants were collected at 72 hr. ELISA was used to measure the antigen-stimulated production of IFN-γ, IL-2, IL-5, IL-1α, IL-1β and IL-17 in the cultures. Values shown represent the mean and error bars represent SD. The data represent one of two independent experiments. Asterisk** denotes 0.001 < p < 0.01, * denotes 0.01< p < 0.05, ns denotes not significant
Recent studies have shown that autoantigen-reactive IL-17-producing T cells, Th17 cells, are the primary pathogenic CD4+ T cells for inducing EAE in mice (20). To determine whether Th17 T cells from IRAK4 KI mice were primarily responsible for the reduction of EAE, we isolated CD4+ T cells from MOG35-55-immunized WT and IRAK4 KI mice and differentiated them into Th17 cells in vitro in the presence of MOG35-55 peptide. The in vitro differentiated MOG35-55-specific Th17 T cells were then adoptively transferred into either WT or IRAK4 KI recipient mice. Compared with the adoptive transferred Th17 cells from WT mice, IRAK4 KI Th17 cells failed to induce EAE in either WT or IRAK4 KI recipient mice (Fig. 3A). To further confirm our findings, we isolated total mRNA from the spinal cord of both WT and IRAK4 KI mice with EAE and measured the IL-17 mRNA expression by real-time PCR. As shown in Fig. 3B, the expression of IL-17 mRNA was diminished in spinal cords of IRAK4 KI mice compared with WT mice. Whereas no difference in the mRNA levels of IL-12-p35 and even slightly higher levels of IL-23 p19 mRNA in IRAK4 KI mice were observed (Fig. 3B). Our results indicate that the IRAK4 KI Th17 cells were defective in inducing EAE and the inactivation of IRAK4 kinase leads to the resistance of EAE in IRAK4 KI mice.
Figure 3. Impaired IL-17 expression in CNS of IRAK4 KI mice.

(A). Differentiated MOG35-55-specific Th17 cells from both MOG35-55-immunized IRAK4 KI and WT mice were adoptively transferred into recipient mice of either WT or IRAK4 KI mice. The progression of disease was scored according to our EAE disease score as described in Materials and Methods. (B). Reduced IL-17 mRNA expression in spinal cord of IRAK4 KI mice with EAE. Spinal cord was used for preparing RNA on day 21 following MOG35-55 immunization of both WT and IRAK4 KI mice. The mRNA expression of indicated genes was assessed by real-time PCR. The mRNA expression of WT mice was set to 100%. The data represent one of two independent experiments with similar results. Asterisk ** denotes 0.001<p < 0.01, *** denotes p<0.001.
Absence of IRAK4 kinase activity leads to reduced expression of Th17 cytokines in differentiated Th17 cells
Naïve CD4+ T cells can be differentiated into Th1, Th2, or Th17 lineages in the presence of different cytokines (13, 16, 31). To address the question of whether IRAK4 kinase activity is involved in Th cell differentiation, we first isolated naïve CD4+ T cells from WT and IRAK4 KI mice and incubated them under conditions to induce Th1, Th2, or Th17 differentiation. Cytokine expression from differentiated Th1 or Th2 cells was indistinguishable between WT and IRAK4 KI cells (data not shown), suggesting that IRAK4 kinase signaling pathway is not involved in these processes. Next, we examined whether the defective IL-17 production in IRAK4 KI mice was due to the impaired differentiation of CD4+ T cells into effector Th17 cells. We purified naïve CD4+ T cells from WT or IRAK4 KI mice and differentiated them into Th17 cells in the presence of IL-6, TGF-β, IL-23, and IL-1β as described in the Materials and Methods. As shown in Fig. 4A, expression of Th17 cytokine mRNAs including IL-17 and IL-17F was dramatically induced in WT Th17 cells compared with Th0 cells. In contrast, the expression of these cytokines in IRAK-4 KI Th17 cells was only slightly induced compared with Th0 cells and was significantly lower as compared with WT Th17 cells. We also tested the ability of IL-23 to further induce or maintain the Th17 cell phenotype in both WT and IRAK4 KI Th17 cells. As shown in Fig. 4B and C, IL-23 treatment of differentiated WT Th17 cells induced expression Th17 cytokines at both the mRNA and protein levels. In contrast, only low level Th17 cytokine expression in IRAK4 KI Th17 cells was observed under the same conditions. Following IL-23 stimulation, the increase in IL-17 and IL-17F mRNA was 12- and 47-fold, respectively, in WT Th17 cells as compared to only 5.4- and 10-fold, respectively, in IRAK4 KI Th17 cells. These data indicate that IL-23 is essential for the maintenance of Th17 cytokine expression. To examine the mechanism of the defective IL-17 production in differentiated Th17 cells from IRAK4 KI mice, we also investigated the expression of the two Th17 specific transcription factors, RORγt and RORα, under the same Th17 differentiation conditions in both WT and IRAK4 KI T cells. Surprisingly, we did not reproducibly observe any dramatic difference in the induced expression of these two transcription factors between WT and IRAK-4 KI Th17 cells (data not shown), suggesting that IRAK4 kinase activity does not play a major role regulating expression of these two lineage specific transcription factors during Th17 differentiation.
Figure 4. Reduced expression of Th17 cytokines in differentiated Th17 cells from IRAK4 KI mice.

Naïve CD4+ T cells were purified from both WT and IRAK4 KI mice and differentiated into Th17 cells with plate-bound anti-CD3, anti-CD28 mAbs in the presence IL-1β, IL-6, and TGF-β for 4 days, or T cells were incubated with only plate-bound mAbs anti-CD3 and anti-CD28 in the presence of neutralizing mAbs to IL-4 and IFNγ (Th0 cells). (A). Total mRNAs were isolated from 4-day differentiated Th17 cells or Th0 cells. Both IL-17 and IL-17F mRNA expression were assessed by real-time PCR. The expression level was normalized to β-actin mRNA. The data represent one of three independent experiments. (B). Differentiated Th17 cells were washed, replated in medium, and incubated in the presence or absence of recombinant IL-23 for 16 hrs. Total mRNA isolation and IL-17/IL-17F mRNA expression was performed as in (A). The data represent one of three independent experiments. (C) Cell supernatants from the same treatment groups from (B) were then filtered into fresh 96-well tissue culture plates, and different cytokine levels were measured by ELISA assay. Values shown represent the mean and error bars represent SD. The data represent one of three independent experiments. Asterisk ** denotes 0.001<p<0.01, *** denotes p < 0.001.
Abolished induction of IL-23 receptor expression and IL-23-induced Stat3 activation in IRAK4 KI Th17 cells
IL-23 receptor is primarily expressed on memory T cells in naïve mice and its expression is greatly increased during Th17 cell differentiation (18, 32). To examine whether the reduced IL-17 expression in IRAK4 KI Th17 cells was due to the impaired IL-23 receptor induction or impaired IL-23 signaling, we differentiated naïve CD4+ T cells from WT and IRAK4 KI mice into Th17 cells and examined IL-23 receptor mRNA expression. As shown in Fig. 5A, compared with Th0 cells, the expression of IL-23 receptor mRNA was dramatically induced in differentiated WT Th17 cells as described previously (18). In contrast, this increase in Th17 cells from IRAK4 KI mice was reduced significantly (Fig. 5A). Furthermore, the expression of IL-23R mRNA was further increased or maintained upon IL-23 treatment of differentiated WT Th17 cells, whereas IL-23 treatment failed to maintain the high expression of IL-23R mRNA in differentiated IRAK4 KI Th17 cells (Fig. 5B), suggesting that one of the mechanisms of stabilizing Th17 polarization by IL-23 is through induction of its own receptor expression. To further determine whether the reduced IL-23R expression affected IL-23 signaling in IRAK4 KI Th17 cells, we next examined the main signaling event of IL-23 receptor, Stat3 activation. As shown in Fig. 5C, differentiated Th17 cells from both WT and IRAK4 KI mice showed equal level of expression of total STAT3, suggesting that IRAK4 was not involved in regulating total STAT3 expression. Upon stimulation with IL-23, an increase of phospho-Stat3 protein was observed only in WT Th17 cells (Fig. 5C). Further quantitative measurement by phospho-Stat3 ELISA showed that the level of phosphorylation of Stat3 in WT Th17 cells was significantly increased by 1.8–2.5-fold. In contrast, no increase in Stat3 phosphorylation was observed following treatment with IL-23 in IRAK4 KI Th17 cells (Fig. 5D). These data suggest IRAK4 kinase plays a critical role in regulating IL-23 receptor expression on differentiated Th17 cells and ultimately affects IL-23-induced Stat3 activation and IL-17 production in Th17 cells.
Figure 5. Reduced IL-23 receptor expression and IL-23-stimulated Stat3 activation in Th17 cells of IRAK4 KI mice.

Purified native CD4+ T cells from both WT and IRAK4 KI mice were differentiated into Th17 cells in vitro with plate-bound anti-CD3 and anti-CD28 mAbs in the presence of IL-1β, IL-6, and TGF-β for 4 days, or T cells were incubated with only plate-bound mAbs anti-CD3 and anti-CD28 in the presence of neutralizing mAbs to IL-4 and IFNγ (Th0 cells). (A) Total mRNA was purified from both Th0 and Th17 cells and the expression of IL-23 receptor mRNA was assessed by real-time PCR and normalized to β-actin mRNA. The data represent one of three independent experiments with similar results. (B) Differentiated Th17 cells were washed and then treated with recombinant human IL-23 for 16 hrs. Total RNA isolation, IL-23R mRNA expression was assessed as in (A) and the data represent one of three independent experiments. (C). Differentiated Th17 cells were washed, starved in serum free medium for 4 hrs and then stimulated with recombinant IL-23 for 15 min. Cell lysates were run on Western blot and probed with mAbs to either total Stat3 and phospho-Stat3 (Y705). Loading control was probed with β-actin mAb. (D). Fold change of phospho-STAT3 in Th17 cell lysate stimulated with IL-23 compared with untreated cell lysate. The phospho-Stat3 was measured by ELISA and the phospho-Stat3 in untreated samples was set to be 1.0. The data represent one of two independent experiments.
Impaired Th17 cytokine production in IRAK4 KI Th17 cells is IL-1-dependent
To examine whether IL-1-mediated signaling is essential for Th17 differentiation and Th17 cytokine expression induced by IL-23 in differentiated Th17 cells, we differentiated naïve CD4 T cells in the presence or absence of the IL-1 antagonist, IL-1RA. Gene expression of Th17 cell signature molecules, IL-17, IL-17F and IL-23 receptor, was measured by real-time PCR following differentiation for 4 days. As shown in Fig. 6A, the expression of IL-17, IL-17F and IL-23R mRNAs was induced as expected (without IL-1RA) upon Th17 differentiation, but was completely inhibited in the presence of IL-1RA, indicating that Th17 differentiation is highly dependent on the IL-1- mediated signaling pathway. To determine whether IL-1 is also involved in IL-23- mediated Th17 cell maintenance, we washed and re-plated differentiated Th17 cells and then stimulated with IL-23 for an additional 16 hrs in the presence or absence of IL-1RA. The levels of IL-17, IL-17F and IL-23R mRNAs in Th17 cells following IL-23 treatment remained high as we expected; however, adding IL-1RA into the medium completely abolished the expression of Th17 cell signature molecules including IL-23R under these same conditions (Fig. 6B). This indicates that IL-1 is critical for Th17 differentiation and IL-23-mediated continuity or stability of Th17 polarization. Together, these data are consistent with our observations in IRAK-4 KI mice as well as in IL-1R1 deficient mice we studied (data not shown) and from the previous studies (25) in which either loss of IRAK4 kinase activity or IL-1 receptor expression led to the complete inhibition of IL-23-induced IL-17 production. In addition, these data support the conclusion that IL-1is an essential component in regulating Th17 differentiation.
Figure 6. IL-1RA blocks Th17 cell differentiation.
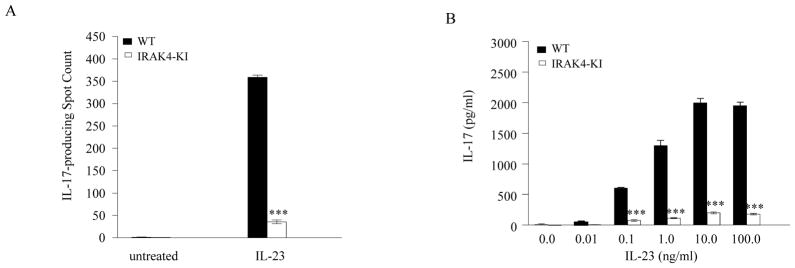
(A). Purified naïve WT CD4+ T cells were differentiated into Th17 cells with plate-bound anti-CD3 and anti-CD28 mAbs, IL-6, and TGF-β in the presence or absence of IL-1RA (1 μg/ml) for 4 days. Total mRNA was then purified and the expression of IL-17, IL-17F, and IL-23R was assessed by real-time PCR and normalized to β-actin mRNA. The data represent one of three independent experiments. (B). A portion of differentiated Th17 cells in the absence of IL-1RA from (A) were washed, re-plated in medium, and incubated in the presence or absence of recombinant IL-23 or IL-23 and IL-1RA for 16 hrs. Total mRNAs were isolated from cells and IL-17, IL-17F, and IL-23R mRNA expression was quantitated by real-time PCR. The expression level was normalized to β-actin mRNA. The data represent one of three independent experiments. Asterisk *** denotes p < 0.001.
Discussion
IRAK4 is a Ser/Thr kinase that plays an essential role in TLR and IL-1 receptor signaling in innate immunity (33–35). The central function of IRAK4 in vivo and the role of its kinase activity in innate immunity have recently been revealed by studying both genetically altered mice and patients with IRAK4 mutations (27–29). However, its function in the adaptive immune response is less clear or even controversial. In this article, we report that IRAK4 plays a critical role in Th17-mediated autoimmune disease by regulating Th17 cell polarization and the response of these cells to IL-23. The loss of IRAK4 kinase activity reduced MOG35-55-induced EAE disease incidence and progression in mice with reduced infiltrating inflammatory cells and abolished IL-17 expression in the CNS (Fig. 1 and 3). IRAK4 kinase activity was shown to be essential for increased expression of IL-23 receptor mRNA in differentiated Th17 cells, which consequently led to the impaired continuous IL-23 receptor expression, Stat3 activation and Th17 cytokine expression by IL-23 in IRAK4 KI Th17 cells. More importantly, the impaired Th17 cytokine expression in IRAK4 KI Th17 cells is completely IL-1 dependent. Neutralizing IL-1 with IL-1RA completely abolished IL-23 receptor expression and Th17 differentiation, further supporting the conclusion that IL-1-IRAK4- mediated signaling pathway is essential for Th17 differentiation. Our results provide a functional mechanism of IL-1-mediated Th17 cytokine production and demonstrate the essential role for IRAK4 kinase activity in controlling production of Th17 cytokine and Th17-mediated autoimmune disease.
The IL-1R-mediated signaling pathway involves a cascade of kinases organized by multiple adapter molecules that form into signaling complexes (35). Interestingly, IRAK4 was found to be abundantly expressed in mouse CD4+ T cells (data not shown), suggesting that IRAK4 may play an important role in T cell function. Recently, Suzuki et al showed that IRAK4 was recruited to T cell lipid rafts upon TCR stimulation resulting in activation of downstream kinases including ZAP70 and PKCθ. They concluded that IRAK4 is essential in TCR-induced T cell signaling (28). In contrast, we show here that both WT and IRAK4 KI mice exhibited normal T cell immune responses to MOG35-55 peptide immunization as demonstrated by similarities in T cell proliferation and cytokine expression. However, IRAK4 KI mice showed reduced expression of IL-17 and were significantly resistant to EAE induction and progression (Fig. 1). More importantly, IRAK4 was selectively involved in regulating IL-17 expression (Fig. 2 and 3), since the antigen-stimulated primed T cells from both WT and IRAK4 KI mice produced similar levels of Th1 and Th2 cytokines. Similarly, anti-CD3 antibody-induced polyclonal stimulation of T cells from WT and IRAK4 KI mice also induced similar numbers of IL-2-producing cells and similar levels of IL-2 protein (data not shown), suggesting that IRAK4-mediated IL-17 expression is specific rather than a general immune suppressive effect in mice lacking IRAK4 kinase activity. Unlike IRAK4 KI mice, we have previously shown that deficiency of PKCθ in mice leads to impaired T cell responses including T cell proliferation and IL-2 production, indicating that IRAK4 unlikely signals directly through PKCθ for T cell function as suggested previously (28, 36). Interestingly, both PKCθ deficient and IRAK4 KI mice exhibited reduced IL-17 production in response to antigen stimulation compared with WT mice (36) (see Fig. 2B), suggesting that both IRAK4 and PKCθ are involved in regulating IL-17 expression in T cells, probably via different pathways.
Th17 cells are the key pathogenic T cells involved in inflammatory and autoimmune diseases and can be differentiated in vitro by several cytokines including TGF-β and IL-6 (37–40). A recent study by Sutton et al shows that IL-23 alone is necessary but not sufficient for inducing IL-17 expression and requires IL-1R signaling (25). As described previously by many others (39, 40), the Th17 cell differentiation conditions we used in which there was no exogenous IL-1 added still leads to Th17 differentiation and Th17 cytokine expression. However, our data suggests that IL-1 is essential for Th17 differentiation (Fig. 6). It is conceivable that the undetectable levels of IL-1 were present in the culture to support optimal Th17 differentiation. The source of IL-1 under these conditions is not known, but we can not exclude the possibility that low levels are present in serum or produced by cells. Consistent with this notion is our finding that recombinant IL-1RA resulted in complete inhibition of Th17 differentiation. Along the similar lines, recent studies suggest that low levels of TGF-β presented in serum and/or produced by impure cell preparations contradict previous claims that TGF-β was not an essential Th17 differentiation factor for human CD4+ T cells (23, 24). Another recent study by Veldhoen et al. also showed that natural agonists for aryl hydrocarbon receptor in culture medium can affect optimal differentiation of Th17 cells, suggesting the possibility that factors present in the experimental assay system could affect directly or indirectly Th17 differentiation (41). In fact, our preliminary data suggest that Th17 cells express mRNA of both IL-1β and IL-1R during differentiation (data not shown). Thus, further experiments will be warranted to identify the sources of IL-1 in our assay system, which will further clarify the role of IL-1 in Th17 differentiation in both humans and mice.
Upon IL-1R/TLR activation, IRAK1 is rapidly phosphorylated by IRAK4, which leads to hyper-phosphorylated IRAK1 via auto-phosphorylation. Unlike IRAK4, however, the importance of its kinase activity has been controversial (42). Previous study by Deng et al. shows that IRAK1 deficient mice are resistant to EAE and T cells exhibit impaired priming, failing to proliferate or secret IFN-γ in response to Ag, regardless of the adjuvant used (43). In contrast, the absence of IRAK4 kinase activity in IRAK4 KI mice led to impaired Th17 differentiation and Th17 cytokine production without affecting T cell priming, Th1 differentiation, or antigen-induced IFN-γ production (Fig. 2 and data not shown). The precise mechanisms for the discrepancy between IRAK1 deficiency and IRAK4 KI mice are unclear. In our IRAK4 KI mice, both IRAK4 and IRAK1 protein are intact; therefore, it is possible that IRAK1 might be activated in the absence of IRAK4 kinase activity, which is essential for the normal T cell priming and IFN-γ production. Alternatively, IRAK1 deficiency could modulate immune response through an impaired adjuvant effect on antigen presenting cells as a result of suboptimal TLR activation. Consistent with this notion, Th1 development was less affected after prolonged Ag immunization with complete adjuvant (43), suggesting that IRAK1 plays an important role in APC function and T cell responses. More recent advances in our understanding of the EAE model indicate that Th17, not Th1 cells are the pathogenic cells triggering disease (19, 20). Therefore, it will be essential to investigate whether the absence of IRAK1 or its kinase activity will also affect Th17 differentiation during EAE induction.
The expression of IL-23 receptor mRNA is increased by IL-6 during Th17 cell differentiation and synergistically enhanced by IL-23, which further promotes Th17 differentiation (45). In the absence of IL-1 or IRAK4 kinase activity, TGF-β and IL-6 were not able to induce the expression of IL-23R on naïve CD4+ T cells (Fig. 5), indicating that the one of the mechanisms of IL-1 pathway is to regulate the expression IL-23 receptor mRNA. Recently, Stritesky et al. showed that IL-1β can augment the ability of IL-23 to maintain the Th17 phenotype in long-term cultures (44). Similar to our observation, they showed that IL-23R expression was upregulated during Th17 differentiation, and subsequently maintained during IL-23 treatment. In addition, Th17 cytokine expression in the IL-23-treated Th17 cells was further augmented by IL-1β. Interestingly, unlike the profound effect of IL-1 signaling on IL-23R expression observed in our IRAK4 KI Th17 cells (Fig. 4 and 5), the expression of IL-23R was not affected by IL-1β treatment in this assay system from this study, and the precise role of IL-1β in these processes was also unclear (44). In our study, the absence of IL-1 signaling by inactivation of IRAK4 kinase (IRAK4 KI mice) or by neutralizing IL-1 function with IL-1RA led to the reduction of IL-23R expression during Th17 differentiation as well as IL-23-mediated Th17 maintenance (Fig. 5 and 6). This differential effect of IL-1 on IL-23R expression between the studies of Stritesky’s and ours could be due to several possibilities, including the pre-existing low levels of IL-1β in the assay or simple due to different assay conditions. In our study, naïve CD4+ T cells lacking IRAK4 kinase activity or IL-1 signaling were used for Th17 differentiation, whereas Stritesky et al. differentiated Th17 cells using naive WT CD4+ T cells which IL-1 signaling would be intact. Therefore, future studies to understand the mechanism of IL-1 signaling pathway in initial Th17 differentiation and later maintenance of Th17 phenotype are warranted.
Previous studies suggest that TGF-β and IL-6 are sufficient to induce the expression of Th17 transcription factors, RORα and RORγt, which are essential for Th17 differentiation and IL-23 receptor expression (38–39, 46). However, there was no reproducible difference in the induction of either RORα or RORγt mRNAs between WT and IRAK4 KI CD4+ T cells under our Th17 differentiation conditions (data not shown), suggesting that IL-1-mediated Th17 cytokine expression might act through an RORα and RORγt-independent pathway. Alternatively, IRAK4 could potentially function downstream of the IL-23 receptor. Inactivative IRAK4 could block IL-23R signaling and lead to inhibition of IL-23-induced Th17 cytokine expression in IRAK4 KI Th17 cells. However, we currently do not have any direct experimental evidence to support this hypothesis. Taken together, our study provides a key link between the IL-1/IL-1R/IRAK4 signaling pathway, expression of IL-23R, and Th17 differentiation. Understanding the precise mechanism by which these signaling components regulate Th17 cell polarization and cytokine expression will require further studies. This work demonstrates that IRAK4 kinase activity is critical in Th17 cytokine production, provides evidence for IL-1 function in regulating Th17 cell differentiation and cytokine expression, and suggests that IRAK4 is a promising target for the treatment of Th17 cell-mediated inflammatory diseases.
Supplementary Material
Acknowledgments
We thank Dr. Jonathon D. Sedgwick for critical reading and discussion and Carol Broderick for immunizing mice, Larry Mann and Shuang Luo for technical support.
The work is also supported by funding from National Multiple Sclerosis Society awarded to Xiaoxia Li (RG 4056-A-1)
References
- 1.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. Review. [DOI] [PubMed] [Google Scholar]
- 2.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. Review. [DOI] [PubMed] [Google Scholar]
- 3.Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–155. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matusevicius D, Kivisäkk P, He B, Kostulas N, Ozenci V, Fredrikson S, Link H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–104. doi: 10.1177/135245859900500206. [DOI] [PubMed] [Google Scholar]
- 5.Kim HR, Kim HS, Park MK, Cho ML, Lee SH, Kim HY. The clinical role of IL-23p19 in patients with rheumatoid arthritis. Scand J Rheumatol. 2007;36:259–264. doi: 10.1080/03009740701286813. [DOI] [PubMed] [Google Scholar]
- 6.Teunissen MB, Koomen CW, de Waal Malefyt R, Wierenga EA, Bos JD. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645–649. doi: 10.1046/j.1523-1747.1998.00347.x. [DOI] [PubMed] [Google Scholar]
- 7.Rahman MS, Yang J, Shan LY, Unruh H, Yang X, Halayko AJ, Gounni AS. IL-17R activation of human airway smooth muscle cells induces CXCL-8 production via a transcriptional-dependent mechanism. Clin Immunol. 2005;115:268–276. doi: 10.1016/j.clim.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 8.Hwang SY, Kim JY, Kim KW, Park MK, Moon Y, Kim WU, Kim HY. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-kappaB- and PI3-kinase/Akt-dependent pathways. Arthritis Res Ther. 2004;6:R120–128. doi: 10.1186/ar1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 10.Chu CQ, Swart D, Alcorn D, Tocker J, Elkon KB. Interferon-gamma regulates susceptibility to collagen-induced arthritis through suppression of interleukin-17. Arthritis Rheum. 2007;56:1145–1151. doi: 10.1002/art.22453. [DOI] [PubMed] [Google Scholar]
- 11.Taylor PC. Antibody therapy for rheumatoid arthritis. Curr Opin Pharmacol. 2003;3:323–328. doi: 10.1016/s1471-4892(03)00032-8. Review. [DOI] [PubMed] [Google Scholar]
- 12.Hofstetter HH, Ibrahim SM, Koczan D, Kruse N, Weishaupt A, Toyka KV, Gold R. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell Immunol. 2005;237:123–130. doi: 10.1016/j.cellimm.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 13.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–944. doi: 10.1038/nri954. Review. [DOI] [PubMed] [Google Scholar]
- 14.McGuirk P, Mills KH. Pathogen-specific regulatory T cells provoke a shift in the Th1/Th2 paradigm in immunity to infectious diseases. Trends Immunol. 2002;23:450–455. doi: 10.1016/s1471-4906(02)02288-3. Review. [DOI] [PubMed] [Google Scholar]
- 15.Bettelli E, Korn T, Kuchroo VK. Th17: the third member of the effector T cell trilogy. Curr Opin Immunol. 2007;19:652–657. doi: 10.1016/j.coi.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stockinger B, Veldhoen M. Differentiation and function of Th17 T cells. Curr Opin Immunol. 2007;19:281–286. doi: 10.1016/j.coi.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Harrington LE, Mangan PR, Weaver CT. Expanding the effector CD4 T-cell repertoire: the Th17 lineage. Curr Opin Immunol. 2006;18:349–356. doi: 10.1016/j.coi.2006.03.017. Review. [DOI] [PubMed] [Google Scholar]
- 18.Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, Watowich SS, Tian Q, Jetten AM, Dong C. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kikly K, Liu L, Na S, Sedgwick JD. The IL-23/Th(17) axis: therapeutic targets for autoimmune inflammation. Curr Opin Immunol. 2006;18:670–675. doi: 10.1016/j.coi.2006.09.008. Review. [DOI] [PubMed] [Google Scholar]
- 20.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thakker P, Leach MW, Kuang W, Benoit SE, Leonard JP, Marusic S. IL-23 is critical in the induction but not in the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:2589–2598. doi: 10.4049/jimmunol.178.4.2589. [DOI] [PubMed] [Google Scholar]
- 22.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and anti29 inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1857. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Volpe E, Servant E, Zollinger R, Bogiatzi S, Hupe P, Barillot B, Soumelis V. A critical function for transforming growth factor-[beta], interleukin 23 and proinflammatory cytokines in driving and modulating human TH-17 responses. Nature Immunol. 2008;9:650–657. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 24.Manel N, Unutmaz D, Littman DR. The differentiation of human TH-17 cells requires transforming growth factor-[beta] and induction of the nuclear receptor RORγt. Nature Immunol. 2008;9:641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203:1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X. IRAK4 in TLR/IL-1R signaling: Possible clinical applications. Eur J Immunol. 2008;38:614–618. doi: 10.1002/eji.200838161. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki N, Suzuki S, Eriksson U, Hara H, Mirtosis C, Chen NJ, Wada T, Bouchard D, Hwang I, Takeda K, Fujita T, Der S, Penninger JM, Akira S, Saito T, Yeh WC. IL-1R-associated kinase 4 is required for lipopolysaccharide-induced activation of APC. J Immunol. 2003;171:6065–6071. doi: 10.4049/jimmunol.171.11.6065. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki N, Suzuki S, Millar DG, Unno M, Hara H, Calzascia T, Yamasaki S, Yokosuka T, Chen N, Elford AR, Suzuki J, Takeuchi A, Mirtsos C, Bouchard D, Ohashi PS, Yeh WC, Saito TA. critical role for the innate immune signaling molecule IRAK-4 in T cell activation. Science. 2006;311:1927–1932. doi: 10.1126/science.1124256. [DOI] [PubMed] [Google Scholar]
- 29.Kawagoe T, Sato S, Jung A, Yamamoto M, Matsui K, Kato H, Uematsu S, Takeuchi O, Akira S. Essential role of IRAK-4 protein and its kinase activity in Toll-like receptor-mediated immune responses but not in TCR signaling. J Exp Med. 2007;204:1013–1024. doi: 10.1084/jem.20061523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim TW, Staschke K, Bulek K, Yao J, Peters K, Oh KH, Vandenburg Y, Xiao H, Qian W, Hamilton T, Min B, Sen G, Gilmour R, Li X. A critical role for IRAK4 kinase activity in Toll-like receptor-mediated innate immunity. Exp Med. 2007;204:1025–1036. doi: 10.1084/jem.20061825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaiko GE, Horva JC, Beagley KW, Hansbro PM. Immunological decision-making: how does the immune system decide to mount a helper T-cell response? Immunology. 2008;123:326–338. doi: 10.1111/j.1365-2567.2007.02719.x. review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Z, Laurence A, O’Shea JJ. Signal transduction pathways and transcriptional regulation in the control of Th17 differentiation. Semin Immunol. 2007;19:400–408. doi: 10.1016/j.smim.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuglstatter A, Villaseñor AG, Shaw D, Lee SW, Tsing S, Niu L, Song KW, Barnett JW, Browner MF. Cutting Edge: IL-1 receptor-associated kinase 4 structures reveal novel features and multiple conformations. J Immunol. 2007;178:2641–2645. doi: 10.4049/jimmunol.178.5.2641. [DOI] [PubMed] [Google Scholar]
- 34.Lasker MV, Gajjar MM, Nair SK. Cutting edge: molecular structure of the IL-1R-associated kinase-4 death domain and its implications for TLR signaling. J Immunol. 2005;175:4175–4179. doi: 10.4049/jimmunol.175.7.4175. [DOI] [PubMed] [Google Scholar]
- 35.Li X, Qin J. Modulation of Toll-interleukin 1 receptor mediated signaling. J Mol Med. 2005;83:258–266. doi: 10.1007/s00109-004-0622-4. review. [DOI] [PubMed] [Google Scholar]
- 36.Tan SL, Zhao J, Bi C, Chen XC, Hepburn DL, Wang J, Sedgwick JD, Chintalacharuvu SR, Na S. Resistance to experimental autoimmune encephalomyelitis and impaired IL-17 production in protein kinase C theta-deficient mice. J Immunol. 2006;176:2872–2879. doi: 10.4049/jimmunol.176.5.2872. [DOI] [PubMed] [Google Scholar]
- 37.Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol. 2007;19:409–417. doi: 10.1016/j.smim.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stockinger B, Veldhoen M. Differentiation and function of Th17 T cells. Curr Opin Immunol. 2007;19:281–286. doi: 10.1016/j.coi.2007.04.005. Review. [DOI] [PubMed] [Google Scholar]
- 39.Mangan PR, Harrington LE, O’Quinn DB, Hems WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 40.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 41.Veldhoen M, Hirota K, Christensen K, O’Garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med. 2009;206:43–49. doi: 10.1084/jem.20081438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gottipati S, Rao NL, Fung-Leung WP. IRAK1: a critical signaling mediator of innate immunity. Cellular Signaling. 2008;20:269–276. doi: 10.1016/j.cellsig.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 43.Deng C, Radu C, Diab A, Tsen MF, Hussain R, Cowdery JS, Racke MK, Thomas JA. IL-1 Receptor-Associated Kinase 1 Regulates Susceptibility to Organ-Specific Autoimmunity. J Immunol. 2003;170:2833–2842. doi: 10.4049/jimmunol.170.6.2833. [DOI] [PubMed] [Google Scholar]
- 44.Stritesky GL, Yeh N, Kaplan MH. IL-23 Promotes Maintenance but Not Commitment to the Th17 Lineage. J Immunol. 2008;181:5948–5955. doi: 10.4049/jimmunol.181.9.5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thakker P, Leach MW, Kuang W, Benoit SE, Leonard JP, Marusic S. IL-23 is critical in the induction but not in the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:2589–2598. doi: 10.4049/jimmunol.178.4.2589. [DOI] [PubMed] [Google Scholar]
- 46.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory help T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 47.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.