

ABSTRACT
Specialization of bacteria in a new niche is associated with genome repertoire changes, and speciation in bacterial specialists is associated with genome reduction. Here, we tested a signature-tagged mutant library of 3,456 Bartonella birtlesii clones to detect mutants that could grow rapidly in vitro. Overall, we found 124 mutants that grew faster than the parental wild-type strain in vitro. We sequenced the genomes of the four mutants with the most rapid growth (formed visible colonies in only 1 to 2 days compared with 5 days for the wild type) and compared them to the parental isolate genome. We found that the number of disrupted genes associated with translation in the 124 rapid-growth clones was significantly higher than the number of genes involved in translation in the full genome (P < 10−6). Analysis of transposon integration in the genome of the four most rapidly growing clones revealed that one clone lacked one of the two wild-type RNA ribosomal operons. Finally, one of the four clones did not induce bacteremia in our mouse model, whereas infection with the other three resulted in a significantly lower bacterial count in blood than that with the wild-type strain.
IMPORTANCE
Here, we show that specialization in a specific niche could be caused by the disruption of critical genes. Most of these genes were involved in translation, and we show that evolution of obligate parasitism bacteria was specifically associated with disruption of translation system-encoding genes.
Introduction
The specialization of bacteria in a new niche is associated (as a cause or a consequence) with genome repertoire changes. When bacteria are allopatric, i.e., they have no opportunity to exchange genes with other organisms (1), genome modifications are restricted to gene duplications and, more commonly, gene mutations or deletions. Allopatric speciation is generally associated with genome reduction, and bacterial specialists, especially pathogens, have smaller genome repertoires than less-specialized bacteria (2). In a stable environment, such as an intracellular environment, many of the genes coming from a free-living lifestyle are no longer needed and are prone to be inactivated and eventually lost (3).
In a seminal work, Louis Pasteur propagated Pasteurella multocida, the etiologic agent of avian cholera, and showed that the fittest bacteria in axenic culture (“agar fitness”) were nonpathogenic, i.e., less fit, in hens. The loss of pathogenicity was later shown to be caused by gene deletions (4). In several pathogens, such as Shigella, Salmonella, Yersinia pestis, or Francisella tularensis, certain genes must be inactivated or deleted for expression of the pathogenicity, and these genes are known as antivirulence genes (5). This concept of evolutionary force driving gene loss is that a gene may be advantageous in one environment but deleterious in another one (5). Comparative genomics has shown that this phenomenon is general and that specialized bacteria, including highly pathogenic bacteria, have fewer genes encoding factors than their epidemic neighbors (6), with the exception of toxins and toxin-antitoxin modules (1). Supporting this hypothesis, Glass et al. (7) used global transposon mutagenesis to generate 100 different mutants of Mycoplasma genitalium with disrupted genes, and 3 mutants grew more rapidly than the native M. genitalium. The deleted genes in these mutants encoded malate dehydrogenase and 2 hypothetical proteins.
To evaluate whether this phenomenon can be generalized to other bacteria, we tested a signature-tagged mutant library of more than 3,000 Bartonella birtlesii clones (8) obtained from the B. birtlesii wild-type reference genome (9) for rapid growth in vitro. One of the rapid-growth mutants had, surprisingly, a deletion of one of the two RNA ribosomal operons present in the B. birtlesii genome. Therefore, we sequenced the original strain and four rapid-growth mutants to identify genome modifications associated with higher “agar” fitness, defined as growth rate increase on a blood agar plate.
RESULTS
Rapid-growing clones of B. birtlesii.
Using a 96-well puncture machine, small volumes (3 to 5 µl) of B. birtlesii mutant clones were plated on 5% sheep’s blood agar. Of the 3,456 clones tested, 124 were able to grow more rapidly (1 to 4 days) than the wild-type strain of B. birtlesii (5 days). A list of these clones is provided in Table S1 in the supplemental material. Among these 124 clones, four (E4, E7, E11, and H12) grew to full size in only one or two days (see Table S1).
Gene sequence analysis.
Using the Genome Walker universal kit and the restriction enzyme DraI, the 124 mutant clones of B. birtlesii were PCR amplified and sequenced using both forward and reverse primers, and the sequences were compared with the B. birtlesii genome sequence (9) via BLAST analysis. The results are shown in Table S1 in the supplemental material. Among the 124 rapid-growth clones, 43 of the disrupted genes could be identified confidently with a known COG (cluster of orthologous group of proteins) function (see Table S1). We found that 16/43 of these genes disrupted in the rapid-growth clones belong to the translation COG (see Fig. S1), including three clones with disruptions of the 16S rRNA and 23S rRNA genes (H12, 43C4, and 43B10, respectively; see Table S1) and one clone with disruption of the 30S ribosomal protein S18/S6 (43A1; see Table S1). Moreover, the number of disrupted genes associated with translation in the rapid-growth clones was significantly higher than that expected by chance based on the number of translation genes in the genome (P < 10−6). Finally, we compare the COG functions of these 43 disrupted genes to the 100 COGs previously found to be conserved in all bacteria (10), and we found that 24 disrupted genes belong to this set of genes, including 16 genes involved in translation. Conversely, none of the remaining 19 disrupted genes belonging to the set of the 100 orthologous genes lost in specialists was associated with translation system (P < 10−6) (see Fig. S1).
Transposon integration in the genome.
Analysis of transposon integration in the genomes of the four most rapidly growing clones compared to the genome of the wild-type strain revealed that the transposon was integrated one or several times in each clone. While the H12 mutant contained one transposon integration, the E11 and E4 mutants contained two integrations each, and the E7 mutant contained three integrations. Figure 1 shows the sites of transposon integration in the wild-type genome and in the disrupted genes of the four clones. The integration sites were similar to those produced by Genome Walker analysis (see Table S1 in the supplemental material) and included the following: integration of the transposon in the 16S rRNA gene of mutant H12; integrations in a noncoding region flanked by a hypothetical protein and a tRNA-methyltransferase, an adenine-specific DNA methyl-transferase, and a hypothetical protein for E7; integrations in an outer membrane efflux protein and in a noncoding region flanked by a filamentous hemagglutinin protein and a hypothetical protein for E11; and integrations in a hypothetical protein and in a noncoding region flanked by phosphoserine aminotransferase and a phage-related lysozyme protein for E4. Figure 2 summarizes the locations of the transposon integrations in the different mutants.
FIG 1 .
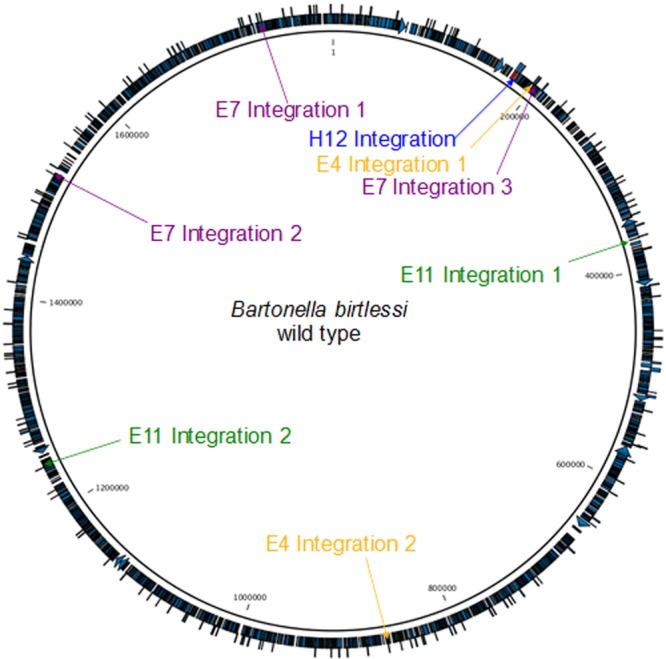
Transposon integration sites in the Bartonella birtlesii genome. The transposon integration events in the H12, E11, E7, and E4 strains are coded in blue, green, violet, and light orange, respectively.
FIG 2 .

Sites of transposon integration and identification of the disrupted genes in the four clones with rapid growth.
Specific phylogenies for adenine-specific DNA methyltransferase (clone E7) and outer membrane efflux protein (clone E11).
The protein sequence of adenine-specific DNA methyltransferase from clone E7 was studied extensively, and 437 full genome sequences available in NCBI were searched for homologs. Proteins with high levels of similarity were found in 420 genomes, and the remaining 17 genomes contained hypothetical proteins with high similarity to this sequence. Interestingly, these hypothetical proteins included the putative adenine modification methyltransferase from the prophages Pseudomonas syringae pv. phaseolicola and Xylella fastidiosa. The outer membrane efflux protein from clone E11 was homologous to outer membrane factor lipoproteins of the NodT family and likely part of a type I secretion system. This gene belongs to an operon in the resistance-nodulation-division (RND) efflux system that is well conserved in the Bartonella genus and Rhizobiales (see Fig. S2 in the supplemental material).
Mouse infection model.
The wild-type strain of B. birtlesii induced bacteremia in our mouse model, but similar to the negative control, clone E11 did not induce bacteremia. Clones E4, E7, and H12 produced a significantly lower bacterial count in the blood (UFC/ml) than the wild-type strain (Fig. 3).
FIG 3 .
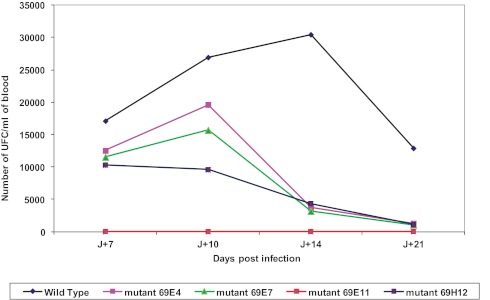
Growth kinetics of rapid-growth clones in the mouse model. Bacteremia in BALB/c mice (n = 5) infected with B. birtlesii wild-type (WT) and mutant strains from day 0 to day 21. *, P < 0.05; **, P < 0.01.
DISCUSSION
In this report, using a signature-tagged mutant library of more than 3,000 Bartonella birtlesii clones, we found that 124 clones were associated with significantly faster growth in axenic medium, i.e., an increased “agar fitness” in this artificial environment that mimicked rapid specialization. These mutants became specialists for in vitro culture on agar. Whole-genome sequencing was done for the four most rapidly growing clones to decipher the specific sites of integration of the transposon in the chromosome. Interestingly we found that the transposon could integrate the chromosome several times for the same clone. This has been also recently reported for a signature-tagged mutant library in Borrelia burgdorferi with an average of 2.68 unique insertions per kilobyte of DNA (11). One of the rapid-growth strains was unable to colonize its former niche—mouse erythrocytes (8)—and the three others had slower growth in mice (lower fitness). This phenomenon resembles the reduced virulence of Pasteurella multocida, the etiologic agent of fowl cholera, after specialization for in vitro culture. This model of evolution has been well described and starts with the principle that gene loss or acquisition required for fitness in one niche (here, the agar fitness) could inhibit the fitness of the microbe in another environment (here, in the animal model). Bacterial specialization involves potentially dramatic gene loss in both extra- and intracellular bacteria. The evolution of specialized bacteria is associated with various types of genome reduction, including decreases in gene number, GC percentage, and the numbers of both complete and intact ribosomal operons (10, 12, 13). Previously, we were surprised by the number of abnormal or split ribosomal operons in specialized bacteria, particularly intracellular bacteria (10). It appeared that the presence of an abnormal ribosomal operon was a triggering event in several groups of specialized bacteria and that this event occurred independently at least eight times. The presence of an abnormal ribosomal operon appeared to be especially common during the emergence of the Rickettsiales but also in Helicobacter pylori, Leptospira species, and the group containing Mycoplasma and Buchnera. This phenomenon is unlikely to be explained by chance. Similarly, in this work, we found that 16 of the genes disrupted in the rapid-growth clones belong to the translation COG, including ribosomal operon disruption in 4 rapidly growing clones. Our first hypothesis was that the number of active genes in a very restrictive niche must be lower than in a widely changing environment, because the translation apparatus does not need to be used extensively. This hypothesis suggests that if the bacteria do not use as many ribosomal operons in the new niche, they are lost (14). Interestingly, analysis of the signature-tagged mutant library recently reported in B. burgdorferi revealed that each of the two 23S rRNA-encoding loci and one of two 5S rRNA genes were disrupted, whereas the single 16S rRNA locus was not (11). Nevertheless, growth rates for these clones were not evaluated in this study (11).
The disruption of the rRNA operon is followed by increased fitness and multiplication (10). We speculated that the better translational machinery allows the low-level translation of nonessential protein, slowing the growth rate of the bacteria. This rescue system may permit the bacteria to face environmental changes and maintain their adaptability, despite its inefficiency in the current niche. Conversely, bacteria that do not produce unused proteins have the direct advantage of saving the cost of protein translation (15). However, they lose their capability to colonize their former murine niche, as demonstrated in the present study. This hypothesis is consistent with the Mayr proximate-ultimate model of evolution (16, 17) in which limiting the translation capabilities provides an immediate growth rate advantage (proximate) but limits the ability to survive in other niches (UltiMate).
Therefore, we speculate that stringently limiting translation is critical for specialization, as the speciation of specialized bacteria is often correlated to ribosomal operon inactivation (10). Indeed, this confirms the hypothesis that the number of rRNA operons is linked to bacterial ecological strategies (1). To the best of our knowledge, this hypothesis suggested that the duplication of the ribosomal operon was the key component in sympatric bacteria. Here, we demonstrated that rRNA operon disruption is a key factor for specialization in a specific niche.
The adenine-specific DNA methylase is an enzyme that methylates specific DNA targets (GANTC for alphaproteobacteria), resulting in a reduction of the thermodynamic stability of DNA (1) and changes in transcription regulation. In Escherichia coli, mutants lacking this gene exhibit increased spontaneous mutations (mutator phenotype), which may explain the rapid gene loss in specialized bacteria that lack this gene. Furthermore, this gene is critical in host-pathogen interactions, and it is missing from several specialized bacteria. Among the disrupted genes, we found efflux proteins associated with increased growth rate. Although the role of these proteins with growth rate is unknown, there are some experimental evidences that expression of the type III secretion system was associated with a growth penalty in a nonhost environment in Salmonella (18). Finally, the number of deleted genes in rapid growers was significantly higher than the number of conserved genes in specialized bacteria (Fig. 3). Genes associated with transcription and those encoding outer membrane proteins are frequently inactivated; interestingly, the same families of genes are specifically inactivated in E. coli that has been cultured for thousands of passages (19).
Based on the mechanistic approach to pathogenicity, bacterial pathogens were thought to have repertoires for accumulating virulence genes (besides toxins), and Shigella was the paradigm of the Red Queen theory, which postulates an arms race for pathogenicity (6, 10).
Comparative genomics has contradicted the previous dogma and has shown that specialists (including pathogens) and specifically Shigella (6, 20) have smaller genome repertoires. Rickettsia (1, 21) and mycobacterial (10) “killer bug” genomes contain a subset of the genes from their neighbors’ “nonkiller” genomes. Finally, it has been clarified that the most obvious changes from nonpathogenic E. coli to pathogenic and specialized Shigella were gene losses (22, 23).
Here, we show that specialization in a specific niche could be caused by the disruption of critical genes. Most of these genes were involved in translation, and we show that evolution of obligate parasitism bacteria was specifically associated with inactivation of translation system-encoding genes and that unique gene inactivation is a major source of specialization.
MATERIALS AND METHODS
Culture of B. birtlesii wild-type and mutant clones.
Wild-type B. birtlesii (IBS 135T, CIP 106691T) (24) and 3,456 signature-tagged mutant B. birtlesii clones (8) were cultured on Columbia agar containing 5% sheep’s blood in a humidified atmosphere containing 5% CO2 at 37°C. These clones were screened for rapid growth using a 96-well puncture machine and compared to the B. birtlesii parental isolate. Bacterial growth was monitored every day until growth appeared, and the number of days required for bacterial growth was recorded for each clone.
Identification of genes disrupted by transposon insertion.
The Genome Walker universal kit (Clontech Laboratories, United States) was used to identify genes disrupted by transposon insertion. Rapid-growth clones of B. birtlesii mutants were enzymatically digested with the DraI restriction enzyme. The regions upstream and downstream of Tn were sequenced, and the obtained sequences were analyzed and identified using BLAST analysis at the NCBI website (http://www.ncbi.nlm.nih.gov/Blast.cgi). Primers specific to the identified genes were used for PCR amplification and sequencing to confirm the identification of the disrupted genes associated with the rapid-growth phenotypes.
Phylogeny of the disrupted genes in rapid-growth clones.
The protein sequences of disrupted genes were analyzed by BLASTP against NR. The first 20 best BLAST hits were retrieved and subjected to multiple alignment using MUSCLE. Multiple alignments were curated using trimAL, and a maximum likelihood phylogeny reconstruction was generated by phyML. Finally, the protein sequences of disrupted genes from rapid-growth clones with known functions were compared to the set of 100 orthologous genes lost in all obligate intracellular bacteria (specialists) (10).
Genome sequencing, assembly, and annotation.
The genomes of four rapid-growth clones of B. birtlesii (clones E4, E11, H12, and E7) were fully sequenced with the 454 pyrosequencing platform (454 Life Sciences, Branford, CT) (25). A library of paired-end fragments was created following the manufacturer’s instructions. This library was sequenced using the GS titanium sequencer. Reads from each strain were assembled into contigs and scaffolds using Newbler 2.53 (454 Life Sciences, Branford, CT). The assembly was verified using the CLC Genomics software (CLC Bio, Massachusetts).
Identification of transposon insertion sites.
Mutant reads were mapped to the wild-type B. birtlesii genome (9) with CLC Genomics software (CLC Bio, Massachusetts) to identify the genes disrupted by the 2,281-bp transposon (8). The sequences upstream and downstream of the transposon insertion were identified by alignment with the wild-type reference genome (9) and verified at the NCBI website (http://www.ncbi.nlm.nih.gov/Blast.cgi).
Bartonella birtlesii mouse infection model.
Thirty-week-old female BALB/c mice from Charles River Laboratories were housed in an animal facility (5 animals/cage) and allowed to acclimate to the facility and the diet for at least 5 days prior to infection. Food and water were provided ad libitum (8).
B. birtlesii wild-type or signature-tagged mutants were grown on agar plates for 5 days. Each strain was suspended in phosphate-buffered saline (PBS) immediately before infection. Five mice were infected with 5 × 107 CFU (10 µl culture with an optical density at 595 nm [OD595] of 1) of each strain by tail vein injection. On days 7, 10, 14, and 21 postinfection, 50 µl blood was taken from the tail vein of each infected mouse. Bacteria were released from erythrocytes by a freeze-thaw cycle and plated on CBA-km medium (mutants) or CBA medium (wild type). After 10 days, bacterial colonies were counted, and the degree of bacteremia was calculated.
Nucleotide sequence accession numbers.
The complete genome sequences of the four Bartonella birtlesii mutants have been submitted to GenBank under the bioproject numbers PRJNA168998 (strain H12), PRJNA168999 (strain E7), PRJNA169001 (strain E11), and PRJNA169101 (strain E4).
SUPPLEMENTAL MATERIAL
Functional categories of the 43 rapid-growth clones with known functions. Download
The RND efflux system operon. Download
Putative functions of disrupted genes in Bartonella birtlesii mutants, as determined by BLASTn.
ACKNOWLEDGMENTS
We thank Linda Hadjadj for technical assistance and American Journal Experts for comments on the manuscript.
This work was funded by the French Centre National de la Recherche Scientifique (CNRS), INRA, and Region Île de France.
D.R., J.M.R., M.V.-T., and C.D. conceived and designed the experiments. M.V.-T., V.M., W.S., D.L.R., and C.R. performed the experiments. D.R., J.M.R., M.V.-T., V.M., W.S., G.G., C.D., D.L.R., and D.R. analyzed the data. C.R., G.G., D.L.R., and V.M. contributed reagents, materials, and analysis tools. J.M.R., M.V.-T., V.M., W.S., C.D., and D.R. wrote the paper.
Footnotes
Citation Rolain JM, Vayssier-Taussat M, Saisongkorh W, Merhej V, Gimenez G, Robert C, Le Rhun D, Dehio C, Raoult D. 2013. Partial disruption of translational and posttranslational machinery reshapes growth rates of Bartonella birtlesii. mBio 4(2):e00115-13. doi:10.1128/mBio.00115-13.
REFERENCES
- 1. Georgiades K, Merhej V, El Karkouri K, Raoult D, Pontarotti P. 2011. Gene gain and loss events in rickettsia and Orientia species. Biol. Direct. 6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tamas I, Klasson L, Canbäck B, Näslund AK, Eriksson AS, Wernegreen JJ, Sandström JP, Moran NA, Andersson SG. 2002. 50 million years of genomic stasis in endosymbiotic bacteria. Science 296:2376–2379 [DOI] [PubMed] [Google Scholar]
- 3. Moran NA. 2002. Microbial minimalism: genome reduction in bacterial pathogens. Cell 108:583–586 [DOI] [PubMed] [Google Scholar]
- 4. Laslett D, Canback B. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32:11–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bliven KA, Maurelli AT. 2012. Antivirulence genes: insights into pathogen evolution through gene loss. Infect. Immun. 80:4061–4070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Georgiades K, Raoult D. 2011. Genomes of the most dangerous epidemic bacteria have a virulence repertoire characterized by fewer genes but more toxin-antitoxin modules. PLoS One 6:e17962 http://dx.doi.org/10.1371/journal.pone.0017962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Glass JI, Assad-Garcia N, Alperovich N, Yooseph S, Lewis MR, Maruf M, Hutchison CA, Smith HO, Venter JC. 2006. Essential genes of a minimal bacterium. Proc. Natl. Acad. Sci. U. S. A. 103:425–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vayssier-Taussat M, Le Rhun D, Deng HK, Biville F, Cescau S, Danchin A, Marignac G, Lenaour E, Boulouis HJ, Mavris M, Arnaud L, Yang H, Wang J, Quebatte M, Engel P, Saenz H, Dehio C. 2010. The Trw type IV secretion system of Bartonella mediates host-specific adhesion to erythrocytes. PLoS Pathog. 6:e1000946 http://dx.doi.org/10.1371/journal.ppat.1000946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rolain JM, Vayssier-Taussat M, Gimenez G, Robert C, Fournier PE, Raoult D. 2012. Genome sequence of Bartonella birtlesii, a bacterium isolated from small rodents Apodemus. J. Bacteriol. 194:4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Merhej V, Royer-Carenzi M, Pontarotti P, Raoult D. 2009. Massive comparative genomic analysis reveals convergent evolution of specialized bacteria. Biol. Direct. 4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin T, Gao L, Zhang C, Odeh E, Jacobs MB, Coutte L, Chaconas G, Philipp MT, Norris SJ. 2012. Analysis of an ordered, comprehensive STM mutant library in infectious Borrelia burgdorferi: insights into the genes required for mouse infectivity. PLoS One 7:e47532 http://dx.doi.org/10.1371/journal.pone.0047532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Andersson JO, Andersson SG. 1999. Insights into the evolutionary process of genome degradation. Curr. Opin. Genet. Dev. 9:664–671 [DOI] [PubMed] [Google Scholar]
- 13. Moran NA, Wernegreen JJ. 2000. Lifestyle evolution in symbiotic bacteria: insights from genomics. Trends Ecol. Evol. 15:321–326 [DOI] [PubMed] [Google Scholar]
- 14. Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35:3100–3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moran NA. 2002. Microbial minimalism: genome reduction in bacterial pathogens. Cell 108:583–586 [DOI] [PubMed] [Google Scholar]
- 16. Laland KN, Sterelny K, Odling-Smee J, Hoppitt W, Uller T. 2011. Cause and effect in biology revisited: is Mayr’s proximate-ultimate dichotomy still useful? Science 334:1512–1516 [DOI] [PubMed] [Google Scholar]
- 17. Mayr E. 1961. Cause and effect in biology. Science 134:1501–1506 [DOI] [PubMed] [Google Scholar]
- 18. Sturm A, Heinemann M, Arnoldini M, Benecke A, Ackermann M, Benz M, Dormann J, Hardt WD. 2011. The cost of virulence: retarded growth of Salmonella Typhimurium cells expressing type III secretion system 1. PLoS Pathog. 7:e1002143 http://dx.doi.org/10.1371/journal.ppat.1002143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barrick JE, Yu DS, Yoon SH, Jeong H, Oh TK, Schneider D, Lenski RE, Kim JF. 2009. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461:1243–1247 [DOI] [PubMed] [Google Scholar]
- 20. Maurelli AT, Fernandez RE, Bloch CA, Rode CK, Fasano A. 1998. Black holes and bacterial pathogenicity: a large genomic deletion that enhances the virulence of Shigella spp. and enteroinvasive Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 95:3943–3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ogata H, Audic S, Renesto-Audiffren P, Fournier PE, Barbe V, Samson D, Roux V, Cossart P, Weissenbach J, Claverie JM, Raoult D. 2001. Mechanisms of evolution in Rickettsia conorii and R. prowazekii. Science 293:2093–2098 [DOI] [PubMed] [Google Scholar]
- 22. Barbagallo M, Di Martino ML, Marcocci L, Pietrangeli P, de Carolis E, Casalino M, Colonna B, Prosseda G. 2011. A new piece of the Shigella pathogenicity puzzle: spermidine accumulation by silencing of the speG gene. PLoS One 6:e27226 http://dx.doi.org/10.1371/journal.pone.0027226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feng Y, Chen Z, Liu SL. 2011. Gene decay in Shigella as an incipient stage of host-adaptation. PLoS One 6:e27754 http://dx.doi.org/10.1371/journal.pone.0027754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bermond D, Heller R, Barrat F, Delacour G, Dehio C, Alliot A, Monteil H, Chomel B, Boulouis HJ, Piémont Y. 2000. Bartonella birtlesii sp. nov. isolated from small mammals (Apodemus spp.). Int. J. Syst. Evol. Microbiol. 50:1973–1979 [DOI] [PubMed] [Google Scholar]
- 25. Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM, Rothberg JM. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Functional categories of the 43 rapid-growth clones with known functions. Download
The RND efflux system operon. Download
Putative functions of disrupted genes in Bartonella birtlesii mutants, as determined by BLASTn.